

Abstract
Prothrombin (FII) is activated to α-thrombin (IIa) by prothrombinase. Prothrombinase is composed of a catalytic subunit, factor Xa (fXa), and a regulatory subunit, factor Va (fVa), assembled on a membrane surface in the presence of divalent metal ions. We constructed, expressed, and purified several mutated recombinant FII (rFII) molecules within the previously determined fVa-dependent binding site for fXa (amino acid region 473–487 of FII). rFII molecules bearing overlapping deletions within this significant region first established the minimal stretch of amino acids required for the fVa-dependent recognition exosite for fXa in prothrombinase within the amino acid sequence Ser478–Val479–Leu480–Gln481–Val482. Single, double, and triple point mutations within this stretch of rFII allowed for the identification of Leu480 and Gln481 as the two essential amino acids responsible for the enhanced activation of FII by prothrombinase. Unanticipated results demonstrated that although recombinant wild type α-thrombin and rIIaS478A were able to induce clotting and activate factor V and factor VIII with rates similar to the plasma-derived molecule, rIIaSLQ→AAA with mutations S478A/L480A/Q481A was deficient in clotting activity and unable to efficiently activate the pro-cofactors. This molecule was also impaired in protein C activation. Similar results were obtained with rIIaΔSLQ (where rIIaΔSLQ is recombinant human α-thrombin with amino acids Ser478/Leu480/Gln481 deleted). These data provide new evidence demonstrating that amino acid sequence Leu480–Gln481: 1) is crucial for proper recognition of the fVa-dependent site(s) for fXa within prothrombinase on FII, required for efficient initial cleavage of FII at Arg320; and 2) is compulsory for appropriate tethering of fV, fVIII, and protein C required for their timely activation by IIa.
Keywords: blood, coagulation factor, prothrombin, thrombin, thrombosis, activation, factor Xa, factor v, mutation, prothrombinase
Introduction
In the presence of a procoagulant membrane surface and divalent metal ions, factor Va (fVa)3 binds factor Xa (fXa) to form prothrombinase. Prothrombinase is the two-subunit enzymatic complex where the non-enzymatic regulatory subunit (fVa) controls the rate and directs cleavage of prothrombin (FII) by the catalytic subunit (fXa) at two spatially distinct sites resulting in timely α-thrombin (IIa) formation at the place of vascular injury (1–3). Cleavage at Arg271 and Arg320 of FII is required to form the active serine protease IIa. The essential IIa molecule bears strong homology with other serine protease enzymes, such as activated protein C (APC), chymotrypsin, and fXa. Several different numberings of IIa residues appear in the literature based on either the chymotrypsin numbering (4) or IIa numbering (5, 6) or the entire FII sequence (7). The latter nomenclature is used herein with the appropriate chymotrypsin numbering in parentheses when required for comparison with the existing data in the literature.
Historically, it has been shown that in the absence of fVa, initial cleavage at Arg271 of FII results in the generation of the inactive intermediate prethrombin-2 and fragment 1·2. Further cleavage of prethrombin-2 at Arg320 generates IIa (prethrombin-2 pathway) (8–21). Concurrent with the appearance of excess fVa during clotting and in the presence of a procoagulant surface, the order of cleavages is reversed, and initial cleavage at Arg320 generates a transient enzymatically active intermediate, meizothrombin, that has much higher catalytic efficiency than IIa toward chromogenic substrates usually employed to assess IIa activity (18, 22–24). Meizothrombin is next cleaved at Arg271 resulting in the generation of IIa and fragment 1·2 (meizothrombin pathway). Although efficient cleavage at each site requires the presence of phospholipids, initial cleavage at Arg320 is entirely fVa-dependent.
In the absence of fVa, the two activation cleavage sites are not readily available, and FII is activated at a slow non-physiological rate by membrane-bound fXa alone. Interactions between fXa and FII are known to exist in the presence and absence of fVa; however, the enhanced activity of fXa within prothrombinase toward both activating cleavage sites is controlled solely by the membrane-bound non-enzymatic cofactor (3, 16, 17, 25, 26). Consequently, the innate process of coagulation rests on specific molecular interactions involved in the fVa-dependent activation of FII by prothrombinase. In relation to fXa alone, the relative rate of IIa formation by prothrombinase is increased by 300,000-fold because of the increase in the rates of both FII cleavages. This increase is mainly associated with a large (3,000-fold) increase in the kcat value of fXa within prothrombinase with a 100-fold decrease in the Km value of the enzyme (16). This substantial increase in enzymatic activity resulting in rapid and physiologically relevant IIa generation at the place of vascular injury is credited through precise and unique interactions of the cofactor with specific amino acids affiliated with both membrane-bound fXa and membrane-bound FII as recently demonstrated (27). Accordingly, the introduction of the non-enzymatic cofactor into prothrombinase equips the organism's coagulation artillery necessary for the explosive arrest of vasculature bleeding.
Factor V (fV) is a large quiescent multidomain (A1-A2-B-A3-C1-C2) protein that circulates in blood at a concentration of 20 nm (28–31). Three sequential cleavages of fV at Arg709, Arg1018, and Arg1545 (29, 32–34) by IIa and/or fXa (35, 36) release the B domain and promote formation of the active cofactor fVa. FII circulates abundantly in blood at a concentration of 1.4 μm as the zymogen form of the serine protease IIa (7, 37). Mature FII protein is composed of a region containing several post-translationally modified γ-carboxyglutamic acid residues (described as the Gla domain, residues 1–46), followed by two Kringle domains (residues 65–143 and 170–248, respectively) and a serine protease domain (residues 272–579, see Fig. 1). FII contains three linkers as follows: linker 1 (residues 47–64) connects the Gla domain to kringle-1; linker 2 (residues 144–169) connects the two kringles; and linker 3 (residues 249–284) connects kringle-2 to the A-chain portion of IIa (7, 38) (Fig. 1).
FIGURE 1.
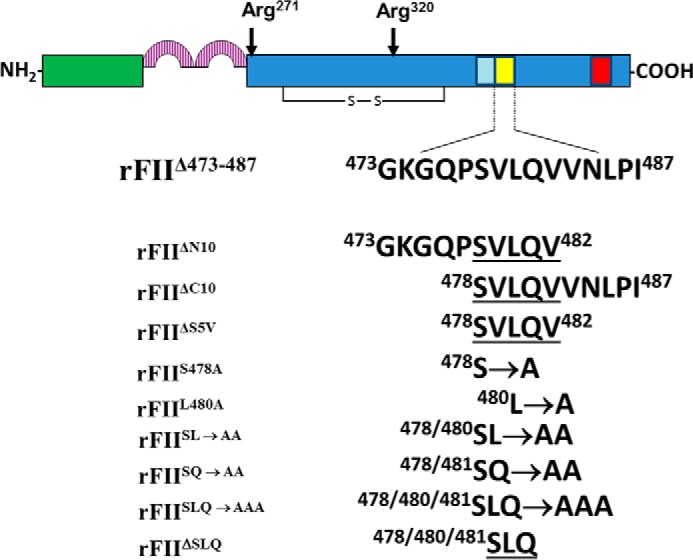
Schematic of FII. FII is converted to IIa through two fXa-catalyzed cleavages at Arg271 and at Arg320 resulting in IIa formation. The red rectangle denotes the fVa-independent site for fXa on FII (57), and the yellow rectangle represents the fVa-dependent site for fXa (56, 95) studied herein. The light blue rectangle denotes the amino acids composing (pro)exosite I (50). All mutants created, stably transfected, purified to homogeneity, and used in the study are shown together with their assigned name used throughout this work.
The necessary fVa-dependent activation of FII by prothrombinase is a widely studied mechanism of coagulation but is still poorly understood. Numerous fVa-binding sites are acknowledged to exist on FII. Earlier investigations have shown the existence of binding sites on FII for fVa in each of the kringle domains (39–41) and within the Gla domain (42). Furthermore, significant protein-protein interactions between the acidic COOH-terminal region of fVa and a region rich in basic amino acids of FII have been inferred and characterized indirectly by employing molecular techniques involving specific hirudin-like ligands and the anion-binding (pro)exosite I (ABE I) of FII derivatives, as well as directly using a specific acidic peptide derived from the COOH-terminal region of the fVa heavy chain and recombinant fVa molecules (43–49). Site-directed mutagenesis of the basic residues in the proenzyme generated a recombinant FII molecule impaired in its ability to be activated by fully assembled prothrombinase (50). Although a crystal structure and a model of fVa have been available for some time now (51, 52), the crucial interaction of the acidic hirudin-like COOH-terminal portion of the heavy chain of the cofactor with FII required for efficient IIa formation was initially ignored because it was missing from the crystal structure of the cofactor (51). This interaction was further discounted without providing any solid evidence (53) despite initial findings by Guinto and Esmon (54) and more recent original findings from our laboratory (47–49). A very recent model of prothrombinase using as a template the crystal structure of prothrombinase from the snake venom of Pseudonaja textilis verified and established the critical role of the acidic COOH-terminal region of fVa heavy chain for optimal rates of FII cleavage at two spatially distinct sites by prothrombinase resulting in timely IIa formation at the place of vascular injury (27, 55).
Additional studies with several recombinant prethrombin-1 molecules, where seven critical basic amino acids within (pro)exosite I were individually changed to glutamic acid, confirmed the interaction of (pro)exosite I with fVa acidic regions (50). Notably, the data revealed that although mutated prethrombin-1 is a poor substrate for prothrombinase, the same molecule was activated by membrane-bound fXa alone with similar rates as wild type prethrombin-1. Supplementary to these studies, Yegneswaran et al. (56), utilizing synthetic peptide derived from a highly conserved region of FII, postulated the existence of an fVa-dependent binding exosite for fXa within the sequence 473–487 of FII (chymotrypsin numbering 149D-163 (4)) that is in close spatial arrangement to (pro)exosite I. The same authors have also identified an fVa-independent site for fXa on prothrombin (amino acids 557–571) (57).
This study was initiated to identify and investigate the identity and role of the minimum required amino acid stretch within sequence 473–487 of FII that is conserved in a wide range of mammalian species and regulates peptide bond specificity and FII activation by prothrombinase in an fVa-dependent manner. Our findings identify for the first time two specific amino acids within FII that have a dual role. They are required for efficient fVa-dependent tethering of fXa needed for timely FII cleavage at Arg320 and IIa formation, while also serving an important role in directing efficient cleavage of IIa's physiological substrates. The latter is a prerequisite for expression of optimal physiological IIa activity.
Experimental Procedures
Materials
Phenylmethylsulfonyl fluoride (PMSF), o-phenylenediamine dihydrochloride, Hepes, Trizma (Tris base), and Coomassie Blue R-250 were purchased from Sigma. fV-deficient plasma was purchased from Research Protein Inc. (Essex Junction, VT). Secondary anti-mouse, anti-sheep, and anti-equine IgG coupled to peroxidase were from Southern Biotechnology Associates, Inc. (Birmingham, AL). l-α-Phosphatidylserine (PS) and l-α-phosphatidylcholine (PC) were from Avanti Polar Lipids (Alabaster, AL). Chemiluminescent reagent ECL Plus, heparin-Sepharose, and Mono Q 5/50 columns were from GE Healthcare. Normal reference plasma and chromogenic substrate Spectrozyme-TH were from American Diagnostica Inc. (Greenwich, CT). S-2238 was from AnaSpec (Fremont, CA); recombiPlasTin used in the clotting assays was purchased from Instrumentation Laboratory Co. (Lexington, MA). The reversible fluorescent IIa inhibitor dansylarginine-N-(3-ethyl-1,5-pentanediyl)amide (DAPA), human plasma-derived protein C, human plasma-derived IIa, human plasma-derived FII, and FII-deficient plasma were purchased from Hematologic Technologies Inc. (Essex Junction, VT). The purified human plasma-derived protein C preparation used contained both heavy chain isoforms that are activated to APC with similar rates as described earlier (58, 59). Human fXa was purchased from Enzyme Research Laboratories (South Bend, IN). The plasmid pZEM229R-lite encoding human recombinant prothrombin (rFII) was a generous gift from Dr. Kathleen Berkner (Cleveland Clinic Foundation, Cleveland, OH). QuikChange® II XL site-directed mutagenesis kit was obtained from Agilent Technologies Genomics (Santa Clara, CA). All molecular biology and tissue culture reagents, specific primers, and medium were obtained from Gibco, Invitrogen, or as indicated. Monoclonal antibodies to fV (αHFVHC17 and αHFVLC9), monoclonal antibody αHFV1 coupled to Sepharose used to purify plasma and recombinant fV molecules, and a polyclonal antibody to FII used for immunoblotting experiments during rFII production were provided by Dr. Kenneth G. Mann (Department of Biochemistry, University of Vermont, Burlington). Plasma factor V (fVplasma) and plasma fVa (fVaplasma) were purified as described previously (60–62).
Construction of rFII Molecules
To investigate the importance of amino acid region 473–487 of the serine protease domain of FII, we first constructed a recombinant mutant FII molecule with this region deleted (rFIIΔ473–487) using Stratagene's QuikChange® site-directed mutagenesis kit and the pZEM229R-lite plasmid. rFIIΔ473–487 was constructed using the mutagenic primers 5′-GAG ACG TGG ACA GCC AAC GTT GTG GAG CGG CCG GTC TGC AAG-3′ (sense) and 5′-CTT GCA GAC CGG CCG CTC CAC AAC GTT GGC TGT CCA CGT CTC-3′ (antisense) (corresponding to the 473GKGQPSVLQVVNLPI487 deletion). The mutation was confirmed by DNA sequencing (DNA Analysis Facility, Department of Molecular Cardiology at The Learner Research Institute, Cleveland Clinic). To further investigate the minimum sequence of amino acids required for the fVa-dependent fXa binding on FII within the region 473–487 of the serine protease domain, several rFII molecules with the mutations denoted as rFIIΔN10 (rFIIΔN10 is recombinant human prothrombin missing amino acids GKGQPSVLQV), rFIIΔC10, rFIIΔS5V, rFIIS478A (rFIIS478A is recombinant human prothrombin with themutation S478A), rFIIL480A, rFIISL→AA, rFIISQ→AA, and rFIISLQ→AAA (where rFIISLQ→AA is recombinant human prothrombin with the mutation S478A/L480A/Q481A) were constructed using Stratagene's QuikChange® site-directed mutagenesis kit and the pZEM229R-lite plasmid. First, overlapping deletions in the region 473–487 were constructed using the mutagenic primers for rFIIΔN10 5′-G ACG TGG ACA GCC AAC GTT GTG AAC CTG CCC ATT GTG GAG-3′ (sense) and 5′-CTC CAC AAT GGG CAG GTT CAC AAC GTT GGC TGT CCA CGT C-3′ (antisense) (corresponding to the 473GKGQPSVLQV482 deletion), whereas mutagenic primers used for rFIIΔC10 were 5′-GTT GGT AAG GGG CAG CCC GTG GAG CGG CCG GTC TGC-3′ (sense) and 5′-GCA GAC CGG CCG CTC CAC GGG CTG CCC CTT ACC AAC-3′ (antisense) (corresponding to the 478SVLQVVNLPI487 deletion). Similarly, the middle deletion of the overlapping mutations rFIIΔS5V was constructed using the mutagenic primers 5′-GTT GGT AAG GGG CAG CCC GTG AAC CTG CCC ATT GTG-3′ (sense) and 5′-CAC AAT GGG CAG GTT CAC GGG CTG CCC CTT ACC AAC-3′ (antisense) (corresponding to the 478SVLQV482 middle deletion). Next, within the sequence 478–482 several point mutations were made based on amino acid solvent exposure and homology within other proteins. The first rFII point alanine mutation rFIIS478A was constructed using the mutagenic primers 5′-GGT AAG GGG CAG CCC GCA GTC CTG CAG GTG-3′ (sense) and 5′-CAC CTG CAG GAC TGC GGG CTG CCC CTT ACC-3′ (antisense) (corresponding to the S478A mutation). The next single point mutation rFIIL480A was constructed using the mutagenic primers 5′-GGG CAG CCC AGT GTC GCG CAG GTG GTG AAC CTG CCC-3′ (sense) and 5′-GGG CAG GTT CAC CAC CTG CGC GAC ACT GGG CTG CCC-3′ (antisense) (corresponding to the L480A mutation). In addition to single point mutations, we constructed two double alanine mutations, rFIISL→AA and rFIISQ→AA, within the stretch 478–482 of FII. For rFIISL→AA, we used the mutagenic primers 5′-GGT AAG GGG CAG CCC GCA GTC GCG CAG GTG GTG AAC CTG-3′ (sense) and 5′-CAG GTT CAC CAC CTG CGC CAC TGC GGG CTG CCC CTT ACC-3′ (antisense) (corresponding to the S478A/L480A mutation). Also, for rFIISQ→AA (where rFIISQ→AA is recombinant human prothrombin with the mutation S478A/Q481A), we used the mutagenic primers 5′-GGT AAG GGG CAG CCC GCA GTC CTG GCG GTG GTG AAC CTG-3′ (sense) and 5′-CAG GTT CAC CAC CGC CAG GAC TGC GGG CTG CCC CTT ACC-3′ (antisense) (corresponding to the S478A/Q481A mutation). Finally, we constructed a rFII molecule with a triple mutation, rFIISLQ→AAA with the mutagenic primers 5′-GGT AAG GGG CAG CCC GCA GTC GCG GCG GTG GTG AAC CTG-3′ (sense) and 5′-CAG GTT CAC CAC CGC CGC GAC TGC GGG CTG CCC CTT ACC-3′ (antisense) (corresponding to the S478A/L480A/Q481A mutation) and an rFII molecule with Ser478/Leu480/Gln481 deleted (rFIIΔSLQ) using the mutagenic primers 5′-GGT AAG GGG CAG CCC GTC GTG GTG AAC CTG CCC-3′ (sense) and 5′-GGG CAG GTT CAC CAC GAC GGG CTG CCC CTT ACC-3′ (antisense). All deletions and point mutations were confirmed by DNA sequencing (DNA Analysis Facility, Department of Molecular Cardiology, The Lerner Research Institute, Cleveland Clinic).
Expression of Wild Type and Mutant rFII Molecules in Mammalian Cells
rFII expression in baby hamster kidney (BHK-21) cells has been described previously in detail (63). Briefly, BHK-21 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with fetal bovine serum (10%), and a streptomycin/penicillin (1%) mixture. Isolated plasmids (4–6 μg) for wild type and mutant rFII molecules were transfected into the BHK-21 cells using a lipid-based transfection reagent, Lipofectamine (Invitrogen), according to the manufacturer's instructions. Following an incubation period of 48 h, DMEM was supplemented with fetal bovine serum (10%), streptomycin/penicillin (1%) mixture, and methotrexate (1 μm) and added to the cells. After 3 weeks of treatment with the selection medium, colonies were isolated, grown, and screened for levels of rFII expression by Western blot analysis using a monoclonal antibody and compared with plasma-derived FII as a standard (1 μg/ml). Identification of the highest secreting rFII clone was further used in large scale protein expression with serum-free Opti-MEM supplemented with ZnCl2 (50 μm), vitamin K1 (10 μg/ml), and penicillin/streptomycin/Fungizone (1% v/v) mixture, and the medium were collected every 2 days for 2–3 weeks. Following collection, the media were stored at −80 °C until the desired amount (usually 4 liters) was obtained and used for purification.
Purification of rFII Molecules
Purification of rFII was performed through a well established protocol previously described in detail (63). Briefly, collected media were thawed, filtered (0.45 μm), and loaded on a tandem column setup of Amberlite XAD2 and Q-Sepharose. Following the complete addition of medium to the two columns, the Q-Sepharose column was separated and washed with TBS (0.02 m Tris, 150 mm NaCl, pH 7.4). The bound material containing rFII on the Q-Sepharose was eluted with 0.02 m Tris, 0.5 m NaCl, pH 7.4. The material was treated with barium citrate, and the isolated pellet was dissolved in a minimum volume of EDTA (0.5 m, pH 7.4). The dissolved pellet was dialyzed twice in fresh TBS (two times, 4 liters) and filtered (0.45 μm) prior to being loaded onto a General Electric FPLC instrument, equipped with a strong anionic exchanger Mono Q 5/50 column. The column was equilibrated in TBS, and a stepwise gradient of calcium (0–50 mm) in TBS was used to isolate fully γ-carboxylated rFII. Tubes containing the rFII molecules were concentrated using an appropriate Millipore Centricon (Billerica, MA), and aliquots were frozen at −80 °C to avoid repeated freeze-thaw cycles. Following purification and before any experiment, all rFII molecules were characterized as extensively described below.
The level of γ-carboxylation of all rFII molecules was determined at the Protein Chemistry Facility, Texas A&M University, by alkaline hydrolysis followed by amino acid analysis as described (64, 65). All purified molecules were found to be properly carboxylated (Table 1). To verify that rFIIWT and rFIIΔ473–487 are processed at the appropriate cleavage sites when incubated with prothrombinase or fXa alone and produced the expected fragments, the recombinant proteins were incubated with PCPS vesicles and fXa in the presence and absence of fVa. Following gel electrophoresis, fragments were identified following NH2-terminal sequencing from PVDF membranes (see below). All fragments derived from the recombinant FII molecules have the expected NH2-terminal sequence following cleavage by either prothrombinase or membrane-bound fXa alone (data not shown).
TABLE 1.
Physical properties, clotting times, and rate of cleavage of various rFII molecules in the presence of a fixed concentration of membrane-bound fXa or in the presence of prothrombinase
| Mutant | mol of Gla/mol of protein | Clotting timea | Rate of FII molecule cleavage by membrane-bound fXa aloneb (nm FII consumed·s−1·fXa−1) | Rate of FII molecules cleavage by prothrombinaseb (nm FII consumed·s−1·fXa−1) |
|---|---|---|---|---|
| s | ||||
| FIIplasma | 10 ± 1 | 12.7 ± 0.12 | 0.13 ± 0.03 (0.94)c | 27.5 ± 4.2 (0.99)c |
| rFIIWT | 9.1 ± 0.9 | 12.2 ± 0.16 | 0.1 ± 0.012 (0.99) | 22.8 ± 3.8 (0.98) |
| rFIIΔ473–487 | 9.5 ± 0.1 | >120d | 0.53 ± 0.013 (0.99) | 1.0 ± 0.26 (0.92) |
| rFIIΔN10 | 12.0 ± 1 | >120d | 0.7 ± 0.03 (0.98) | 1.5 ± 0.34 (0.94) |
| rFIIΔC10 | 12.5 ± 1 | >120d | 0.35 ± 0.04 (0.99) | 1.3 ± 0.09 (0.99) |
| rFIIΔS5V | 11 ± 1 | > 120d | 0.8 ± 0.03 (0.99) | 1.8 ± 0.12 (0.99) |
| rFIIS478A | 10.8 ± 1 | 11.75 ± 0.32 | 0.25 ± 0.015 (0.99) | 24.9 ± 4.6 (0.97) |
| rFIIL480A | 10.1 ± 1 | 29.8 ± 0.41 | 0.6 ± 0.02 (0.99) | 28.8 ± 2.8 (0.98) |
| rFIISL→AA | 9.5 ± 0.9 | 28.7 ± 0.35 | 0.63 ± 0.03 (0.99) | 18.2 ± 3.7 (0.99) |
| rFIISQ→AA | 10 ± 1 | 23.2 ± 0.3 | 0.23 ± 0.025 (0.98) | 35.9 ± 3.8 (0.99) |
| rFIISLQ→AAA | 10.6 ± 1 | 116.2 ± 0.65 | 0.6 ± 0.04 (0.99) | 1.23 ± 0.12 (0.98) |
| rFIIΔSLQ | 10.6 ± 1.1 | >120d | 0.22 ± 0.04 (0.95) | 1.84 ± 0.15 (0.99) |
a Clotting times were determined using FII-deficient plasma as described under “Experimental Procedures” in quadruplicate.
b The rates of rFII consumption were obtained following scanning densitometry of gels studying rFII activation. Some of the gels used are shown in Figs. 3–5. The final rate of rFII consumption in the presence of membrane-bound fXa or prothrombinase was calculated using the apparent first-order rate constant, k (s−1), obtained directly from the graph following plotting of the data as described under “Experimental Procedures.”
c The numbers in parentheses represent the goodness of fit (R2) to the equation representing first-order exponential decay using the software Prizm from where the first-order rate constant was obtained.
d No clotting time could be detected following a 120-s incubation time period.
The fact that the rFIIΔ473–487 molecule contains a 15-amino acid deletion was verified by cDNA sequencing. However, in view of the surprising and unexpected data presented herein, it was important to confirm the existence of the deletion in the purified recombinant protein. This was accomplished by mass spectrometry. Briefly, following activation of rFIIΔ473–487 and FIIplasma by prothrombinase, aliquots were analyzed in triplicate under reducing conditions on an SDS-12% PAGE. Following staining/destaining, the B-chain of ΙΙa was excised from the gel, and the proteins were reduced and alkylated with iodoacetamide. Digestion (in gel) was accomplished with porcine trypsin. Analysis of the resulting peptides was performed with an α-cyanohydroxycinnamic acid (matrix) Kratos Axima CFR MALDI-TOF mass spectrometer (reflector mode; 25,000 accelerating voltage) in the Protein Chemistry Laboratory, Texas A & M University, under the direction of Dr. Larry Dangott. The data obtained were compared with the peptide map following digestion of the B chain of IIa obtained from ExPASy/Swiss-Prot and verified the existence of the deletion in rFIIΔ473–487. Similar experimental work performed with some other mutant molecules demonstrated that the rFII molecules described herein are fully carboxylated, can be appropriately processed by prothrombinase and fXa alone, and do indeed contain the expected deletion/mutations.
Gel Electrophoresis, Western Blotting, and Amino Acid Sequence from PVDF Membranes
SDS-PAGE was performed according to the method of Laemmli (66), using 9.5% gels following reduction with 2% β-mercaptoethanol. Screening for high levels of rFII-secreting clones was performed by Western blotting using PVDF according to a modified method initially described by Towbin et al. (67). Successfully transferred proteins were visualized by chemiluminescence using ECL Plus reagents following incubation with a polyclonal antibody specific to prethrombin-1. In some experiments, proteins were transferred to PVDF membranes and stained with Coomassie Blue, and NH2-terminal sequencing analysis was performed at the Biomolecular Resource Facility at the University of Texas Medical Branch (Galveston TX) as described previously in detail by our laboratory (68).
Studies of the Pathway for FII Activation by Gel Electrophoresis
The investigation of the activation rates of plasma-derived and of all rFII molecules, cleavage and activation by fXa alone or prothrombinase was performed according to a protocol previously described by our laboratory using plasma-derived FII or rFII (47, 49, 69). The calculation of the rates of all FII molecules consumption by fXa alone or by prothrombinase were performed as described previously with the software Prizm (GraphPad) (47, 49, 69).
Kinetic Titrations of Prothrombinase
To investigate the kinetic constants (Km and kcat) of prothrombinase, assays with a set amount of plasma-derived fVa and fXa (as described in the legend to the figures) were executed as described by our laboratory in many instances (47, 49, 69, 70). The initial rate of IIa generation was analyzed with the software Prizm (GraphPad) according to the Michaelis-Menten equation, and all final numbers reported are derived directly from the graphs. Each experiment used to report final numbers was run at least in duplicate, and the goodness of fit (R2) for every model tested is provided under the “Results.” The change in transition-state stabilization free energy, which measures the effect of the mutations in rFII, was calculated for the double and triple mutants as extensively detailed in the literature and previously reported by our laboratory (71–77).
Recombinant Thrombin Activity
rFII molecules were converted to rIIa by 1 nm prothrombinase. Full conversion of rFII to rIIa under these conditions was verified by gel electrophoresis. The chromogenic substrate S-2238 was used to assess rIIa activity by employing serial dilutions of the enzyme in Tris-NaCl buffer in the presence of 0.1% PEG 8000. The final concentrations of S-2238 used in the reactions were 0.94, 1.87, 3.75, 7.50, 15, and 60 μm. The reaction was started by the addition of 4 nm rIIa. The data were obtained at 1 min using a SpectraMax M2 plate reader (Molecular Devices). The optical density was automatically adjusted for a 1-cm pathlength, and the Vmax was calculated from the optical density using the established extinction coefficient of S-2238 at room temperature (78) following plotting of the data to the Michaelis-Menten equation using the software Prizm (GraphPad).
Activation of fV and fVIII by rIIa
rFII molecules were converted to rIIa by 1 nm prothrombinase. Full conversion of rFII to rIIa under the conditions described was assessed by gel electrophoresis. The resulting wild type and mutant rIIa were assessed for their ability to cleave and activate the cofactors over time, by SDS-PAGE. Reaction mixtures containing either 500 nm plasma-derived human fV or recombinant human fVIII were diluted in Tris-NaCl buffer in the presence of Ca2+. The final concentration of rIIa in the mixtures was 4 nm.
Activation of Protein C by Plasma-derived IIa or rIIa
rFII molecules described herein were converted to IIa by 1 nm prothrombinase. Full conversion of rFII to rIIa under the conditions described was assessed by gel electrophoresis. The resulting IIa molecules were assessed for their ability to cleave and activate protein C in the presence of thrombomodulin and PCPS vesicles according to a procedure previously described (79) in Tris-buffered saline with Ca2+. Protein C activation was verified by SDS-PAGE under reducing conditions. The final concentration of IIa in all mixtures was 8 nm. Gels were stained with Coomassie Brilliant Blue.
FII Clotting Assay
To assess the function of all FII molecules in whole plasma, a clotting assay using FII-deficient plasma was employed. The clotting assay was performed as described previously (80), and the time needed for formation of a fibrin clot was monitored at 37 °C using a Diagnostica Stago STart® 4 hemostasis analyzer as described previously (80). The analyzer was set up to automatically measure the time to clot up to 120 s.
Structural Analysis
To evaluate the structural features of the Ser478, Leu480, and Gln481 residues, crystal structures of FII and IIa were superimposed and compared. The three human FII crystal structures that have been reported, show similar conformations for the residues of interest and neighboring regions; the highest resolution of these structures was chosen for detailed analysis (38). From the many human IIa crystal structures that are available, several representative examples in different bound states were compared and found to have similar conformations for the region containing the residues of interest. A high resolution structure of unbound IIa was chosen as the representative structure for detailed analysis (81). The program COOT was used to inspect structural features and determine distances (82). AREAIMOL (83–85) was used to calculate the solvent-accessible surface areas for specific residues, and molecular figures were prepared with the PyMOL Molecular Graphics System, version 1.5.0.4 (Schrödinger, LLC).
Results
rFII Expression
To evaluate the minimal amino acid sequence necessary for the fVa-dependent FII activation by prothrombinase within the 473–487 critical amino acid stretch of FII, we stably transfected BHK-21 cell lines according to a previously well defined protocol (63) with several constructs. Mutant rFII molecules prepared were as follows: rFIIΔ473–487 (missing residues 473GKGQPSVLQVVNLPI487), rFIIΔN10 (missing amino acid residues 473GKGQPSVLQV482), and rFIIΔC10 (missing amino acid residues 478SVLQVVNLPI487) (Fig. 1). These three mutant molecules have five amino acids in common (478SVLQV482). We thus proceeded to construct, stably express, and purify to homogeneity rFIIΔS5V (a rFII molecule missing amino acids 478SVLQV482) (Fig. 1). Preliminary experiments with rFIIΔS5V demonstrated that the mutant molecule had no clotting activity similarly to rFIIΔ473–487, rFIIΔN10, and rFIIΔC10. We next proceeded to make single, double, and triple alanine substitutions within this significant region. The following recombinant mutant molecules were made: rFIIS478A (S478A), rFIIL480A (L480A), rFIISL→AA (S478A/L480A), rFIISQ→AA (S478A/Q481A), and rFIISLQ→AAA (S478A/L480A/Q481A) (Fig. 1). It is important to note that rIIaS478A (where rIIaS478A is recombinant human α-thrombin with the mutation S478A) was previously tested and found to behave as rIIaWT (5, 6). We have thus used this mutation as an internal control for all double and triple alanine substitutions of rFII. Finally, and to verify the results obtained with rFIISLQ→AAA, we have also made an rFII molecule with these three amino acids deleted (rFIIΔSLQ). In all experiments, results with the mutant molecules were compared with results obtained with rFIIWT or FIIplasma.
Prothrombin Time
The ability of FII and all rFII molecules to be activated under physiological conditions and to promote fibrin clot formation was first assessed using prothrombin times (PTs) (Fig. 2). The results shown in Table 1 demonstrate that although FIIplasma, rFIIWT, and rFIIS478A had comparable clotting times of 12.7, 12.2, and 11.7 s, respectively, rFIIL480A (where rIIaL480A is recombinant human α-thrombin with the mutation L480A) exhibited a minimal but significant prolonged PT of ∼30 s (Fig. 2). Surprisingly, although rFIISL→AA (where rFIISL→AA is recombinant human prothrombin with the mutation S478A/L480A) and rFIISQ→AA had slow but comparable PTs of ∼30 s, the triple mutant rFIISLQ→AAA was severely ineffective in fibrin clot formation (PT ∼116 s), whereas rFIIΔSLQ (where rFIIΔSLQ is recombinant human prothrombin with amino acids Ser478/Leu480/Gln481 deleted) had a PT around 140 s (data not shown). In contrast, rFIIΔ473–487, rFIIΔN10, rFIIΔC10, and rFIIΔS5V were unable to induce clotting under the conditions described. These functional data demonstrate that either rFIISLQ→AAA or rFIIΔSLQ cannot get activated to rIIa in a timely fashion, or that rIIaSLQ→AAA and/or rIIaΔSLQ formed are catalytically impaired because of the mutations, or both. Because previous data have shown that the S478A transition in IIa is of no consequence for its chromogenic and proteolytic activity (5, 6), overall these results demonstrate for the first time that both Leu480 and Gln481 have a profound effect on IIa generation and/or IIa activity during fibrin clot formation or both.
FIGURE 2.
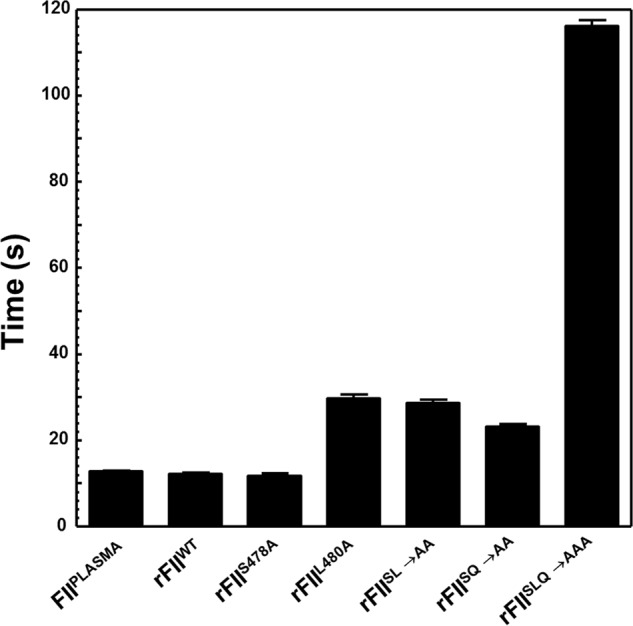
Clotting activity of all forms of FII. The average clotting time found in four different measurements in FII-deficient plasma is shown for all FII/rFII molecules identified at the bottom of the graph.
Activation of rFII Molecules
To ascertain the effect of region 473–487 of FII on its ability to be activated by membrane-bound fXa alone, in the absence of fVa, we assessed the pattern of activation by gel electrophoresis over a 2-h time period (Fig. 3). Fig. 3A shows a control experiment and demonstrates that FIIplasma activation by membrane-bound fXa alone proceeds following initial cleavage at Arg271, through the intermediate prethrombin-2 with very slow gradual appearance of the B-chain of IIa because of inefficient rate of cleavage at Arg320. Surprisingly, with the removal of amino acids 473–487 from prothrombin (Fig. 3B), there is acceleration of rFIIΔ473–487 consumption through initial cleavage at Arg271 that is evident by the prompt appearance of prethrombin-2. Additional examinations of the intensity of the B-chain of thrombin reveal a substantially delayed cleavage at Arg320 of rFIIΔ473–487 compared with rFIIWT resulting in insignificant IIa generation. Scanning densitometry of the gels shown in Fig. 3, A and B, showed that the rate of rFIIΔ473–487 consumption by membrane-bound fXa is ∼4-fold increased compared with the rate of cleavage of rFIIplasma under similar experimental conditions (Table 1). These data suggest that amino acid sequence 473–487 of prothrombin provides a potential obstruction for efficient initial cleavage of prothrombin at Arg271 by membrane-bound fXa alone in the absence of fVa.
FIGURE 3.

SDS-PAGE analyses of FIIplasma and rFIIΔ473–487 activation by membrane-bound fXa alone or prothrombinase. A, rFIIplasma (1.4 μm) in the presence of PCPS vesicles, DAPA, and membrane-bound fXa alone (5 nm). B, rFIIΔ473–487 (1.4 μm) in the presence of PCPS vesicles, DAPA, and membrane-bound fXa alone (5 nm). C, rFIIplasma (1.4 μm) in the presence of PCPS vesicles, DAPA, and prothrombinase (1 nm fXa and 20 nm fVa). D, rFIIΔ473–487 (1.4 μm) in the presence of PCPS vesicles, DAPA, and prothrombinase (1 nm fXa and 20 nm fVa). Aliquots were withdrawn at various time intervals and treated as described (47, 69). M represents the lane with molecular weight markers (from top to bottom): 98,000, 64,000, 50,000, and 36,000, respectively. Lanes 1–19 show samples from the reaction mixture before (0 min) the addition of fXa and 20, 40, 60, 80, 100, 120, 150, 180, 210, and 240 s and 5, 6, 10, 20, 30, 60, 90, and 120 min, respectively, after the addition of fXa. Following scanning densitometry as described under “Experimental Procedures,” the data representing FII consumption as a function of time (s) were plotted using non-linear regression analysis according to the equation representing a first-order exponential decay and the rates of FII consumption using the apparent first-order rate constant, k (s−1), obtained directly from the fitted data, were calculated as described (47) and are reported in Table 1. FII-derived fragments are identified to the right of A–D as follows: FII, prothrombin (amino acid residues 1–579); P1, prethrombin-1 (amino acid residues 156–579); F1·2-A, fragment 1·2-A chain (amino acid residues 1–320); F1·2, fragment 1·2 (amino acid residues 1–271); P2, prethrombin-2 (amino acid residues 272–579); B, B-chain of IIa (amino acid residues 321–579); F1, fragment 1 (amino acid residues 1–155).
To further improve our understanding of the fundamental role of amino acid region 473–487 for FII activation by prothrombinase, we studied the pattern of FII activation by fully assembled prothrombinase with gel electrophoresis over a 2-h time period. A control experiment (Fig. 3C) demonstrates that under the conditions used the activation of FIIplasma proceeds efficiently following initial cleavage at Arg320, through the enzymatically active intermediate meizothrombin, as confirmed by the appearance of fragment 1·2-A. Rapid cleavage of this fragment at Arg271 leads to the formation of IIa. In contrast, activation of rFIIΔ473–487 under similar experimental conditions is significantly delayed through the same pathway as verified by the late appearance of the B-chain of IIa (Fig. 3D). Scanning densitometry of the gels shown in Fig. 3, C and D, showed that rFIIΔ473–487 is consumed with a rate that is ∼27-fold slower compared with the rate of FIIplasma consumption or ∼23-fold slower compared with the rate of rFIIWT consumption under the experimental conditions used (Table 1). These data suggest that under conditions of saturating amounts of fVa with respect to fXa, amino acid sequence 473–487 of FII plays a preeminent role because it is required for fast and efficient initial cleavage of FII at Arg320 by prothrombinase.
To further investigate the effect of the deletions and point mutations on rFII cleavage and activation by membrane-bound fXa alone, we studied rFII activation by gel electrophoresis of all mutants detailed in Fig. 1. Fig. 4A shows a control experiment and demonstrates that rFIIWT activation by membrane-bound fXa proceeds typically following initial cleavage at Arg271, as its plasma counterpart through the intermediate prethrombin-2 with very slow and minimal appearance of the B chain of IIa because of a nonproductive rate of cleavage at Arg320. With the removal of amino acids 478–482 from rFIIΔS5V (Fig. 4B), there is acceleration of rFIIΔ478–482 consumption by fXa alone through initial cleavage at Arg271 that is evident by the rapid appearance of prethrombin-2. The fact that only trace amounts of B-chain of IIa are apparent under the conditions employed suggests a substantially deferred rate of cleavage at Arg320 of the deletion mutant compared with cleavage of rFIIWT resulting in insignificant amounts of IIa generation. Scanning densitometry of similar gels shown in Fig. 4, A and B, showed that the rate of consumption of all rFII molecules by membrane-bound fXa alone is ∼2.3–8-fold increased compared with the rate of cleavage of rFIIWT or FIIPLASMA (Fig. 4C and Table 1). However, although with rFIIS478A, rFIIL480A, rFIISL→AA, and rFIISQ→AA minimal amounts of the B-chain of IIa are formed (datanot shown), when studying rFIIΔN10, rFIIΔC10, rFIIΔS5V, rFIISLQ→AAA, and rFIIΔSLQ activation, there is accumulation of prethrombin-2 with no significant amounts of B-chain of IIa generated suggesting impaired cleavage of prethrombin-2 at Arg320 by membrane-bound fXa (Fig. 4, B and D). These data confirm our findings with rFIIΔ473–487 (Fig. 3) and reveal that the dipeptide Leu480–Gln481 within the 15-amino acid stretch 473–487 of FII appears to be responsible for the similar effects observed with rFIIΔN10, rFIIΔC10, rFIIΔS5V, rFIISLQ→AAA, and rFIIΔSLQ when studying rFII molecular activation by membrane-bound fXa alone in the absence of fVa (Fig. 4D and Table 1).
FIGURE 4.

SDS-PAGE analyses of rFII molecule activation by membrane-bound fXa alone. A, rFIIWT (1.4 μm) in the presence of PCPS vesicles, DAPA, and membrane bound fXa alone (5 nm); B, rFIIΔS5V (1.4 μm) in the presence of PCPS vesicles, DAPA, and membrane-bound fXa alone (5 nm). Aliquots were withdrawn at various time intervals and treated as described (47, 69). M represents the lane with molecular weight markers (from top to bottom): 98,000, 64,000, 50,000, and 36,000, respectively. Lanes 1–19 show samples from the reaction mixture before (0 min) the addition of fXa and 20, 40, 60, 80, 100, 120, 150, 180, 210, and 240 s and 5, 6, 10, 20, 30, and 60, 90, and 120 min respectively, after the addition of fXa. C, the two gels shown in A and B together with similar gels obtained with all rFII studied were scanned, and rFII consumption was recorded as described under “Experimental Procedures.” Following scanning densitometry and normalization to the initial FII concentration, the data representing rFII consumption as a function of time (seconds) were plotted using non-linear regression analysis according to the equation representing a first-order exponential decay using the software Prizm (GraphPad, San Diego). Prothrombinase was assembled with rFIIWT (filled circles; R2 0.98), rFIIΔC10 (filled squares; R2 0.98), rFIIΔN10 (filled triangles; R2 0.99), rFIIΔS5V (filled inverse triangles; R2 0.99), rFIIS478A (filled diamonds; R2 0.99), rFIIL480A (open squares; R2 0.99), rFIISQ→AA (open circles; R2 0.98), rFIISL→AA (open triangles; R2 0.99), and rFIISLQ→AAA (open inverse triangles; R2 0.99). The rates of rFII consumption shown in C, using the apparent first-order rate constant, k (s−1) obtained directly from the fitted data, were calculated as reported (47), and the data are shown in Table 1. D, schematic representation of fragments derived following FII activation by membrane-bound fXa alone. The red arrow indicates impaired cleavage at Arg320 in rFIIΔC10 (filled squares), rFIIΔN10 (filled triangles), rFIIΔS5V (filled inverse triangles), and rFIISLQ→AAA (open inverse triangles) resulting in prethrombin-2 accumulation. FII-derived fragments are identified to the right of each panel, according to the description provided in the legend of Fig. 3.
To improve our understanding of the essential role of amino acids Leu480 and Gln481 for FII activation, we studied the pattern of all rFII molecules activation shown in Fig. 1 by fully assembled prothrombinase (i.e. in the presence of an excess of fVa) by gel electrophoresis over a 2-h time period (Fig. 5). A control experiment (Fig. 5A) demonstrates that under the conditions used, rFIIWT proceeds as its plasma counterpart following initial cleavage at Arg320, through the enzymatically active intermediate meizothrombin, as confirmed by the appearance of fragment 1·2-A. Rapid cleavage of this fragment at Arg271 leads to the formation of rIIa. Similar results were found when using rFIIS478A (Fig. 5B) demonstrating that the S478A transition alone is of no consequence for timely FII activation by prothrombinase. In contrast, activation of rFIIΔS5V and rFIISLQ→AAA under similar experimental conditions was significantly delayed through the same pathway as verified by the lingering of fragment 1·2-A at the late time points and the late appearance of the B-chain of rIIa (Fig. 5, C and D). Similar results were obtained with rFIIΔSLQ (Table 1). A systematic analysis of the activation of all rFII mutant molecules by prothrombinase using similar experimental procedures, followed by scanning densitometry of the gels and calculation of the rate of rFII consumption, revealed the existence of two groups as follows: a group of molecules represented by FIIplasma, rFIIWT, and rFIIS478A (also containing rFIIL480A, rFIISL→AA, and rFIISQ→AA) that are efficiently activated by prothrombinase; and a second group of proteins represented by rFIIΔS5V and rFIISLQ→AAA (including rFIIΔN10, rFIIΔC10, and rFIIΔSLQ) that are activated by fully assembled prothrombinase with a rate that is ∼13–18-fold slower than the rate observed with rFIIWT (Fig. 6A, inset, and Table 1). The data suggest that under conditions of saturating amounts of fVa with respect to fXa, the dipeptide Leu480–Gln481 of prothrombin plays a leading role during FII activation because it is required for fast and efficient initial cleavage at Arg320 by prothrombinase (Fig. 6B, the deficient step is represented by the red arrow).
FIGURE 5.

SDS-PAGE analyses of rFII molecule activation by prothrombinase. A, rFIIWT (1.4 μm) in the presence of PCPS vesicles, DAPA, and prothrombinase (1 nm fXa and 20 nm fVa). B, rFIIS478A same conditions as in A. C, rFIIΔS5V same conditions as in A. D, rFIISLQ→AAA same conditions as in A. Aliquots were withdrawn at various time intervals and treated as described (47, 69). Same time points as described in the legend to Fig. 4. FII-derived fragments are identified to the right of each panel, according to the description provided in the legend of Fig. 3.
FIGURE 6.
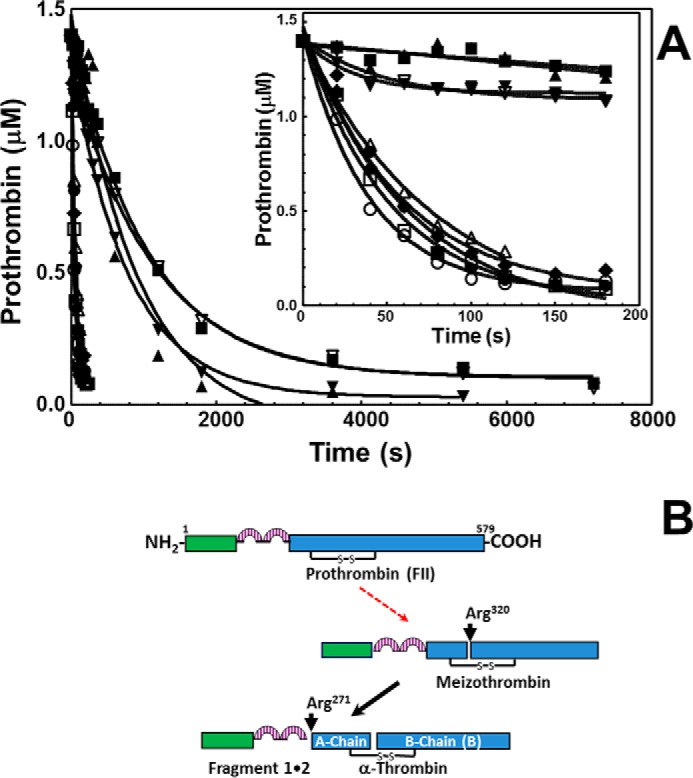
Analyses of the rates of activation of rFII by prothrombinase. A, gels shown in Fig. 5, together with similar gels obtained with all rFII studied, were scanned, and rFII consumption was recorded as described under “Experimental Procedures.” Following scanning densitometry, the numbers were normalized to the initial concentration of rFII studied, and the data representing rFII consumption as a function of time (seconds) were plotted using non-linear regression analysis according to the equation representing a first-order exponential decay using the software Prizm (GraphPad, San Diego). rFIIWT (filled circles; R2 0.98), rFIIΔC10 (filled squares; R2 0.99), rFIIΔN10 (filled triangles; R2 0.94), rFIIΔS5V (filled inverse triangles; R2 0.99), rFIIS478A (filled diamonds; R2 0.97), rFIIL480A (open squares; R2 0.98), rFIISQ→AA (open circles; R2 0.99), rFIISL→AA (open triangles; R2 0.99), and rFIISLQ→AAA (open inverse triangles; R2 0.99) are shown. The inset shows the progress of the reaction during the first 180 s. The rates of rFII consumption using the apparent first-order rate constant k (s−1), obtained directly from the fitted data, were calculated as reported (47) and shown in Table 1. B, schematic representation of fragments derived following rFII activation by prothrombinase in the presence of excess fVa with respect to fXa. The red arrow indicates impaired cleavage (at Arg320) in rFIIΔC10 (filled squares), rFIIΔN10 (filled triangles), rFIIΔS5V (filled inverse triangles), and rFIISLQ→AAA (open inverse triangles).
Kinetic Analyses of the Activation of rFII Molecules
To understand the effect of the S478A/L480A/Q481A substitutions on the activity of prothrombinase in activating the rFII molecules, we first examined the rates of rIIa formation from all rFII molecules under similar experimental conditions. Historically, this method was designed to identify any deficiency in fVa or fXa as part of prothrombinase in cleaving and activating FII and is measured indirectly by using IIa generation as a reporting probe with a chromogenic substrate. The comprehensive kinetic data for several mutants are shown in Fig. 7 with the kinetic constants derived directly from the fitted data reported in Table 2. The combined findings demonstrate that whereas the single and double alanine substitutions rFII mutants are activated by prothrombinase similarly, providing kinetic constants comparable with the wild type or plasma FII molecules, kinetic analyses of prothrombinase activation of rFIISLQ→AAA demonstrate a modest 2.7-fold decrease in the kcat and a large 21-fold increase in the Km value of the reaction. Similar experiments studying rFIIΔSLQ activation by fully assembled prothrombinase revealed a 29-fold increase in Km with a concomitant 16-fold decrease in the kcat values of the reaction. A direct comparison between the data obtained with rFIISLQ→AAA with the data obtained with rFIIΔSLQ strongly suggest an important contribution of the backbone structure of the peptide bond between these three amino acids for efficient rFII activation by prothrombinase.
FIGURE 7.
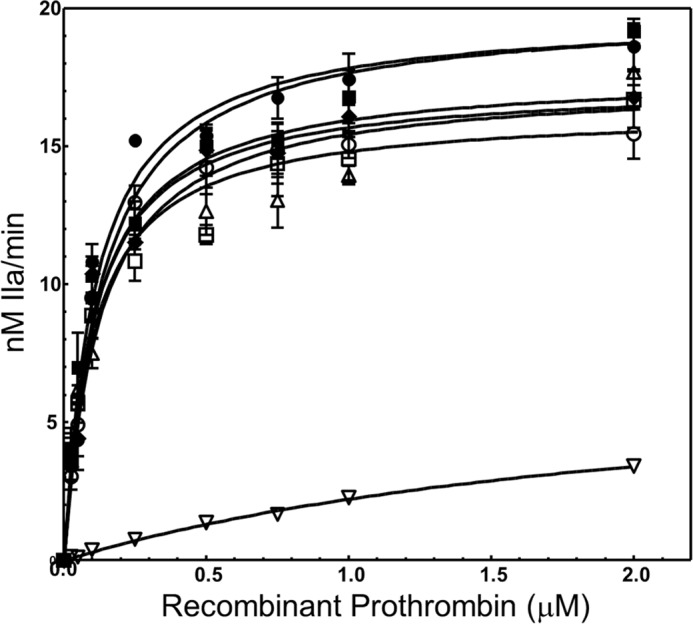
Determination of kinetic parameters of prothrombinase catalyzing cleavage and activation of various FII molecules. IIa generation experiments were conducted as described under “Experimental Procedures” by varying the substrate concentration and using a chromogenic substrate. The solid lines represent the nonlinear regression fit of the data using Prizm GraphPad software according to the Henri Michaelis-Menten equation (Vo = Vmax·[FII]/Km + [FII]) to yield the Km and kcat (kcat = Vmax/Etot, where Etot is the concentration of fully assembled prothrombinase, in this case is 10 pm, Table 2). Prothrombinase activity with various rFII molecules is shown as follows: rFIIWT filled circles; FIIPLASMA filled squares; rFIIS478A filled diamonds; rFIIL480A open squares; rFIISL→AA open triangles; rFIISQ→AA open circles; rFIISLQ→AAA open inverse triangles. Kinetic constants reported in the text and in Table 2 were extracted directly from the fitted data shown herein.
TABLE 2.
Kinetic constants of plasma FII and various rFII mutant molecules activation by prothrombinase
| FII species | Kma | kcata,b | R2/points/titrationsc | kcat/Km | Decreased |
|---|---|---|---|---|---|
| μm | min−1 | (m−1·s−1)·108 | -fold | ||
| FIIplasma | 0.13 ± 0.02 | 1992 ± 68 | 0.93/30/3 | 2.5 | |
| rFIIWT | 0.11 ± 0.015 | 1976 ± 57 | 0.97/20/2 | 2.9 | |
| rFIIWTe | 0.11 ± 0.02 | 2054 ± 84 | 0.97/10/1 | 3.1 | |
| rFIIΔ473–487 | NPf | ||||
| rFIIΔN10 | NP | ||||
| rFIIΔC10 | NP | ||||
| rFIIΔS5V | NP | ||||
| rFIIS478A | 0.10 ± 0.013 | 1764 ± 45 | 0.97/20/2 | 2.9 | 1.0 |
| rFIIL480A | 0.10 ± 0.015 | 1630 ± 49 | 0.96/20/2 | 2.7 | 1.1 |
| rFIISL→AA | 0.12 ± 0.02 | 1734 ± 65 | 0.92/30/3 | 2.4 | 1.2 |
| rFIISQ→AA | 0.10 ± 0.015 | 1727 ± 52 | 0.94/30/3 | 2.9 | 1.0 |
| rFIISLQ→AAA | 2.3 ± 0.5 | 730 ± 101 | 0.96/36/4 | 0.053 | 55 |
| rFIIΔSLQe | 3.2 ± 1.5 | 128 ± 34 | 0.96/9/1 | 0.0067 | 463 |
a The Km and kcat values of prothrombinase assembled with saturating concentrations of recombinant fVa molecules were determined as described under “Experimental Procedures” according to the Michaelis-Menten equation using the software Prizm with several different preparations of rFII molecules (representative experiments are shown in Fig. 7). Kinetic constants were derived directly from the fitted data.
b kcat = Vmax/[enzyme]; the enzyme concentration of prothrombinase (fXa·fVa complex on the membrane surface in the presence of Ca2+) under the conditions employed herein was 10 pm.
c R2 is the goodness of fit of the data points to the Michaelis-Menten equation using the software Prizm. Points and titrations studied represent 10 measurements/graph for all experiments (up to 4 μm plasma-derived FII or rFII molecules) except experiments with rFIISLQ→AAA (nine measurements/graph, up to 2 μm prothrombin) and with rFIIΔSLQ (nine measurements/graph, up to 4 μm prothrombin).
d The -fold decrease is the ratio of the second-order rate constant (kcat/Km) of prothrombinase catalyzing rFIIWT activation compared with the second-order rate constant of prothrombinase catalyzing activation of all other rFII molecules.
e Experiments with these two preparations of recombinant molecules were performed in parallel with same reagents (fXa and PCPS vesicles). The results shown are representative of four separate titrations with three different preparations of rFIIΔSLQ compared with either rFIIWT or FIIplasma.
f NP is no plot; data could not be plotted to the Michaelis-Menten equation using the software Prizm.
To quantify the interaction between the two sets of double mutations (S478A/L480A and S478A/Q481A) and to confirm their apparent synergistic detrimental effect on prothrombinase function for activation of rFIISLQ→AAA, we have further calculated the difference in free energy of the transition state analog (ΔΔGint) for the triple mutant as described previously by our laboratory (75, 76). The large positive value of ΔΔGint (+2.4 kcal/mol) for the combination of the mutations at Leu480 and Gln481 together with the sizable 55-fold decrease in the second-order rate constant of prothrombinase for rFIISLQ→AAA activation signify that there is a deficiency in recognition between prothrombinase and rFIISLQ→AAA. These findings solidify our previous conclusion that these substitutions are detrimental to the activation of rFII bearing the triple amino acid substitution by fully assembled prothrombinase. However, it is important to note that it is also possible that rIIaSLQ→AAA may also be deficient in its own catalytic activity as observed with rIIaΔSLQ, and the effect observed with rFIISLQ→AAA activation may be likewise due to the deficiency of rIIa in cleaving the chromogenic substrate. Thus, although we cannot yet assign the poor performance of prothrombinase in cleaving rFIISLQ→AAA solely to a deficiency in recognizing the mutated substrate, and because the S478A transition is of no consequence for either rFIIS478A activation or rIIaS478A activity, the overall data presented thus far suggest that amino acid sequence Leu480–Gln481 may have a dual effect in properly directing prothrombinase recognition of FII, as well as providing the resulting enzyme with the appropriate surface required for proper substrate tethering and cleavage. However, it is also possible that these two amino acids are allosterically involved in both prothrombinase interactions with FII as well as the expression of the enzymatic activity of IIa.
Analyses of the Activity of rIIa Molecules
Although many investigations have identified the specific amino acid residues from FII/IIa participating in either prothrombinase recognition or IIa activity toward its physiological substrates, respectively, few studies have shown that identical residues are involved in both FII recognition by prothrombinase and IIa activity. To understand the effect of the deletions/mutations on IIa activity, we further assessed the amidolytic and biological activity of selected rIIa molecules generated herein toward the chromogenic substrate S-2238 and toward thrombin's natural substrates, fV, fVIII, and protein C.
To understand the effect of the mutations on the amidolytic activity of IIa, we determined the kinetic constants for the hydrolysis of S-2238 by the rIIa molecules under steady state conditions. The data shown in Table 3 reveal the following: 1) rIIaWT produced under the conditions described by our laboratory has similar activity as previously found with other rIIaWT preparations, and 2) rIIaS478A has similar catalytic efficiency (kcat/Km) as rIIaWT, as demonstrated previously (6, 86). In addition, we also found that whereas rIIaSLQ→AAA was devoid of activity toward S-2238, rIIaSQ→AA (where rIIaSQ→AA is recombinant human α-thrombin with the mutation S478A/Q481A) has similar amidolytic activity as rIIaWT, whereas rIIaL480A and rIIaSL→AA (where rIIaSL→AA is recombinant human α-thrombin with the mutation S478A/L480A) are the most deficient in S-2238 hydrolysis among the single and double alanine mutants when compared with rIIaWT or rIIaS478A (Table 3). The combined data clearly demonstrate that amino acid Leu480 plays an important role during the expression of IIa amidolytic activity and that the integrity of amino acid sequence Leu480–Gln481 is required for optimal expression of this activity.
TABLE 3.
Kinetic constants of wild-type and selected rIIa mutant molecules toward S-2238
| α-Thrombin species | Kma | kcatb | R2c | kcat/Km |
|---|---|---|---|---|
| μm | s−1 | (m−1· s−1)·106 | ||
| rIIaWT | 4.7 ± 1.8 | 22.7 ± 2.7 | 0.93 | 4.8 |
| rIIaS478A | 8.5 ± 1.5 | 36.5 ± 2.4 | 0.98 | 4.3 |
| rIIaL480A | 8.9 ± 1.7 | 15.5 ± 1.5 | 0.98 | 1.7 |
| rIIaSL→AA | 7.7 ± 3.0 | 14.7 ± 2.0 | 0.92 | 1.9 |
| rIIaSQ→AA | 10.8 ± 4.2 | 33.1 ± 4.9 | 0.93 | 3.0 |
| rIIaSLQ→AAA | NPd | NP | NP |
a The Km value for S-2238 is listed for wild-type rIIa and selected rIIa mutants and determined as described under “Experimental Procedures” according to the Michaelis-Menten equation using the software Prizm. Kinetic constants shown were derived directly from the fitted data.
b kcat = Vmax/[enzyme]; the Vmax was calculated as described under “Experimental Procedures,” and the enzyme concentrations of rIIa was 4 nm for all experiments shown.
c R2 is the goodness of fit of the data points to the Michaelis-Menten equation using the software Prizm.
d NP is no plot; no data could be plotted to the Michaelis-Menten equation using the software Prizm.
The data shown in Figs. 8 and 9 demonstrate that whereas rIIaWT and rIIaS478A cleave and activate fV and fVIII with similar rates (Figs. 8, A and C, and 9, A and C), rIIaΔC10 (rIIaΔC10 is recombinant human prothrombin missing amino acids SVLQVVNLPI), and rIIaΔS5V are totally deficient in cleaving both cofactor molecules over a 3-h incubation period (Figs. 8, B and H, and 9, B and H). These data are in complete agreement with our findings shown in Table 1, explaining the fact that rFIIΔC10 and rFIIΔS5V are devoid of clotting activity, and further attest to the crucial dual role of the dipeptide Leu480–Gln481 during coagulation. Further analyses of the single or double mutants reveal a slight differentiation in cleavage and activation of the two cofactors by the various rIIa molecules. Although rIIaL480A and rIIaSL→AA appear devoid of activity toward fV (Fig. 8, D and E), both molecules slowly cleave fVIII at the Arg372- and Arg1689-activating cleavage sites (Fig. 9, D and E) (87–90). Similarly, although rIIaSLQ→AAA has no apparent activity toward fV (Fig. 8G) over a 3-h time period, the mutant enzyme cleaves fVIII slowly at the non-activating Arg740 cleavage site (Fig. 9G). Finally, although rIIaSQ→AA cleaves fV efficiently at Arg709 to produce the heavy chain of fV and an Mr 220,000 intermediate (Fig. 8F), the enzyme is also efficient in cleaving fVIII at the Arg372- and Arg1689-activating cleavage sites (Fig. 9F). These two cofactors have strategic functions within the amplified coagulation response to vascular damage and must be activated to perform accordingly within their respective enzymatic complexes. The combined data explain the impaired procoagulant activity of rFIISLQ→AAA (Fig. 2), which is deficient in producing large amounts of rIIa in a timely fashion (Fig. 5D). However, even when rIIaSLQ→AAA is generated, the recombinant enzyme is deficient in activating the pro-cofactors.
FIGURE 8.

Activation of plasma-derived fV by rIIa. Plasma-derived fV (500 nm) was incubated with rIIa (4 nm) as described under “Experimental Procedures.” At selected time intervals, aliquots of the mixtures were removed, mixed with 2% SDS, heated for 5 min at 90 °C, and analyzed on a 4–12% SDS-PAGE followed by staining with Coomassie Blue. Lane 1 in all panels depicts aliquots of the mixture withdrawn from the reaction before the addition of rIIa. Lanes 2–8 represent aliquots of the reaction mixture withdrawn at 10, 20, 30, 45, 60, 120, and 180 min. The positions of all fV fragments and of the heavy (HC) and light chains (LC) of the active cofactor are indicated on the right. Fragments a and b of fV are identified as previously described (80). The rIIa molecule used each time is indicated under each panel.
FIGURE 9.

Activation of recombinant fVIII by rIIa. rfVIII (500 nm) was incubated with rIIa (4 nm) as described under “Experimental Procedures.” At selected time intervals, aliquots of the mixtures were removed, mixed with 2% SDS, heated for 5 min at 90 °C, and analyzed on a 4–12% SDS-PAGE followed by staining with Coomassie Blue. Lane 1 in all panels depicts aliquots of the mixture withdrawn from the reaction before the addition of rIIa. Lanes 2–8 represent aliquots of the reaction mixture withdrawn at 10, 20, 30, 45, 60, 120, and 180 min. The positions of all rfVIII fragments are indicated on the right. Fragments from rfVIII are identified as previously demonstrated (80). The rIIa molecule used each time is indicated under each panel.
We next assessed the capability of the rIIa molecules in the presence of thrombomodulin to activate protein C and produce APC. Fig. 10 shows the results of such an experiment and demonstrates that although rIIaΔS5V (rIIaΔS5V is recombinant human α-thrombin with region SVLQV deleted) cannot cleave and activate protein C, rIIaSLQ→AAA has small but significant activity generating minute amounts of APC (Fig. 10, lanes 8 and 9), which in turn can cleave fV at Arg506/Arg306 and produce the characteristic Mr 30,000 fragment (data not shown) (68, 91). All other rIIa mutant molecules tested, for APC generation, have similar activities as rIIaWT or plasma-derived IIa under the condition described (Fig. 10).
FIGURE 10.
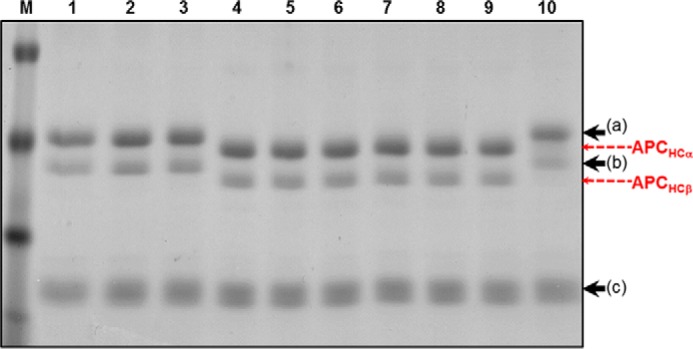
Activation of protein C by rIIa. Plasma-derived protein C (80 nm) was incubated with rIIa (8 nm), thrombomodulin, and PCPS as described under “Experimental Procedures.” Following a 3-h incubation period, each individual solution was dried with a vacuum concentrator, resuspended in Tris buffer, mixed with 2% SDS and 2% β-mercaptoethanol, heated for 5 min at 90 °C, and analyzed on a 5–15% SDS-PAGE followed by staining with Coomassie Blue. Lane 1, protein C alone no IIa; lane 2, protein C alone, no IIa incubated with buffer for 3 h; lane 3, protein C and rIIaΔS5V; lane 4, protein C and plasma-derived IIa; lane 5, protein C and rIIaWT; lane 6, protein C and rIIaS478A; lane 7, protein C and rIIaL480A; lane 8, protein C and rIIaSL→AA; lane 9, protein C and rIIaSQ→AA; and lane 10, protein C and rIIaSLQ→AAA. Positions of protein C and APC heavy and light chain fragments are indicated at the right (a/b heavy chains, and c light chain). The two heavy chains of protein C in plasma (a and b) have been identified earlier, differ by one glycosylation site, and have been extensively studied (58, 59).
These data demonstrate a differential requirement of IIa for cleavage and activation of both the pro-cofactor molecules and protein C and attest to the sensitive requirements of fV for cleavage and activation by IIa. Overall these results demonstrate that amino acids Leu480 and Gln481 within the serine protease domain of FII serve a dual purpose, and thus both are required for efficient cleavage at Arg320 by prothrombinase and may be involved in the presentation of an obligatory exosite for timely fV, fVIII, and protein C activation by IIa.
Discussion
Our data demonstrate that amino acid region 473–487 of FII is required for timely activation of FII through the meizothrombin pathway. Although prior work using synthetic peptides suggested that this region of the cofactor may contain an fVa-dependent fXa-binding site for FII, the data presented herein with recombinant FII molecules provide for the first time a mechanistic interpretation of these findings and identify the crucial amino acids from this sequence responsible for the effect observed (56).
To elucidate the number and identity of the required amino acids within amino acid sequence 473–487 of FII, we constructed, expressed, purified to homogeneity, and studied several rFII molecules with deletions and point mutations within this important regulatory region. We first investigated the effects of the 15-amino acid deletion with rFIIΔ473–487, followed by experiments with rFII molecules containing overlapping deletions within this segment (rFIIΔN10 and rFIIΔC10 and rFIIΔS5V and rFIIΔSLQ). Several rFII molecules bearing single mutations (rFIIS478A and rFIIL480A), double mutations (rFIISL→AA and rFIISQ→AA), and a triple mutation (rFIISLQ→AAA) were subsequently made. Membrane-bound fXa cleaves FII sequentially at Arg271 followed by Arg320, forming small amounts of IIa. Under these conditions, the activation of the deletion mutants rFIIΔ473–487, rFIIΔN10, rFIIΔC10, rFIIΔS5V, the triple alanine mutant, and the deletion mutant (rFIISLQ→AAA and rFIIΔSLQ) resulted in a modest increase of the rate of activation. In addition, activation of these six rFII molecules by fXa alone resulted in accumulation of prethrombin-2, with very little IIa formed. In contrast, activation of all these rFII mutants by fully assembled prothrombinase is significantly delayed. The combined data suggest that amino acids Leu480 and Gln481 within region 473–487 of FII either represent or are responsible for the presentation of an fVa-dependent site for fXa on FII, which is essential for optimal rate of cleavage at Arg320, which in turn is required for timely IIa formation at the place of vascular injury.
The autolysis loop of APC bears strong homology with the FII sequence 473–487 (chymotrypsin numbering 149D-163) (92). Replacement of several basic amino acids from this homologous region in APC by site-directed mutagenesis to alanine demonstrated the ability of this exosite to interact with its substrate fVa and to differentiate between the Arg506 and Arg306 cleavage sites (93). Yegneswaran et al. (56) using synthetic peptides provided initial evidence that sequence 473–487 of FII is able to disrupt prothrombinase assembly only in an fVa-dependent manner. Chen et al. (50) identified a sequence within pro-exosite I of prothrombin containing basic residues Arg35, Lys36, Arg67, Lys70, Arg73, Arg75, and Arg77 (chymotrypsin numbering), which is in close spatial proximity to region 473–487 of FII. These investigations revealed that following replacement of all basic residues from pro-exosite I with Glu, there was a significant effect on fXa within prothrombinase when compared with fXa alone in cleaving and activating FII, suggesting that these amino acids are specific fVa-dependent recognition sites for fXa on FII. Further kinetic studies by Chen et al. (50) using the hirudin COOH-terminal peptide (hirugen) showed that the peptide inhibited wild type prethrombin-1 activation by prothrombinase, whereas hirugen had no inhibitory effect on the activation of the mutated zymogen lacking the basic residues in pro-exosite I by fXa alone. The combined studies of Chen et al. (50) and Yegneswaran et al. (56) suggest the requirement of both sites for optimum productive interaction of prothrombinase with FII and timely IIa formation.
Research with discontinuous assays using a chromogenic substrate for IIa revealed that when fVa is incorporated into the prothrombinase complex, the resulting Km value of the reaction was decreased by 100-fold (corresponding to a 100-fold increase in affinity of prothrombinase for FII as compared with the affinity of fXa alone for the substrate), whereas the catalytic efficiency (kcat) of fXa was increased by 3,000-fold resulting in a 300,000-fold overall increase in the activity of prothrombinase (second-order rate constant) for FII compared with the activity of fXa alone toward FII (16). The significant increase in affinity of prothrombinase for its substrate is attributed to tighter binding of the enzymatic complex to FII because of its localization on the membrane surface by fVa. The longstanding hypothesis that fVa “localizes and positions” FII in an optimum position for efficient catalysis by fXa consistent with the classical role of a cofactor for catalysis was recently confirmed by computational studies with prothrombinase by Shim et al. (27). These studies demonstrated that the acidic COOH-terminal portion of the heavy chain of fVa that is contiguous to the A2 domain of fVa is essential in its ability to interact and snare the serine protease domain of FII. In that manner, this acidic amino acid sequence reposition the Arg320 cleavage site at an optimum position for timely cleavage by fXa and FII activation at the site of vascular injury as earlier suggested (54) and more recently experimentally demonstrated by our laboratory with synthetic peptides and recombinant fVa molecules mutated at these specific sites (45, 47, 48).
We show that following removal of the amino acid sequence 473–487 from FII, prothrombinase loses the ability to efficiently form IIa because of impaired fVa-dependent cleavage of FII by fXa at Arg320. One easy explanation of these results was that elimination of such a huge portion of the molecule results in significant structural changes of the molecule that in turn have deleterious effects on FII molecular conformation resulting in deficient prothrombinase activity. Despite the fact that rFIIΔ473–487 was activated following the same pathways as rFIIWT in the presence or absence of fVa, albeit with different rates, and in the absence of a crystal structure of rFIIΔ473–487, there was still doubt about the structural integrity and function of a molecule bearing such a large deletion. Experiments using more modest overlapping deletions (with rFIIΔN10, rFIIΔC10, and rFIIΔS5V) as well as with a triple alanine mutant (rFIISLQ→AAA) and a triple deletion mutant (rFIIΔSLQ) demonstrated that these molecules are also hindered in their fVa-dependent cleavage at Arg320 to a similar level as rFIIΔ473–487 (Table 1 and Fig. 6). These data provide original evidence demonstrating that the minimal sequence of FII required for the 3,000-fold increase in the catalytic efficiency of prothrombinase, as defined ∼36 years ago (16) for efficient cleavage of FII by prothrombinase at Arg320, is carried at least partially by amino acid sequence Leu480–Gln481 of FII. The findings presented herein silence the notion that the effect seen with rFIIΔ473–487 may be due to a structural change of the mutant molecule rather than to specific amino acid(s) missing from rFIIΔ473–487.
The kinetic findings presented herein revealed comparable Km and kcat constants for prothrombinase when rFII molecules bearing the single and double alanine mutations were used as substrate. However, when rFIISLQ→AAA was the substrate for prothrombinase in the same discontinuous assay, there was a significant 21-fold increase in the Km value and a modest 2.7-fold decrease in the kcat of the enzyme. Similar results were obtained with rFIIΔSLQ. Furthermore, rFIISLQ→AAA and rFIIΔSLQ were also found to be substantially deficient in clot formation in an assay using FII-deficient plasma, whereas rIIaSLQ→AAA was also deficient in S-2238 hydrolysis. rIIaSLQ→AAA was also impaired in cleaving fV, fVIII, and to a lesser extent protein C. These data dovetail nicely with results obtained with rIIaΔS5V and rIIaΔ473–487. We can thus hypothesize that the substantial increase in the Km of prothrombinase toward rFIISLQ→AAA is due to a deficiency in prothrombinase in recognizing the mutant molecule because of the lack of Leu480–Gln481, whereas the decrease in enzymatic activity of the resulting rIIaSLQ→AAA molecule is also the result of the absence of these two important amino acid side chains. Additional data with rFIIΔSLQ provide further evidence of the crucial role of amino acids Leu480–Gln481 and the peptide bond backbone between these two amino acids because, when these residues are completely eliminated, the Km value of the reaction increases by 32-fold, and the kcat value decreases by 16-fold (Table 2). Keeping in mind that the S478A substitution is of no consequence on both rFII activation and rIIa function, these results provide strong evidence in favor of the dual role of amino acids Leu480 and Gln481. Moreover, these amino acids are required by prothrombinase to efficiently promote cleavage of FII at Arg320 and are also required by IIa for optimum amidolytic activity as well as to proficiently cleave and activate fV, fVIII, and protein C. Finally, the possibility that elimination of these two residues from rFII results in an allosteric transition of the amino acids around/within the active site of rIIa, thus modifying the critical distances between the specific residues of the catalytic triad resulting in impaired catalysis, cannot be eliminated.
A comparison of crystal structures of FII, meizothrombin, IIa, prethrombin-1, and prethrombin-2 was carried out to identify structural differences in/near the Gly473–Ile487 segment comprising the fVa-dependent fXa-binding site. These residues adopt similar conformations in all of the crystal structures, with the NH2-terminal residues Gly473–Gln476 being quite solvent-accessible or flexible, and residues Pro477–Ile487 being variable in their degree of solvent exposure. Residue Ile487 is significantly more exposed in prothrombin (accessible surface area of >30 Å2 compared with ≤10 Å2 in meizothrombin and thrombin), as well as the adjacent Pro486 (accessible surface area of ∼15–30 Å2 reducing to <10 Å2 in meizothrombin and thrombin). The amount of solvent exposure of Ile487 and Pro486 appears to be heavily influenced by the flanking loops encompassing residues Ala446–Tyr454 and Lys511–Ser525, which adopt different conformations upon FII activation (Fig. 11). Recently, Pozzi et al. (94) used the crystal structure of Gla-domainless FII with active site S525A to demonstrate that fVa has recognition sites in close proximity to Arg320 (Arg15 chymotrypsin numbering). These sites create a strong electrostatic potential due to a number of basic residues described by Chen et al. (50). Through analysis of this published crystal structure, we have located this basic region to be in the vicinity of the Leu480–Gln481 amino acid sequence of FII that we found to be required for efficient initial cleavage at Arg320 by prothrombinase. It is noteworthy that a very recent study by Pozzi et al. (38) demonstrated a crucial role for linker 2 for the rate of activation of FII by prothrombinase and suggested that this region may be involved in the interaction of FII with the cofactor. These data are in complete accord with data showing that fragment 1, more precisely the kringle 2 region, is involved in the interaction of fVa as part of prothrombinase with FII (39, 41). Finally, a close comparison of crystal structures of FII and IIa revealed that residues Ser478, Leu480, and Gln481 adopt similar conformations in both structures. The Ser478 side chain is exposed on the surface of both molecules, whereas the Leu480 side chain is surrounded by other residues and is not accessible to solvent. Gln481 is partially solvent-exposed in both FII and IIa. The Ser478/Leu480/Gln481 residues are near ABE-I (Fig. 11), but >15 Å from the catalytic Ser525 residue, and even more distant from ABE-II.
FIGURE 11.
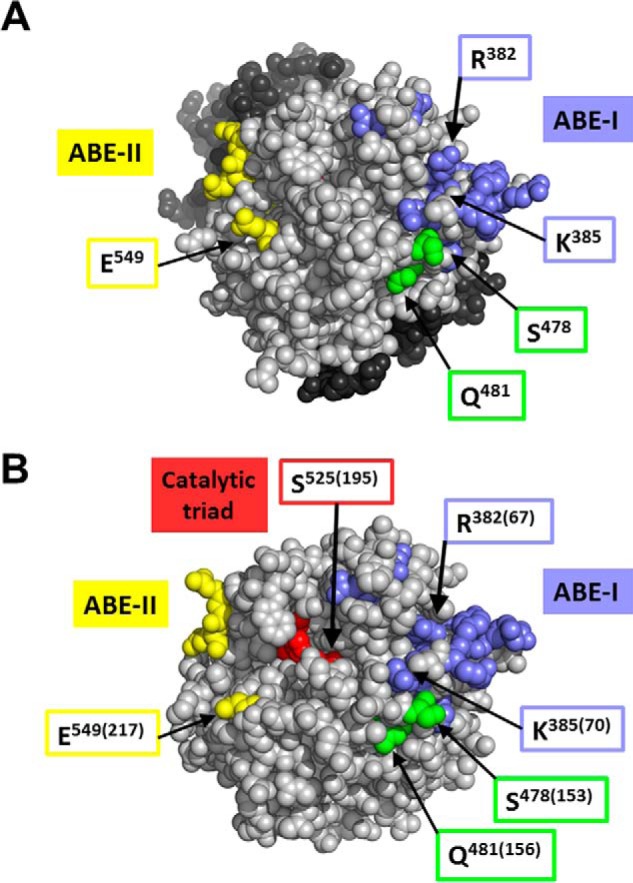
Location of exosites in FII and thrombin structures. Representation of the high resolution crystal structures of FIIWT (A) and IIa (B). A, space-filling representation of human FII (38). Residues Ser478, Leu480, and Gln481 are colored green; ABE-I and ABE-II residues (6, 50) are purple-blue and yellow, respectively; the amino acids composing the catalytic triad are not solvent-accessible and thus not visible. Other catalytic domain residues are in light gray, and those in fragment-1 and fragment-2 are in dark gray. B, space-filling representation of IIa (81). Residues are colored as for FII with the addition of catalytic triad residues His363, Asp419, and Ser525 (red), which are partially solvent-accessible. In parentheses are the corresponding numbers according to the chymotrypsin numbering of IIa (4). Distances between the active Ser525 side chain hydroxyl and several other amino acids of interest are as follows: 17 Å to Gln481 OE1/NE2; 15 Å to Glu549 OE1; 17 Å to Arg382 NH2; 18 Å to Lys385 NH2. The polar atoms at the end of the side chains were used as a reference because these would presumably be involved in intermolecular interactions.
In conclusion, in this study we provide evidence for the dual effect of amino acids Leu480 and Gln481 of FII. Future mutagenesis studies within the amino acids uncovered herein, paired with selected mutations within pro-exosite-I and/or pro-exosite-II of FII, should be able to elucidate the intermolecular communications within FII, required for both optimal fVa-dependent activation of FII and subsequent IIa catalytic activity toward its numerous physiological substrates. Finally, our results provide evidence for the production of large quantities of rFIIΔS5V, rFIISLQ→AAA, or rFIIΔSLQ that could be used as therapeutic agents because these molecules would compete with the natural substrate in vivo, when infused in individuals with prothrombotic tendencies.
Author Contributions
J. R. W. designed, performed, and analyzed most experiments and participated in the writing of the paper; J. H. designed, performed, and analyzed some of the experiments; V. C. Y. designed and produced the structural pictures of prothrombin and thrombin shown in Fig. 11; M. K. conceived and coordinated the study and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Ken Mann and Dr. Tom Orfeo from the Department of Biochemistry, University of Vermont, for providing monoclonal antibodies to factor V and the polyclonal antibody to FII; Dr. Kathy Berkner from the Department of Molecular Cardiology at the Cleveland Clinic for providing the plasmid with FII cDNA and for critical advice during expression and purification of rFII molecules studied herein; and Greg Guzzo for expert technical assistance during the purification of rFII molecules. We acknowledge Dr. Lisa Regan from Bayer Healthcare for the recombinant factor VIII and Dr. Larry Dangott and Virginia Johnson from the Protein Chemistry Laboratory at Texas A & M University for mass spectrometry and γ-carboxyglutamic acid analysis of the mutant recombinant FII molecules. We also thank Dr. Alex Kurosky and Dr. Bo Xu from the Biomolecular Resource Facility at the University of Texas Medical Branch for NH2-terminal sequencing of proteolytic fragments from rFII. Finally, we thank Dr. Susan Kennedy-Kalafatis for helpful advice and for critical reading of the manuscript.
This work was supported in part by Doctoral Dissertation Research Expense Award and Fellowship Program (to J. R. W.), by a Molecular Medicine Graduate Program Fellowship (to J. R. W.), by funds from the Center for Gene Regulation in Health and Disease at Cleveland State University (to M. K.), and funds from the Division of Research and Graduate Studies at Cleveland State University (to M. K.). The authors declare that they have no conflicts of interest with the contents of this article.
- fVa
- factor Va
- fV
- factor V
- FII
- prothrombin
- fXa
- factor Xa
- IIa
- α-thrombin
- r
- recombinant
- PC
- l-α-phosphatidylcholine
- PCPS
- small unilamellar phospholipids vesicles composed of 75% PC and 25% l-α-phosphatidylserine (w/w)
- rFIIWT
- recombinant wild type human prothrombin
- rIIaWT
- recombinant wild type human α-thrombin
- fVIII
- factor VIII
- DAPA
- dansylarginine-N-(3-ethyl-1,5-pentanediyl)amide
- ABE
- anion-binding (pro)exosite
- APC
- activated protein C
- TP
- prothrombin time.
References
- 1. Kalafatis M., Egan J. O., van't Veer C., Cawthern K. M., and Mann K. G. (1997) The regulation of clotting factors. Crit. Rev. Eukaryot. Gene Expr. 7, 241–280 [DOI] [PubMed] [Google Scholar]
- 2. Mann K. G., and Kalafatis M. (2003) Factor V: a combination of Dr Jekyll and Mr Hyde. Blood 101, 20–30 [DOI] [PubMed] [Google Scholar]
- 3. Mann K. G., Nesheim M. E., Hibbard L. S., and Tracy P. B. (1981) Assembly of the prothrombinase complex. Ann. N.Y. Acad. Sci. 370, 378–388 [DOI] [PubMed] [Google Scholar]
- 4. Bode W., Mayr I., Baumann U., Huber R., Stone S. R., and Hofsteenge J. (1989) The refined 1.9 A crystal structure of human α-thrombin: interaction with d-Phe-Pro-Arg chloromethylketone and significance of the Tyr-Pro-Pro-Trp insertion segment. EMBO J. 8, 3467–3475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Myles T., Yun T. H., Hall S. W., and Leung L. L. (2001) An extensive interaction interface between thrombin and factor V is required for factor V activation. J. Biol. Chem. 276, 25143–25149 [DOI] [PubMed] [Google Scholar]
- 6. Myles T., Yun T. H., and Leung L. L. (2002) Structural requirements for the activation of human factor VIII by thrombin. Blood 100, 2820–2826 [DOI] [PubMed] [Google Scholar]
- 7. Degen S. J., and Davie E. W. (1987) Nucleotide sequence of the gene for human prothrombin. Biochemistry 26, 6165–6177 [DOI] [PubMed] [Google Scholar]
- 8. Mann K. G., Bajaj S. P., Heldebrant C. M., Butkowski R. J., and Fass D. N. (1973) Intermediates of prothrombin activation. Semin. Haematol. 6, 479–493 [PubMed] [Google Scholar]
- 9. Heldebrant C. M., Butkowski R. J., Bajaj S. P., and Mann K. G. (1973) The activation of prothrombin. II. Partial reactions, physical and chemical characterization of the intermediates of activation. J. Biol. Chem. 248, 7149–7163 [PubMed] [Google Scholar]
- 10. Owen W. G., Esmon C. T., and Jackson C. M. (1974) The conversion of prothrombin to thrombin. I. Characterization of the reaction products formed during the activation of bovine prothrombin. J. Biol. Chem. 249, 594–605 [PubMed] [Google Scholar]
- 11. Esmon C. T., Owen W. G., and Jackson C. M. (1974) The conversion of prothrombin to thrombin. II. Differentiation between thrombin- and factor Xa-catalyzed proteolyses. J. Biol. Chem. 249, 606–611 [PubMed] [Google Scholar]
- 12. Esmon C. T., and Jackson C. M. (1974) The conversion of prothrombin to thrombin. III. The factor Xa-catalyzed activation of prothrombin. J. Biol. Chem. 249, 7782–7790 [PubMed] [Google Scholar]
- 13. Bajaj S. P., Butkowski R. J., and Mann K. G. (1975) Prothrombin fragments. Ca2+ binding and activation kinetics. J. Biol. Chem. 250, 2150–2156 [PubMed] [Google Scholar]
- 14. Downing M. R., Butkowski R. J., Clark M. M., and Mann K. G. (1975) Human prothrombin activation. J. Biol. Chem. 250, 8897–8906 [PubMed] [Google Scholar]
- 15. Butkowski R. J., Elion J., Downing M. R., and Mann K. G. (1977) Primary structure of human prethrombin 2 and α-thrombin. J. Biol. Chem. 252, 4942–4957 [PubMed] [Google Scholar]
- 16. Rosing J., Tans G., Govers-Riemslag J. W., Zwaal R. F., and Hemker H. C. (1980) The role of phospholipids and factor Va in the prothrombinase complex. J. Biol. Chem. 255, 274–283 [PubMed] [Google Scholar]
- 17. Nesheim M. E., and Mann K. G. (1983) The kinetics and cofactor dependence of the two cleavages involved in prothrombin activation. J. Biol. Chem. 258, 5386–5391 [PubMed] [Google Scholar]
- 18. Krishnaswamy S., Church W. R., Nesheim M. E., and Mann K. G. (1987) Activation of human prothrombin by human prothrombinase. Influence of factor Va on the reaction mechanism. J. Biol. Chem. 262, 3291–3299 [PubMed] [Google Scholar]
- 19. Brufatto N., and Nesheim M. E. (2003) Analysis of the kinetics of prothrombin activation and evidence that two equilibrating forms of prothrombinase are involved in the process. J. Biol. Chem. 278, 6755–6764 [DOI] [PubMed] [Google Scholar]
- 20. Boskovic D. S., Bajzar L. S., and Nesheim M. E. (2001) Channeling during prothrombin activation. J. Biol. Chem. 276, 28686–28693 [DOI] [PubMed] [Google Scholar]
- 21. Bianchini E. P., Orcutt S. J., Panizzi P., Bock P. E., and Krishnaswamy S. (2005) Racheting of the substrate from the zymogen to proteinase conformations directs the sequential cleavage of prothrombin by prothrombinase. Proc. Natl. Acad. Sci. U.S.A. 102, 10099–10104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morita T., and Iwanaga S. (1981) Prothrombin activator from Echis carinatus venom. Methods Enzymol. 90, 303–311 [Google Scholar]
- 23. Doyle M. F., and Haley P. E. (1993) Meizothrombin: active intermediate formed during prothrombin-catalyzed activation of prothrombin. Methods Enzymol. 222, 299–312 [DOI] [PubMed] [Google Scholar]
- 24. Doyle M. F., and Mann K. G. (1990) Multiple active forms of thrombin. Relative activities of meizothrombins. J. Biol. Chem. 265, 10693–10701 [PubMed] [Google Scholar]
- 25. Nesheim M. E., Taswell J. B., and Mann K. G. (1979) The contribution of bovine factor V and factor Va to the activity of prothrombinase. J. Biol. Chem. 254, 10952–10962 [PubMed] [Google Scholar]
- 26. Esmon C. T., Owen W. G., and Jackson C. M. (1974) A plausible mechanism for prothrombin activation by factor Xa, factor Va, phospholipid, and calcium ions. J. Biol. Chem. 249, 8045–8047 [PubMed] [Google Scholar]
- 27. Shim J. Y., Lee C. J., Wu S., and Pedersen L. G. (2015) A model for the unique role of factor Va A2 domain extension in the human ternary thrombin-generating complex. Biophys. Chem. 199, 46–50 [DOI] [PubMed] [Google Scholar]
- 28. Mann K. G., Nesheim M. E., and Tracy P. B. (1981) Molecular weight of undegraded plasma factor V. Biochemistry 20, 28–33 [DOI] [PubMed] [Google Scholar]
- 29. Nesheim M. E., Foster W. B., Hewick R., and Mann K. G. (1984) Characterization of factor V activation intermediates. J. Biol. Chem. 259, 3187–3196 [PubMed] [Google Scholar]
- 30. Esmon C. T., Owen W. G., Duiguid D. L., and Jackson C. M. (1973) The action of thrombin on blood clotting factor V: conversion of factor V to a prothrombin-binding protein. Biochim. Biophys. Acta 310, 289–294 [DOI] [PubMed] [Google Scholar]
- 31. Tracy P. B., Eide L. L., Bowie E. J., and Mann K. G. (1982) Radioimmunoassay of factor V in human plasma and platelets. Blood 60, 59–63 [PubMed] [Google Scholar]
- 32. Suzuki K., Dahlbäck B., and Stenflo J. (1982) Thrombin-catalyzed activation of human coagulation factor V. J. Biol. Chem. 257, 6556–6564 [PubMed] [Google Scholar]
- 33. Kane W. H., and Majerus P. W. (1981) Purification and characterization of human coagulation factor V. J. Biol. Chem. 256, 1002–1007 [PubMed] [Google Scholar]
- 34. Keller F. G., Ortel T. L., Quinn-Allen M. A., and Kane W. H. (1995) Thrombin-catalyzed activation of recombinant human factor V. Biochemistry 34, 4118–4124 [DOI] [PubMed] [Google Scholar]
- 35. Orfeo T., Brufatto N., Nesheim M. E., Xu H., Butenas S., and Mann K. G. (2004) The factor V activation paradox. J. Biol. Chem. 279, 19580–19591 [DOI] [PubMed] [Google Scholar]
- 36. Schuijt T. J., Bakhtiari K., Daffre S., Deponte K., Wielders S. J., Marquart J. A., Hovius J. W., van der Poll T., Fikrig E., Bunce M. W., Camire R. M., Nicolaes G. A., Meijers J. C., and van't Veer C. (2013) Factor Xa activation of factor V is of paramount importance in initiating the coagulation system. Lessons from a tick salivary protein. Circulation 128, 254–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Butenas S., van't Veer C., and Mann K. G. (1999) “Normal” thrombin generation. Blood 94, 2169–2178 [PubMed] [Google Scholar]
- 38. Pozzi N., Chen Z., Pelc L. A., Shropshire D. B., and Di Cera E. (2014) The linker connecting the two kringles plays a key role in prothrombin activation. Proc. Natl. Acad. Sci. U.S.A. 111, 7630–7635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kotkow K. J., Deitcher S. R., Furie B., and Furie B. C. (1995) The second Kringle domain of prothrombin promotes factor Va-mediated prothrombin activation by prothrombinase. J. Biol. Chem. 270, 4551–4557 [DOI] [PubMed] [Google Scholar]
- 40. Deguchi H., Takeya H., Gabazza E. C., Nishioka J., and Suzuki K. (1997) Prothrombin Kringle 1 domain interacts with factor Va during the assembly of prothrombinase complex. Biochem. J. 321, 729–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bukys M. A., Orban T., Kim P. Y., Nesheim M. E., and Kalafatis M. (2008) The interaction of fragment 1 of prothrombin with the membrane surface is a prerequisite for optimum expression of factor Va cofactor activity within prothrombinase. Thromb. Haemost. 99, 511–522 [PubMed] [Google Scholar]
- 42. Blostein M. D., Rigby A. C., Jacobs M., Furie B., and Furie B. C. (2000) The Gla domain of human prothrombin has a binding site for factor Va. J. Biol. Chem. 275, 38120–38126 [DOI] [PubMed] [Google Scholar]
- 43. Anderson P. J., Nesset A., Dharmawardana K. R., and Bock P. E. (2000) Role of pro-exosite I in factor Va-dependent substrate interactions of prothrombin activation. J. Biol. Chem. 275, 16435–16442 [DOI] [PubMed] [Google Scholar]
- 44. Monteiro R. Q., and Zingali R. B. (2002) Bothrojaracin, a pro-exosite I ligand, inhibits factor Va-accelerated prothrombin activation. Thromb. Haemost. 87, 288–293 [PubMed] [Google Scholar]
- 45. Beck D. O., Bukys M. A., Singh L. S., Szabo K. A., and Kalafatis M. (2004) The contribution of amino acid region ASP695-TYR698 of factor V to pro-cofactor activation and factor Va function. J. Biol. Chem. 279, 3084–3095 [DOI] [PubMed] [Google Scholar]
- 46. Kalafatis M., Beck D. O., and Mann K. G. (2003) Structural requirements for expression of factor Va activity. J. Biol. Chem. 278, 33550–33561 [DOI] [PubMed] [Google Scholar]
- 47. Bukys M. A., Kim P. Y., Nesheim M. E., and Kalafatis M. (2006) A control switch for prothrombinase: characterization of a hirudin-like pentapeptide from the COOH terminus of factor Va heavy chain that regulates the rate and pathway for prothrombin activation. J. Biol. Chem. 281, 39194–39204 [DOI] [PubMed] [Google Scholar]
- 48. Hirbawi J., Bukys M. A., Barhoover M. A., Erdogan E., and Kalafatis M. (2008) Role of the acidic hirudin-like COOH-terminal amino acid region of factor Va heavy chain in the enhanced function of prothrombinase. Biochemistry 47, 7963–7974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hirbawi J., Vaughn J. L., Bukys M. A., Vos H. L., and Kalafatis M. (2010) Contribution of amino acid region 659–663 of factor Va heavy chain to the activity of factor Xa within prothrombinase. Biochemistry 49, 8520–8534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen L., Yang L., and Rezaie A. R. (2003) Proexosite-1 on prothrombin is a factor Va-dependent recognition site for the prothrombinase complex. J. Biol. Chem. 278, 27564–27569 [DOI] [PubMed] [Google Scholar]
- 51. Adams T. E., Hockin M. F., Mann K. G., and Everse S. J. (2004) The crystal structure of activated protein C-inactivated bovine factor Va: implications for cofactor function. Proc. Natl. Acad. Sci. U.S.A. 101, 8918–8923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Orban T., Kalafatis M., and Gogonea V. (2005) Completed three-dimensional model of human coagulation factor Va. Molecular dynamics simulations and structural analyses. Biochemistry 44, 13082–13090 [DOI] [PubMed] [Google Scholar]
- 53. Toso R., and Camire R. M. (2006) Role of hirudin-like factor Va heavy chain sequences in prothrombinase function. J. Biol. Chem. 281, 8773–8779 [DOI] [PubMed] [Google Scholar]
- 54. Guinto E. R., and Esmon C. T. (1984) Loss of prothrombin and of factor Xa-factor Va interactions upon inactivation of factor Va by activated protein C. J. Biol. Chem. 259, 13986–13992 [PubMed] [Google Scholar]
- 55. Lechtenberg B. C., Murray-Rust T. A., Johnson D. J., Adams T. E., Krishnaswamy S., Camire R. M., and Huntington J. A. (2013) Crystal structure of the prothrombinase complex from the venom of Pseudonaja textilis. Blood 122, 2777–2783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yegneswaran S., Mesters R. M., Fernández J. A., and Griffin J. H. (2004) Prothrombin residues 473–487 contribute to factor Va binding in the prothrombinase complex. J. Biol. Chem. 279, 49019–49025 [DOI] [PubMed] [Google Scholar]
- 57. Yegneswaran S., Mesters R. M., and Griffin J. H. (2003) Identification of distinct sequences in human blood coagulation factor Xa and prothrombin essential for substrate and cofactor recognition in the prothrombinase complex. J. Biol. Chem. 278, 33312–33318 [DOI] [PubMed] [Google Scholar]
- 58. Miletich J. P., and Broze G. J. (1990) β Protein C is not glycosylated at asparagine 329. J. Biol. Chem. 265, 11397–11404 [PubMed] [Google Scholar]
- 59. Simioni P., Kalafatis M., Millar D. S., Henderson S. C., Luni S., Cooper D. N., and Girolami A. (1996) Compound prothein C deficiency resulting in the presence of only the β-form of protein C in plasma. Blood 88, 2101–2108 [PubMed] [Google Scholar]
- 60. Nesheim M. E., Katzmann J. A., Tracy P. B., and Mann K. G. (1981) Factor V. Methods Enzymol. 80, 249–274 [DOI] [PubMed] [Google Scholar]
- 61. Kalafatis M., Krishnaswamy S., Rand M. D., and Mann K. G. (1993) Factor V. Methods Enzymol. 222, 224–236 [DOI] [PubMed] [Google Scholar]
- 62. Kalafatis M., and Beck D. O. (2002) Identification of a binding site for blood coagulation factor Xa on the heavy chain of factor Va. Amino acid residues 323–331 of factor V represent an interactive site for activated factor X. Biochemistry 41, 12715–12728 [DOI] [PubMed] [Google Scholar]
- 63. Côté H. C., Stevens W. K., Bajzar L., Banfield D. K., Nesheim M. E., and MacGillivray R. T. (1994) Characterization of a stable form of human meizothrombin derived from recombinant prothrombin (R155A, R271A, and R284A). J. Biol. Chem. 269, 11374–11380 [PubMed] [Google Scholar]
- 64. Price P. A. (1983) Analysis for γ-carboxyglutamic acid. Methods Enzymol. 91, 13–17 [DOI] [PubMed] [Google Scholar]
- 65. Przysiecki C. T., Staggers J. E., Ramjit H. G., Musson D. G., Stern A. M., Bennett C. D., and Friedman P. A. (1987) Occurrence of β-hydroxylated asparagine residues in non-vitamin K-dependent proteins containing epidermal growth factor-like domains. Proc. Natl. Acad. Sci. U.S.A. 84, 7856–7860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 67. Towbin H., Staehelin T., and Gordon J. (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. U.S.A. 76, 4350–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kalafatis M., Rand M. D., and Mann K. G. (1994) The mechanism of inactivation of human factor V and human factor Va by activated protein C. J. Biol. Chem. 269, 31869–31880 [PubMed] [Google Scholar]
- 69. Bukys M. A., Blum M. A., Kim P. Y., Brufatto N., Nesheim M. E., and Kalafatis M. (2005) Incorporation of factor Va into prothrombinase is required for coordinated cleavage of prothrombin by factor Xa. J. Biol. Chem. 280, 27393–27401 [DOI] [PubMed] [Google Scholar]
- 70. Singh L. S., Bukys M. A., Beck D. O., and Kalafatis M. (2003) Amino acids Glu323, Tyr324, Glu330, and Val331 of factor Va heavy chain are essential for expression of cofactor activity. J. Biol. Chem. 278, 28335–28345 [DOI] [PubMed] [Google Scholar]
- 71. Ackers G. K., and Smith F. R. (1985) Effects of site specific amino acid modification on protein interactions and biological function. Annu. Rev. Biochem 54, 597–629 [DOI] [PubMed] [Google Scholar]
- 72. Wells J. A. (1990) Additivity of mutational effects in proteins. Biochemistry 29, 8509–8517 [DOI] [PubMed] [Google Scholar]
- 73. LiCata V. J., and Ackers G. K. (1995) Long-range, small magnitude nonadditivity of mutational effects in proteins. Biochemistry 34, 3133–3139 [DOI] [PubMed] [Google Scholar]
- 74. Mildvan A. S., Weber D. J., and Kuliopulos A. (1992) Quantitative interpretation of double mutations of enzymes. Arch. Biochem. Biophys. 294, 327–340 [DOI] [PubMed] [Google Scholar]
- 75. Barhoover M. A., Orban T., Beck D. O., Bukys M. A., and Kalafatis M. (2008) Contribution of amino acid region 334–335 from factor Va heavy chain to the catalytic efficiency of prothrombinase. Biochemistry 47, 6840–6850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Barhoover M. A., Orban T., Bukys M. A., and Kalafatis M. (2008) Cooperative regulation of the activity of factor Xa within prothrombinase by discrete amino acid regions from factor Va heavy chain. Biochemistry 47, 12835–12843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wiencek J. R., Na M., Hirbawi J., and Kalafatis M. (2013) Amino acid region 1000–1008 of factor V is a dynamic regulator for the emergence of procoagulant activity. J. Biol. Chem. 288, 37026–37038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Witting J. I., Miller T. M., and Fenton J. W. 2nd. (1987) Human α and γ thrombin specificity with tripeptide p-nitroanilide substrates under physiologically relevant conditions. Thromb. Res. 46, 567–574 [DOI] [PubMed] [Google Scholar]
- 79. Koike H., Okuda D., and Morita T. (2003) Mutations in the autolytic loop-2 and at Asp554 of human prothrombin that enhance protein C activation by meizothrombin. J. Biol. Chem. 278, 15015–15022 [DOI] [PubMed] [Google Scholar]
- 80. Bukys M. A., Orban T., Kim P. Y., Beck D. O., Nesheim M. E., and Kalafatis M. (2006) The structural integrity of anion binding exosite-I of thrombin is required and sufficient for timely cleavage and activation of factor V and factor VIII. J. Biol. Chem. 281, 18569–18580 [DOI] [PubMed] [Google Scholar]
- 81. Figueiredo A. C., Clement C. C., Zakia S., Gingold J., Philipp M., and Pereira P. J. (2012) Rational design and characterization of d-Phe-Pro-d-Arg-derived direct thrombin inhibitors. PLoS ONE 7, e34354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lee B., and Richards F. M. (1971) The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 55, 379–400 [DOI] [PubMed] [Google Scholar]
- 85. Saff E. B., and Kuijlaars A. B. (1997) Distributing many points on a sphere. The Mathematical Intelligencer 19, 5–11 [Google Scholar]
- 86. Rezaie A. R. (1998) Reactivities of the S2 and S3 subsite residues of thrombin with the native and heparin-induced conformers of antithrombin. Protein Sci. 7, 349–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Eaton D. L., Wood W. I., Eaton D., Hass P. E., Hollingshead P., Wion K., Mather J., Lawn R. M., Vehar G. A., and Gorman C. (1986) Construction and characterization of an active factor VIII variant lacking the central one-third of the molecule. Biochemistry 25, 8343–8347 [DOI] [PubMed] [Google Scholar]
- 88. Esmon C. T., and Lollar P. (1996) Involvement of thrombin anion-binding exosites 1 and 2 in the activation of factor V and factor VIII. J. Biol. Chem. 271, 13882–13887 [DOI] [PubMed] [Google Scholar]
- 89. Fay P. J., and Smudzin T. M. (1992) Characterization of the interaction between the A2 subunit and A1/A3-C1-C2 dimer in human factor VIIIa. J. Biol. Chem. 267, 13246–13250 [PubMed] [Google Scholar]
- 90. Toole J. J., Pittman D. D., Orr E. C., Murtha P., Wasley L. C., and Kaufman R. J. (1986) A large region (approximately equal to 95 kDa) of human factor VIII is dispensable for in vitro procoagulant activity. Proc. Natl. Acad. Sci. U.S.A. 83, 5939–5942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kalafatis M., and Mann K. G. (1993) Role of the membrane in the inactivation of factor Va by activated protein C. J. Biol. Chem. 268, 27246–27257 [PubMed] [Google Scholar]
- 92. Mesters R. M., Heeb M. J., and Griffin J. H. (1993) Interactions and inhibition of blood coagulation factor Va involving residues 311–325 of activated protein C. Protein Sci. 2, 1482–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Gale A. J., Heeb M. J., and Griffin J. H. (2000) The autolysis loop of activated protein C interacts with factor Va and differentiates between the Arg506 and Arg306 cleavage sites. Blood 96, 585–593 [PubMed] [Google Scholar]
- 94. Pozzi N., Chen Z., Gohara D. W., Niu W., Heyduk T., and Di Cera E. (2013) Crystal Structure of prothrombin reveals conformational flexibility and mechanism of activation. J. Biol. Chem. 288, 22734–22744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Yegneswaran S., Fernández J. A., Griffin J. H., and Dawson P. E. (2002) Factor Va increases the affinity of factor Xa for prothrombin: A binding study using a novel photoactivable thiol-specific fluorescent probe. Chem. Biol. 9, 485–494 [DOI] [PubMed] [Google Scholar]