

Abstract
Synthesis of the second messenger cAMP activates a variety of signaling pathways critical for all facets of intracellular regulation. Protein kinase A (PKA) is the major cAMP-responsive effector. Where and when this enzyme is activated has profound implications on the cellular role of PKA. A-Kinase Anchoring Proteins (AKAPs) play a critical role in this process by orchestrating spatial and temporal aspects of PKA action. A popular means of evaluating the impact of these anchored signaling events is to biochemically interfere with the PKA–AKAP interface. Hence, peptide disruptors of PKA anchoring are valuable tools in the investigation of local PKA action. This article outlines the development of PKA isoform-selective disruptor peptides, documents the optimization of cell-soluble peptide derivatives, and introduces alternative cell-based approaches that interrogate other aspects of the PKA–AKAP interface.
Keywords: AKAP, cAMP signaling, Protein kinase A (PKA), Compartmentalization, Anchoring proteins, RIAD, Ht31, AKAP-IS, STAD, Rselect
1 Introduction
A-K inase Anchoring Proteins (AKAPs) play a fundamental role in the spatial and temporal regulation of protein kinase A (PKA), yet how these protein–protein interactions influence normal and pathological signaling in the cell is just beginning to be understood. Although AKAPs differ greatly in sequence, subcellular localization, and repertoire of enzyme binding partners, they all share the defining commonality of a direct interaction with the regulatory subunits (RI or RII) of the PKA holoenzyme [1]. PKA anchoring proceeds through an amphipathic helix that inserts into a customized groove formed by the docking and dimerization (D/D) of R-subunit protomers [2 – 4]. When tethered to AKAPs, the PKA holoenzyme is spatially restricted with access to a limited number of cellular substrates (Fig. 1). This offers a mechanism to selectively promote cellular events that proceed through the ubiquitous second messenger molecule cAMP [5, 6]. However, this PKA-binding module denotes only one facet of AKAP action as other regions of the anchoring protein interact with additional enzymes to integrate other second messenger signals within distinct multivalent assemblies [7 – 9]. Accordingly, these signaling complexes can include other kinases, protein phosphatases, adenylyl cyclases, phosphodiesterases, and target substrates [10 – 14].
Fig. 1.
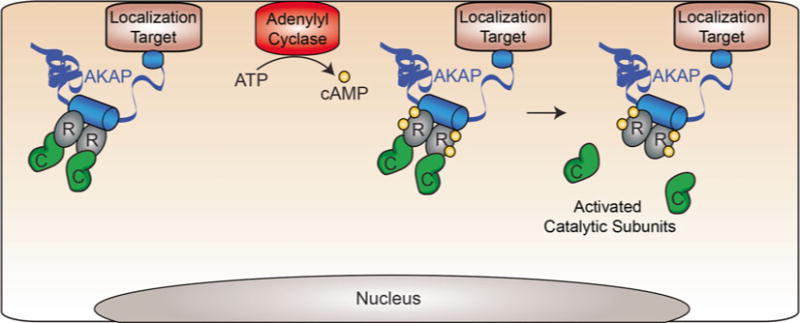
Signaling through AKAP complexes. When intracellular concentrations of cAMP are low, the PKA holoenzyme complex is largely bound to AKAPs within the cell. AKAPs are localized to various intracellular sites including the plasma membrane and organelles, thereby concentrating PKA to particular locations within the cell. Upon stimulation, intracellular cAMP levels rise. Each R-subunit of PKA binds two cAMP molecules and undergoes an allosteric conformational change to release the activated catalytic subunits
The complexity of this cellular system is further compounded by the utilization of four distinct regulatory subunit isoforms of PKA: RI (RIα and RIβ) and RII (RIIα and RIIβ) which differ in tissue distribution, cAMP sensitivity, and AKAP-mediated localization. These additional layers finely tune when and where PKA activity is applied [15]. The vast majority of AKAPs selectively bind the RII isoform; however, a limited number of dual-specific AKAPs can also interact with RI [4, 16, 17]. Due to the spatial and temporal nature of interactions with AKAPs, uncovering the intricacies of AKAP-mediated signaling events has proven to be a substantial challenge. To complicate matters further, the human genome encodes about fifty AKAPs and most cell types express at least 10–15 different anchoring proteins [18]. Added to this, most anchoring proteins are expressed as families of alternatively spliced transcripts [19, 20]. This degree of complexity makes it difficult to elucidate each of their individual roles. However, one strategy to study the role of anchoring in these signaling events is to selectively displace PKA subtypes from the AKAP platform. Accordingly, numerous isoform-specific disruptors have been developed (Fig. 2; Table 1) [21, 22]. Although these compounds are valuable tools to study AKAP–PKA signaling, the major drawback is that these inhibitors will nonspecifically inhibit all AKAP interactions with either the RI or RII isoforms by binding to and occluding the anchoring site on the regulatory subunits.
Fig. 2.

Engineered peptide disruptors of AKAP complexes. Isoform-selective disruptors were developed to have specificity of targeting toward either the RI or RII isoform of PKA. Despite considerable sequence divergence between the different disruptor peptides, they all share the common feature of forming an amphipathic helix with a largely hydrophobic binding interface (shown in gray) that complements the binding surface of the D/D domain of the R-subunits. Asterisks represent incorporation of the nonnatural amino acid (S)-2-(4′-pentenyl)alanine to form an all-hydrocarbon bridge within the sequence
Table 1.
PKA inhibitor compounds for inhibition of AKAP-mediated signaling
| PKA inhibitors | Mechanism of action |
|---|---|
| PKI peptide | Blocks the catalytic site of PKA |
| H89 | ATP-competitive inhibitor of PKA |
| Rp-cAMPS | Prevents cAMP binding to R-subunits |
1.1 RII-Selective Disruptors of AKAP Complexes
The first AKAP disruptor peptide, Ht31, was derived from the PKA-anchoring domain of AKAP-Lbc [23]. In this study, a 23-amino-acid amphipathic helix was identified from a screen seeking to find peptide antagonists of PKA anchoring. The discovery of this peptide set the precedence for defining canonical docking interactions between AKAPs and RII. Although Ht31 has limited cell permeability, chemical modification of the peptide was performed to increase its overall hydrophobicity [24]. The addition of stearic acid to the N-terminus of the peptide was found to greatly enhance cellular permeability. However, there may be concern that the conjugation of such a lipid moiety contributes to retention of Ht31 in cell membranes. Stearated forms of Ht31 and the negative proline analog control (St-Ht31 and St-Ht31P) are widely available as commercial reagents.
Since there is considerable amino acid divergence between the PKA-anchoring helices of various AKAPs, a bioinformatics approach was used to identify an RII-specific consensus sequence [25]. This sequence was then optimized by peptide array screening to identify a more potent RII inhibitor peptide, AKAP-in silico (AKAP-IS) [25]. This peptide was shown to have improved affinity for RII as compared to Ht31 peptide. The K d value of AKAP-IS is less than 1 nM for RII, while it has a Kd value for RI in the mid-high nM range. The initial AKAP-IS peptide was not cell permeable and also had limited solubility in aqueous solution. However, a subsequent modification introduced a TAT sequence at the N-terminus of AKAP-IS to greatly improve cell permeability for cell-based experiments [26]. Despite the hydrophilicity of the TAT sequence, the conjugated peptide, TAT–AKAP-IS, is still highly hydrophobic and requires solubilization in an aqueous 10 % DMSO solution. Using a structure-based approach, AKAP-IS was further optimized to improve the affinity and selectivity to yield SuperAKAP-IS [4]. In order to achieve this, the crystal structure of the AKAP docking site on RIIα was solved either alone or in complex with the inhibitor peptide AKAP-IS [4]. The identification of key residues involved in binding to the RII isoform and the use of further peptide screening arrays allowed for the design of a peptide disruptor with significantly enhanced RII selectivity that had fourfold higher affinity for RII and approximately 12-fold less affinity for RI as compared to AKAP-IS .
Based on the biological observation that AKAP18 has a high affinity for RIIα and that an N-terminally truncated form, AKAP18δ, has an even higher affinity, a new class of disruptor peptides was derived [27]. This class of peptides demonstrated high affinity for RIIα with dissociation constants as low as 0.4 nM. Analysis of sequence divergence between these peptides helped to further define important residues for engagement with the RII docking site. Analogous to Ht31, the AKAP18δ peptides were also modified with the addition of a stearate moiety in order to promote cellular uptake.
Within the last 5 years, small molecules were developed to disrupt AKAP–RII interactions [28, 29]. Very large, relatively flat surfaces, such as the protein–protein interaction interface between the amphipathic helix of an AKAP and the RII D/D docking site, are notoriously difficult to target using small molecule approaches. These small molecule scaffolds are an exciting new area for further investigation. Although these different compounds have limited potency (IC50 = 20–40 μM), this is a promising starting point for compound optimization using a small molecule targeting approach. Moreover, development of more selective small molecule scaffolds could yield anchoring disruptors with improved efficacy as they may evade some of the shortcomings inherent in peptides including limited cell permeability, low stability, and loss of secondary structural folds in solution.
Perhaps the most promising development in anchoring disruptor peptides is the recent introduction of Stapled AKAP Disruptor (STAD) peptides. Chemically modified RII-specific AKAP disruptors were developed where nonnatural amino acids were incorporated into A-kinase binding (AKB) sequences to bestow small-molecule-like properties onto the peptide sequences [21]. Synthetic libraries were designed based on previously identified AKB or AKB-like sequences, where nonnatural olefinic amino acids were incorporated and cyclized so as to conformationally constrain an alpha-helical fold. This chemical modification was previously shown to promote cellular permeability and proteolytic stability to peptides [30]. The STAD peptides developed in this study are highly cell permeable and effectively block interactions between AKAPs and RII inside cells. The incorporation of the peptide “staple” introduced significant hydrophobicity to an already hydrophobic sequence, so the addition of a small PEG-3 linker was added to the N-terminus to notably improve water solubility for cell-based experiments. The rapid cellular uptake, resistance to degradation, and relatively long half-lives in cells of the STAD peptides provide a more flexible platform for studying dynamic AKAP signaling events under a variety of conditions.
All of the PKA-anchoring disruptor reagents discussed thus far have been patterned after an AKAP motif. Recently, a phage selection procedure was employed that exploits high-resolution structural information to engineer RII D/D domain mutants that are selective for a particular AKAP [31]. Competitive selection screening revealed RII sequences (RSelect) that were preferential for interaction with an individual AKAP. Biochemical and cell-based experiments validated the efficacy of RSelect mutants for AKAP2 and AKAP18. This new class of engineered proteins based on the reciprocal surface of the AKAP–PKA interaction has the potential to be used to dissect the contributions of different AKAP-targeted pools of PKA and aid in the design of compounds targeting these subset populations.
1.2 RI-Selective Disruptors of AKAP Complexes
Although numerous RII-specific AKAP disruptors have been identified, designing peptides for RI-selective interactions has proven to be more elusive. The first RI-selective peptide inhibitors were identified through peptide array screening nearly a decade after the design of Ht31 [32]. The prototype used for the peptide array was derived from the A-kinase binding (AKB) domain of AKAP10 [32]. Although the crystal structure of the AKB domain of RI was not solved at the time, the minimal sequence required and surface residue interactions involved in docking to RI were described through systematic analysis. Based on this study, the AKB binding site on RI was shown to involve multiple interactions with charged residues, while the analogous binding site on RII was shown to largely provide a hydrophobic patch for AKB binding. A major limitation of the peptides identified in this study, as with many unmodified peptides, is that they lack cell permeability and therefore require transfection or genetic encoding in order to characterize their activity in cells.
Subsequent studies employed a bioinformatics approach coupled with peptide array screening to yield the RI-selective peptide, RIAD [33]. The binding sequences from several dual-specific AKAPs were used as a starting point to steer toward RI specificity. RIAD was found to have a notably improved binding affinity for RI as well as greater specificity for RI over RII. While the RIAD peptide alone was not cell permeable, the C-terminal addition of 11 arginine residues afforded this property. While transfection can result in artifacts and compensatory expression changes within the cell, the cell-permeable version of RIAD was utilized to illustrate disruption of RI-specific AKAP interactions in intact, non-modified cells. RIAD analogs were later developed that incorporated non-natural and natural amino acids into the sequence to improve proteolytic stability [34]. However, cell permeability of the RIAD analogs remains an issue.
The crystal structure of the docking/dimerization (D/D) domain of RIα was solved in recent years [35]. Numerous structural differences were identified between RI and RII that dictate engagement with various AKB sequences including the depth of the binding groove, the presence of a disulfide bridge within the binding site, and a shift of registry for binding by the AKB sequence. These structural insights will undoubtedly lead to the development of optimized peptide-based or synthetic scaffolds that can discriminate against RII interactions while maintaining high-affinity binding with RI. Additional selectivity in RI anchoring may involve a separate RI-binding interface that is upstream of the amphipathic helix. A distinct region upstream of the docking helix was identified on RI-specific AKAPs [36]. This RI-specific region (RISR) was also shown to disrupt RI binding and may serve as an additional targeting site for RI-specific disruption.
1.3 cAMP-Stimulating Conditions
As a means to interrogate AKAP signaling events in cell-based studies, multiple strategies can be applied to stimulate increased levels of intracellular cAMP (Table 2). While some reagents stimulate cAMP to physiological levels, many cause inappropriately high concentrations of cAMP. Forskolin is perhaps the most widely used stimulator of cAMP production by activating adenylyl cyclase (AC) activity. To date, nearly 10,000 citations list the use of forskolin as a PKA activator. Forskolin is a diterpene natural product isolated from Coleus forskohlii [37] and was found to stimulate cAMP concentrations in diverse tissue types in a reversible manner [38]. Eight of the nine membrane-bound isoforms of AC are stimulated by forskolin [39], with AC9 being the exception [40]. Further, the potency of stimulation varies among the different isoforms [41]. Since expression and regulation of the AC isoforms vary among cell and tissue types, the extent of forskolin-induced stimulation of cAMP can vary considerably and often to levels that are not physiologically relevant [39]. However, since forskolin acts as an agonist for the majority of the AC isoforms, it is considered to be a general, potent stimulator of intracellular cAMP across diverse cell types.
Table 2.
cAMP-stimulating agents for activation of AKAP complexes
| cAMP-stimulating agents | Mechanism of action |
|---|---|
| Forskolin | Activates adenylyl cyclases |
| IBMX | Inhibits PDEs |
| Isoproterenol | Indirectly activates adenylyl cyclases |
| PGE2 | Indirectly activates adenylyl cyclases |
| DB-cAMP | Activates PKA |
Another approach for increasing intracellular cAMP levels is through inhibition of phosphodiesterase (PDE) activity. A nonspecific PDE inhibitor, 3-isobutyl-1-methylxanthine (IBMX), was first identified from a panel screen of various xanthine derivatives to have inhibitory effects on PDEs [42]. IBMX is a moderately potent inhibitor against the majority of PDE isoforms but appears to have no effect on PDE8 or PDE9 [43]. Due to its broad inhibitory activity on PDEs, IBMX is routinely used in conjunction with an AC-stimulating agent such as forskolin to further increase overall intracellular cAMP concentrations. Additional caution must be taken when interpreting results from experiments that use a forskolin/IBMX cocktail to stimulate PKA as this combination treatment stimulates cAMP production to supraphysiological levels and prolongs the second messenger response well beyond its normal time course.
A much more physiologically relevant means to stimulate cAMP production is through activation of β1- and β2-adrenergic receptors by isoproterenol (isoprenaline) [44]. Isoproterenol is a synthetic catecholamine that acts as an agonist for this subclass of G protein-coupled receptors (GPCRs). Upon stimulation of β-adrenergic receptors, Gs proteins are activated inside cells, thereby leading to stimulation of AC activity. After isoproterenol stimulation, cAMP levels rise significantly, but then fall back to near background levels and are resistant to further stimulation even in the presence of persistent isoproterenol treatment [45]. Although β-adrenergic receptors are widely expressed in a variety of cells and isoproterenol can elicit a notable effect on cAMP levels, isoproterenol-stimulated cAMP production is useful for short time-course studies but is not effective as a cAMP-stimulating agent for sustainable periods.
Another physiologically relevant method for cAMP stimulation involves prostaglandin E2 (PGE2). PGE2 is a hormonelike biological compound that binds to a subclass of G protein-coupled receptors called the E prostanoids (EP) [46]. Both EP2 and EP4 can stimulate cAMP production, but EP4 is broadly expressed while EP2 is only expressed in limited tissue/cell types [47]. Although isoproterenol and PGE2 activate different classes of GPCRs, both compounds ultimately lead to activation of Gs proteins and ACs.
A chemical strategy to induce cAMP-sensitive signaling was developed using the cell-permeable cAMP analog, dibutyryl cyclic adenosine monophosphate (DB-cAMP). Although the compound enters the cell in an inactive form, hydrolysis of one of the butyrate groups permits the compound to activate PKA [48]. The butyrate cleavage product was also found to cause significant, unintended secondary effects in cells including differentiation, activation of cell signaling pathways, and growth inhibition [48]. Subsequent mono-butyrated analogs of DB-cAMP are now available that have reduced hydrolysis and therefore have limited off-target effects caused by the butyrate side product [49]. Although there are clear advantages of these DB-cAMP analogs, it remains unclear whether they are resistant to all of the cAMP phosphodiesterase subtypes that exist in a typical cell, in particular PDE8, PDE10, and PDE11 [50].
1.4 PKA Inhibitors
Over 40 years ago, a protein inhibitor of PKA was identified [51]. Protein kinase inhibitor (PKI) is expressed as three isoforms [52] that differ in expression in different tissues and cells and has an affinity for PKA in the sub-micromolar range [53]. A short, 20-amino-acid sequence was identified as the inhibitory component of PKI, and a synthetic peptide spanning this sequence was shown to act as a highly selective, potent inhibitor of PKA [54 – 56]. Multiple analogs derived from this 20-mer sequence were synthesized and tested so as to define the residues that are critical for its inhibitory activity [57 – 59]. This sequence was also found to be highly specific for PKA with no inhibitory effect on PKG [58]. PKI acts as a substrate mimic to block the catalytic site on PKA, thereby preventing substrate phosphorylation [60]. This mechanism provides greater target specificity of PKI for PKA; however, at high concentrations of PKI treatment, off-target effects have been documented [61]. A variety of PKI inhibitor peptide analogs are commercially available that have a high affinity for PKA and are recognized to have exquisite specificity for PKA at lower concentrations.
H89 is an isoquinoline-based small molecule that was derived from an earlier inhibitor, H8 [62]. While H8 targeted both PKA and PKG, H89 was found to be a potent inhibitor of PKA but also had weak antagonistic activity against several other kinases including PKG, PKC, casein kinases I and II, and CamKII [63]. H89 acts as a competitive inhibitor of ATP binding to occupy and prevent substrate phosphorylation. While H89 is an effective inhibitor of PKA, numerous off-target effects have been documented including disruption of various intracellular signaling pathways and inhibition of a significant number of kinases, including some that were inhibited at greater levels than PKA [64]. Although H89 is among the most commonly used of all PKA inhibitors, caution should be used in interpretation of results due to its numerous off-target effects.
Cyclic nucleotide analogs such as Rp-cAMPS (adenosine-3′,5′-cyclic monophosphorothioate Rp-isomer) have also been used as an inhibitory agent for PKA. Rp-cAMPS is cell permeable and acts as an antagonist of cAMP to prevent activation of PKA by binding to the cAMP-binding sites on the regulatory subunits of PKA [65, 66]. This cAMP analog also demonstrates resistance to hydrolysis by phosphodiesterases. Although Rp-cAMPS has limited cell permeability, newer versions such as Rp-8-Br-cAMPS and Rp-8-Cl-cAMPS are recognized to have improved permeability and greater potency [67]. Yet, since additional signaling elements bind cAMP, it is possible that these analogs may also have other cellular targets aside from PKA-R and can thereby cause unintended secondary effects.
1.5 Practical Considerations
There are clear advantages of screening peptide disruptors in vitro prior to their use for cellular analysis. This is particularly relevant since cross talk is extremely common in kinase signaling cascades and AKAP-specific signaling events are not fully elucidated. Strategies that specifically interrogate the physical interaction between AKAPs and the D/D binding groove of the R-subunits provide a clearer route to identify disruptors. These interactions can be further validated using competitive binding experiments using a known disruptor such as Ht-31 or RIAD to confirm binding to the same interaction surface. A common strategy used for screening is fluorescence polarization where binding of potential disruptors is measured in solution using an increasing concentration of disruptor. This will provide dissociation constant for each disruptor and is a critical first step before entering more complex, cell-based experiments. Outlined below is a protocol that we have used to develop and characterize these anchoring disruptor peptides.
2 Materials
3 Methods
For each protein concentration to be tested, prepare an 80 μL solution of fluorescently labeled peptide (diluted in assay buffer; see Note 1) to a microcentrifuge tube at a concentration of 20 nM. The peptide should be clear in solution with no visible precipitate (see Note 2). Once the protein is added, the final concentration will be 10 nM.
In a separate microcentrifuge tube, create serial dilution stocks of RI or RII in assay buffer at a 2 × concentration (see Note 3). The final protein concentration should be tested to as high a concentration as possible to reach a plateau of nonbinding. For the D/D domains of RI and RII, higher concentrations should reach 50–100 μM. The protein should be tested over a serial dilution range down to 0.1 nM or lower, depending on the affinity of the peptide. The protein should be serially diluted two- to tenfold over this concentration range. For each protein concentration, prepare 80 μL of stock solution.
Combine the peptide solution with each solution of the different protein concentrations (80 μL protein and 80 μL peptide) and gently invert multiple times to ensure proper mixing (see Note 4). Add 50 μL of the solution to each of three wells.
Repeat step 3 for each protein concentration tested.
As a negative control lacking protein, combine 80 μL peptide (20 nM) diluted in assay buffer with 80 μL assay buffer. Invert to mix and plate 50 μL per well into three wells.
As a positive control, a known AKAP inhibitor peptide can be used (see Note 5). Combine 80 μL of the control peptide (20 nM in assay buffer) with either RI or RII over the same concentration range (80 μL per concentration). For each concentration, plate 50 μL in triplicate.
Store the plate in the dark at room temperature for 30–60 min before reading (see Note 6).
Read the plate using absorbance/emission values that are suitable for the peptide label and obtain FP values for each well.
Convert polarization values to anisotropy to determine the relative KD values for each inhibitor peptide tested.
Acknowledgments
This work was supported, in whole or in part, by National Institutes of Health Grants 1K22CA154600 to EJK, and DK105542 and DK054441 to JDS.
Footnotes
Many AKAP disruptor peptides are extremely hydrophobic. To facilitate solubility in buffer, concentrated peptide stock solutions are prepared in DMSO, often in the range of 1–10 mM. Tenfold serial dilutions of the peptide in buffer are performed to accurately reach a 10 nM final concentration.
If the peptide precipitates out of solution, sonication of the solution may promote solubility of the peptide. As an alternative, a minimal amount of peptide-solubilizing agent may need to be added to the media (e.g., DMSO).
To maintain protein stability in solution, the protein dilutions are prepared on ice using freshly thawed protein.
Inverting the solution should be performed with great care to minimize the introduction of bubbles in solution, which will interfere with fluorescence readings. If bubbles are present, they may be removed by low-speed centrifugation of the microplate.
A standard AKAP disruptor and its corresponding negative control that is commercially available are Ht31 and Ht31P.
Longer or shorter incubation times may be necessary for optimal results. As an initial trial, the plate can be analyzed every 30 min over a 2-h time course to identify an optimal time point for analysis.
References
- 1.Scott JD, Pawson T. Cell signaling in space and time: where proteins come together and when they’re apart. Science. 2009;326:1220–1224. doi: 10.1126/science.1175668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carr DW, Stofko-Hahn RE, Fraser ID, et al. Interaction of the regulatory subunit (RII) of cAMP-dependent protein kinase with RII-anchoring proteins occurs through an amphipathic helix binding motif. J Biol Chem. 1991;266:14188–14192. [PubMed] [Google Scholar]
- 3.Newlon MG, Roy M, Morikis D, et al. A novel mechanism of PKA anchoring revealed by solution structures of anchoring complexes. EMBO J. 2001;20:1651–1662. doi: 10.1093/emboj/20.7.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gold MG, Lygren B, Dokurno P, et al. Molecular basis of AKAP specificity for PKA regulatory subunits. Mol Cell. 2006;24:383–395. doi: 10.1016/j.molcel.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 5.Welch EJ, Jones BW, Scott JD. Networking with AKAPs: context-dependent regulation of anchored enzymes. Mol Interv. 2010;10:86–97. doi: 10.1124/mi.10.2.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Skroblin P, Grossmann S, Schafer G, et al. Mechanisms of protein kinase A anchoring. Int Rev Cell Mol Biol. 2010;283:235–330. doi: 10.1016/S1937-6448(10)83005-9. [DOI] [PubMed] [Google Scholar]
- 7.Dessauer CW. Adenylyl cyclase – A-kinase anchoring protein complexes: the next dimension in cAMP signaling. Mol Pharmacol. 2009;76:935–941. doi: 10.1124/mol.109.059345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanderson JL, Dell’Acqua ML. AKAP signaling complexes in regulation of excitatory synaptic plasticity. Neuroscientist. 2011;17:321–336. doi: 10.1177/1073858410384740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diviani D, Dodge-Kafka KL, Li J, et al. A-kinase anchoring proteins: scaffolding proteins in the heart. Am J Physiol Heart Circ Physiol. 2011;301:H1742–H1753. doi: 10.1152/ajpheart.00569.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klauck TM, Faux MC, Labudda K, et al. Coordination of three signaling enzymes by AKAP79, a mammalian scaffold protein. Science. 1996;271:1589–1592. doi: 10.1126/science.271.5255.1589. [DOI] [PubMed] [Google Scholar]
- 11.Coghlan VM, Hausken ZE, Scott JD. Subcellular targeting of kinases and phosphatases by association with bifunctional anchoring proteins. Biochem Soc Trans. 1995;23:592–596. doi: 10.1042/bst0230592. [DOI] [PubMed] [Google Scholar]
- 12.Dodge KL, Khouangsathiene S, Kapiloff MS, et al. mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. EMBO J. 2001;20:1921–1930. doi: 10.1093/emboj/20.8.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tasken KA, Collas P, Kemmner WA, et al. Phosphodiesterase 4D and protein kinase a type II constitute a signaling unit in the centrosomal area. J Biol Chem. 2001;276:21999–22002. doi: 10.1074/jbc.C000911200. [DOI] [PubMed] [Google Scholar]
- 14.Bauman AL, Soughayer J, Nguyen BT, et al. Dynamic regulation of cAMP synthesis through anchored PKA-adenylyl cyclase V/VI complexes. Mol Cell. 2006;23:925–931. doi: 10.1016/j.molcel.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor SS, Yang J, Wu J, et al. PKA: a portrait of protein kinase dynamics. Biochim Biophys Acta. 2004;1697:259–269. doi: 10.1016/j.bbapap.2003.11.029. [DOI] [PubMed] [Google Scholar]
- 16.Herberg FW, Maleszka A, Eide T, et al. Analysis of A-kinase anchoring protein (AKAP) interaction with protein kinase A (PKA) regulatory subunits: PKA isoform specificity in AKAP binding. J Mol Biol. 2000;298:329–339. doi: 10.1006/jmbi.2000.3662. [DOI] [PubMed] [Google Scholar]
- 17.Aye TT, Mohammed S, van den Toorn HW, et al. Selectivity in enrichment of cAMP- dependent protein kinase regulatory subunits type I and type II and their interactors using modified cAMP affinity resins. Mol Cell Proteomics. 2009;8:1016–1028. doi: 10.1074/mcp.M800226-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lester LB, Coghlan VM, Nauert B, et al. Cloning and characterization of a novel A-kinase anchoring protein. AKAP 220, association with testicular peroxisomes. J Biol Chem. 1996;271:9460–9465. doi: 10.1074/jbc.271.16.9460. [DOI] [PubMed] [Google Scholar]
- 19.Trotter KW, Fraser ID, Scott GK, et al. Alternative splicing regulates the subcellular localization of A-kinase anchoring protein 18 isoforms. J Cell Biol. 1999;147:1481–1492. doi: 10.1083/jcb.147.7.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang LJ, Durick K, Weiner JA, et al. Identification of a novel protein kinase A anchoring protein that binds both type I and type II regulatory subunits. J Biol Chem. 1997;272:8057–8064. doi: 10.1074/jbc.272.12.8057. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Ho TG, Bertinetti D, et al. Isoform-selective disruption of AKAP-localized PKA using hydrocarbon stapled peptides. ACS Chem Biol. 2014;9:635–642. doi: 10.1021/cb400900r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scott JD, Dessauer CW, Tasken K. Creating order from chaos: cellular regulation by kinase anchoring. Annu Rev Pharmacol Toxicol. 2013;53:187–210. doi: 10.1146/annurev-pharmtox-011112-140204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carr DW, Hausken ZE, Fraser ID, et al. Association of the type II cAMP-dependent protein kinase with a human thyroid RII-anchoring protein. Cloning and characterization of the RII-binding domain. J Biol Chem. 1992;267:13376–13382. [PubMed] [Google Scholar]
- 24.Vijayaraghavan S, Goueli SA, Davey MP, et al. Protein kinase A-anchoring inhibitor peptides arrest mammalian sperm motility. J Biol Chem. 1997;272:4747–4752. doi: 10.1074/jbc.272.8.4747. [DOI] [PubMed] [Google Scholar]
- 25.Alto NM, Soderling SH, Hoshi N, et al. Bioinformatic design of A-kinase anchoring protein-in silico: a potent and selective peptide antagonist of type II protein kinase A anchoring. Proc Natl Acad Sci U S A. 2003;100:4445–4450. doi: 10.1073/pnas.0330734100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Faruque OM, Le-Nguyen D, Lajoix AD, et al. Cell-permeable peptide-based disruption of endogenous PKA-AKAP complexes: a tool for studying the molecular roles of AKAP-mediated PKA subcellular anchoring. Am J Physiol Cell Physiol. 2009;296:C306–C316. doi: 10.1152/ajpcell.00216.2008. [DOI] [PubMed] [Google Scholar]
- 27.Hundsrucker C, Krause G, Beyermann M, et al. High-affinity AKAP7delta-protein kinase A interaction yields novel protein kinase A-anchoring disruptor peptides. Biochem J. 2006;396:297–306. doi: 10.1042/BJ20051970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Christian F, Szaszak M, Friedl S, et al. Small molecule AKAP-protein kinase A (PKA) interaction disruptors that activate PKA interfere with compartmentalized cAMP signaling in cardiac myocytes. J Biol Chem. 2011;286:9079–9096. doi: 10.1074/jbc.M110.160614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schafer G, Milic J, Eldahshan A, et al. Highly functionalized terpyridines as competitive inhibitors of AKAP-PKA interactions. Angew Chem Int Ed Engl. 2013;52:12187–12191. doi: 10.1002/anie.201304686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verdine GL, Hilinski GJ. Stapled peptides for intracellular drug targets. Methods Enzymol. 2012;503:3–33. doi: 10.1016/B978-0-12-396962-0.00001-X. [DOI] [PubMed] [Google Scholar]
- 31.Gold MG, Fowler DM, Means CK, et al. Engineering A-kinase anchoring protein (AKAP)-selective regulatory subunits of protein kinase A (PKA) through structure-based phage selection. J Biol Chem. 2013;288:17111–17121. doi: 10.1074/jbc.M112.447326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burns-Hamuro LL, Ma Y, Kammerer S, et al. Designing isoform-specific peptide disruptors of protein kinase A localization. Proc Natl Acad Sci U S A. 2003;100:4072–4077. doi: 10.1073/pnas.2628038100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carlson CR, Lygren B, Berge T, et al. Delineation of type I protein kinase A-selective signaling events using an RI anchoring disruptor. J Biol Chem. 2006;281:21535–21545. doi: 10.1074/jbc.M603223200. [DOI] [PubMed] [Google Scholar]
- 34.Torheim EA, Jarnaess E, Lygren B, et al. Design of proteolytically stable RI-anchoring disruptor peptidomimetics for in vivo studies of anchored type I protein kinase A-mediated signalling. Biochem J. 2009;424:69–78. doi: 10.1042/BJ20090933. [DOI] [PubMed] [Google Scholar]
- 35.Sarma GN, Kinderman FS, Kim C, et al. Structure of D-AKAP2:PKA RI complex: insights into AKAP specificity and selectivity. Structure. 2010;18:155–166. doi: 10.1016/j.str.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jarnaess E, Ruppelt A, Stokka AJ, et al. Dual specificity A-kinase anchoring proteins (AKAPs) contain an additional binding region that enhances targeting of protein kinase A type I. J Biol Chem. 2008;283:33708–33718. doi: 10.1074/jbc.M804807200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhat SV, Bajwa BS, Dornauer H, de Souza NJ. Structure and stereochemistry of new labdane diterpenoids from Coleus forskohlii Briq. Tetrahedron Lett. 1977;19:1669–1672. [Google Scholar]
- 38.Seamon KB, Padgett W, Daly JW. Forskolin: unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proc Natl Acad Sci U S A. 1981;78:3363–3367. doi: 10.1073/pnas.78.6.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patel TB, Du Z, Pierre S, et al. Molecular biological approaches to unravel adenylyl cyclase signaling and function. Gene. 2001;269:13–25. doi: 10.1016/s0378-1119(01)00448-6. [DOI] [PubMed] [Google Scholar]
- 40.Premont RT, Matsuoka I, Mattei MG, et al. Identification and characterization of a widely expressed form of adenylyl cyclase. J Biol Chem. 1996;271:13900–13907. doi: 10.1074/jbc.271.23.13900. [DOI] [PubMed] [Google Scholar]
- 41.Dessauer CW, Scully TT, Gilman AG. Interactions of forskolin and ATP with the cytosolic domains of mammalian adenylyl cyclase. J Biol Chem. 1997;272:22272–22277. doi: 10.1074/jbc.272.35.22272. [DOI] [PubMed] [Google Scholar]
- 42.Beavo JA, Rogers NL, Crofford OB, et al. Effects of xanthine derivatives on lipolysis and on adenosine 3′,5′-monophosphate phosphodiesterase activity. Mol Pharmacol. 1970;6:597–603. [PubMed] [Google Scholar]
- 43.Omori K, Kotera J. Overview of PDEs and their regulation. Circ Res. 2007;100:309–327. doi: 10.1161/01.RES.0000256354.95791.f1. [DOI] [PubMed] [Google Scholar]
- 44.Robison GA, Butcher RW, Sutherland EW. Cyclic AMP. Annu Rev Biochem. 1968;37:149–174. doi: 10.1146/annurev.bi.37.070168.001053. [DOI] [PubMed] [Google Scholar]
- 45.Shear M, Insel PA, Melmon KL, et al. Agonist-specific refractoriness induced by isoproterenol. Studies with mutant cells. J Biol Chem. 1976;251:7572–7576. [PubMed] [Google Scholar]
- 46.Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem. 2007;282:11613–11617. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- 47.Zeng L, An S, Goetzl EJ. EP4/EP2 receptor-specific prostaglandin E2 regulation of interleukin-6 generation by human HSB.2 early T cells. J Pharmacol Exp Ther. 1998;286:1420–1426. [PubMed] [Google Scholar]
- 48.Schwede F, Maronde E, Genieser H, et al. Cyclic nucleotide analogs as biochemical tools and prospective drugs. Pharmacol Ther. 2000;87:199–226. doi: 10.1016/s0163-7258(00)00051-6. [DOI] [PubMed] [Google Scholar]
- 49.Zorn M, Maronde E, Jastorff B, et al. Differential effects of two structurally related N6-substituted cAMP analogues on C6 glioma cells. Eur J Cell Biol. 1993;60:351–357. [PubMed] [Google Scholar]
- 50.Beavo JA, Brunton LL. Cyclic nucleotide research – still expanding after half a century. Nat Rev Mol Cell Biol. 2002;3:710–718. doi: 10.1038/nrm911. [DOI] [PubMed] [Google Scholar]
- 51.Walsh DA, Ashby CD, Gonzalez C, et al. Krebs EG: purification and characterization of a protein inhibitor of adenosine 3′,5′-monophosphate-dependent protein kinases. J Biol Chem. 1971;246:1977–1985. [PubMed] [Google Scholar]
- 52.Dalton GD, Dewey WL. Protein kinase inhibitor peptide (PKI): a family of endogenous neuropeptides that modulate neuronal cAMP-dependent protein kinase function. Neuropeptides. 2006;40:23–34. doi: 10.1016/j.npep.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 53.Demaille JG, Peters KA, Fischer EH. Isolation and properties of the rabbit skeletal muscle protein inhibitor of adenosine 3′,5′-monophosphate dependent protein kinases. Biochemistry. 1977;16:3080–3086. doi: 10.1021/bi00633a006. [DOI] [PubMed] [Google Scholar]
- 54.Scott JD, Fischer EH, Demaille JG, et al. Identification of an inhibitory region of the heat-stable protein inhibitor of the cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1985;82:4379–4383. doi: 10.1073/pnas.82.13.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scott JD, Fischer EH, Takio K, et al. Amino acid sequence of the heat-stable inhibitor of the cAMP-dependent protein kinase from rabbit skeletal muscle. Proc Natl Acad Sci U S A. 1985;82:5732–5736. doi: 10.1073/pnas.82.17.5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheng HC, van Patten SM, Smith AJ, et al. An active twenty-amino-acid-residue peptide derived from the inhibitor protein of the cyclic AMP-dependent protein kinase. Biochem J. 1985;231:655–661. doi: 10.1042/bj2310655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scott JD, Glaccum MB, Fischer EH, et al. Primary-structure requirements for inhibition by the heat-stable inhibitor of the cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1986;83:1613–1616. doi: 10.1073/pnas.83.6.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Glass DB, Cheng HC, Kemp BE, et al. Differential and common recognition of the catalytic sites of the cGMP-dependent and cAMP-dependent protein kinases by inhibitory peptides derived from the heat-stable inhibitor protein. J Biol Chem. 1986;261:12166–12171. [PubMed] [Google Scholar]
- 59.Cheng HC, Kemp BE, Pearson RB, et al. A potent synthetic peptide inhibitor of the cAMP-dependent protein kinase. J Biol Chem. 1986;261:989–992. [PubMed] [Google Scholar]
- 60.Reed J, De Ropp JS, Trewhella J, et al. Conformational analysis of PKI(5–22)amide, the active inhibitory fragment of the inhibitor protein of the cyclic AMP-dependent protein kinase. Biochem J. 1989;264:371–380. doi: 10.1042/bj2640371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Glass DB, Feller MJ, Levin LR, et al. Structural basis for the low affinities of yeast cAMP-dependent and mammalian cGMP-dependent protein kinases for protein kinase inhibitor peptides. Biochemistry. 1992;31:1728–1734. doi: 10.1021/bi00121a021. [DOI] [PubMed] [Google Scholar]
- 62.Hidaka H, Inagaki M, Kawamoto S, et al. Isoquinolinesulfonamides, novel and potent inhibitors of cyclic nucleotide dependent protein kinase and protein kinase C. Biochemistry. 1984;23:5036–5041. doi: 10.1021/bi00316a032. [DOI] [PubMed] [Google Scholar]
- 63.Chijiwa T, Mishima A, Hagiwara M, et al. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino) ethyl] - 5 - isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J Biol Chem. 1990;265:5267–5272. [PubMed] [Google Scholar]
- 64.Murray AJ. Pharmacological PKA inhibition: all may not be what it seems. Sci Signal. 2008;1:re4. doi: 10.1126/scisignal.122re4. [DOI] [PubMed] [Google Scholar]
- 65.Rothermel JD, Stec WJ, Baraniak J, et al. Inhibition of glycogenolysis in isolated rat hepatocytes by the Rp diastereomer of adenosine cyclic 3′,5′-phosphorothioate. J Biol Chem. 1983;258:12125–12128. [PubMed] [Google Scholar]
- 66.Rothermel JD, Jastorff B, Botelho LH. Inhibition of glucagon-induced glycogenolysis in isolated rat hepatocytes by the Rp diastereomer of adenosine cyclic 3′,5′-phosphorothioate. J Biol Chem. 1984;259:8151–8155. [PubMed] [Google Scholar]
- 67.Gjertsen BT, Mellgren G, Otten A, et al. Novel (Rp)-cAMPS analogs as tools for inhibition of cAMP-kinase in cell culture. Basal cAMP-kinase activity modulates interleukin-1 beta action. J Biol Chem. 1995;270:20599–20607. doi: 10.1074/jbc.270.35.20599. [DOI] [PubMed] [Google Scholar]
- 68.Kinderman FS, Kim C, von Daake S, et al. A dynamic mechanism for AKAP binding to RII isoforms of cAMP-dependent protein kinase. Mol Cell. 2006;24:397–408. doi: 10.1016/j.molcel.2006.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Banky P, Newlon MG, Roy M, et al. Isoform-specific differences between the type Ialpha and IIalpha cyclic AMP-dependent protein kinase anchoring domains revealed by solution NMR. J Biol Chem. 2000;275:35146–35152. doi: 10.1074/jbc.M003961200. [DOI] [PubMed] [Google Scholar]