

INTRODUCTION
Helicobacter pylori (Hp) plays an important role in the upper digestive tract diseases. It can be divided into two main groups (toxic and non-toxic Hp) according to the production of vacuolating cytotoxin (VacA). The toxic bacteria also produce cytotoxin associated protein A (CagA) which might have something to do with the transcription, folding, transportation or the function of VacA. Studies showed that CagA positive Hp (CagA+Hp) accounted for more than 50% of all kinds of Hp, and peptic ulcer and gastric cancer were closely related to their infection[1-7]. Therefore, detection of the infection of CagA+Hp is of great significance. This study was carried out to express CagA gene in E.coli and develop an immuno-assay for the rapid detection of CagA+Hp.
MATERIAL AND METHODS
Material
pMC3, plasmid that contains most parts of the 5’-end of CagA was kindly given by Dr. Tummuru (Vanderbilt University, USA); pBV220 (preserved in our institute); superdex HR75 (Pharmacia, USA); Wizard plus milipres DNA purification kits (Pr omega, USA); ELISA detecting kits for anti-CagA antibody (Jing Ying Biotech Company, Shanghai); thermolyne PCR Amplitron (USA); Hermle Z323K refrigerated high speed centrifuge (Germany); DU-640-nucleic acid protein analyser (Bechman, USA ); 373 Auto-DNA sequencer, (Bechman Company, USA); bio-Rad mini-protein II electrophoresis (Joss Bio-Lab Company); sera from patients and healthy people (Xijing Hospital, Xi’an).
Methods
CagA gene cloning The oligonucleotide primers were designed and synthesized to amplify a 2544 bp fragment at the 5’-end of CagA according to its sequence[8]. The primers were CagAp1 (5’-TCGCCC-GGGATGACTAACGAAACTATTGACC-3’) and CagAp2 (5’-CAGGTCGACTTAGACTAGGG-TTCCGTTCACAC-3’). At the 5’-end of each primer, there was a restriction endo-nuclease site (SmaI or SalI), which was favorable for the cloning of PCR products. Fifty microlitre polymerase chain reaction mixture contained 0.4 μmol/L primers, 0.2 mmol/L dNTPs, 2 units Taq-DNA polymerase, 1 ng pMC3, 100 mmol/L Tris-HCl, 50 mmol/L KCl, and 15 mmol/L MgCl2. The CagA was amplified by denaturing at 94 °C for 1 min, annealing at 55 °C for 1 min, and extending at 72 °C for 3 min, and the procedures were repeated 30 times. The PCR product was analyzed by 15g/L agarose gel containing 0.5mg/L ethidium bromide (EB). The bands were examined under ultraviolet light and recovered by low melt point agarose gel. By preparing the pUC19 with alkaline dissolving method, we coloned the CagA gene fragment into it for sequencing.
CagA gene expression First, we digested pUC19/ CagA with SmaI and SalI separately, and cloned the 2544 bp fragment into pBV220, but this gene fragment failed to express in DH5α. We then digested the pUC19/CagA with Eco-RI and Bam-HI, and recovered the 5’-end fragment of CagA (FCagA, 854 bp), which was then subcloned into vector pBV220 to form pBV220/FCagA. After transforming the competent DH5α with the pBV220/FCagA, the clones that could grow in Luria-Bertani (LB) plate containing ampicillinum (100 mg/L) were selected and cultured in LB solution at 30 °C overnight. We diluted (1:100) the cultured mixture of a positive clone in LB and let it grow at 30 °C till the late stage of logarithmic. Then we immediately raised the temperature to 42 °C and kept for 4 h. The heat induced fragment of CagA (FCagA) was analyzed by 12% sodiumdodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE).
Purification of FCagA Differential test showed that most of the FCagA existed in inclusion body, of which only 2% was in the supernatant after the lysate was centrifuged at 12000 × g for 10 min. The soluble protein was precipitated by 50% saturated ammonium sulphate and resuspended in phosphate buffered saline (PBS) containing 1.3 mmol/L NaH2PO4, 11 mmol/L Na2HPO4 and 140 mmol/L NaCl. Three millilitre mixture was applied to Superdex HR75 column (100 mL volume bed), and eluted with PBS in a HPLC protein purification system. The active peak was collected after being tested by dot immonogold filtration assay (DIGFA), with staphylococci protein A as the second antibody and coloidgold as the marker[9]. The solution was then lyophilized and dialysed against PBS to eliminate extra salt.
Rapid detection anti-CagA antibody by DIGFA The coloidgold and immunogold were prepared according to the published articles[10,11]. The purified FCagA was immobilized on the nitroce-llulose membrane. To block n on-specific conjugate sites, the membrane was immerged in 5% bovine serum album in (BSA) for 2 h, then washed with PBS three times and laid to dry automati cally. Later, we assembled the DIGFA detecting kits. After a drop of serum was a dded to the membrane and passed through it, the immunogold was added. If the serum contained anti-CagA antibody, there would be a red dot on the membrane, othe rwise, there would be no dot.
RESULTS
Cloning and sequencing of CagA
With pMC3 used as template, CagAp1 and CagAp2 as the primers, the 5’-end of CagA was amplified by polymerase chain reaction. The PCR products were analyzed under ultraviolet light after 1.5% agarose gel electrophoresis (Figure 1). We then cloned the 2544 bp CagA fragment into pUC19 for sequencing. The results showed that the PCR product was indeed a portion sequence of CagA, from which the 854 bp upstream fragment of CagA was achieved when digested with Bam-HI (Figure 2).
Figure 1.
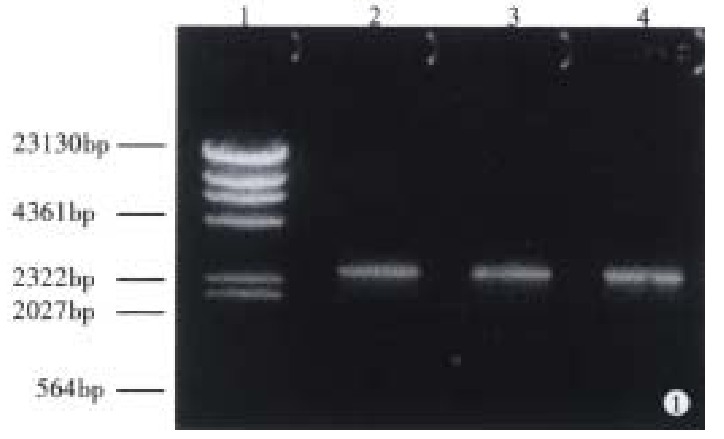
Analysis of the PCR product of CagA by agarose gel electrophoresis. 1: λ DNA/Hind-III markers; 2: PCR product of CagA.
Figure 2.
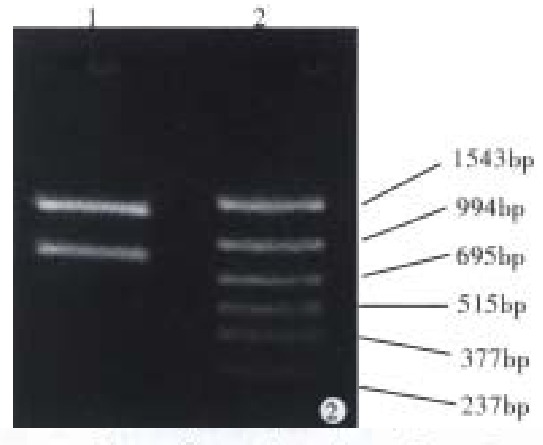
Analysis of CagA fragment by agarose gel electrophoresis. 1: CagA fragments digested by Bam-HI; 2: PCR markers.
Expression of CagA and purification of its products
Subcloning FCagA into vector pBV220 and transforming DH5α with pBV220/FCagA permitted the expression of FCagA with a MW of 38 kDa when the culture temperature reached 42 °C (Figure 3). The new protein mainly existed in inclusion body with only 2% soluble. When the supernatant of the bacteriumlysate was precipitated with ammonium sulphate and the FCagA was purified by Superdex HR75 column, only one protein band of 38 kDa could be seen when the collected fraction correspondent to the active peak was analyzed by 12% SDS PAGE (Figure 4).
Figure 3.
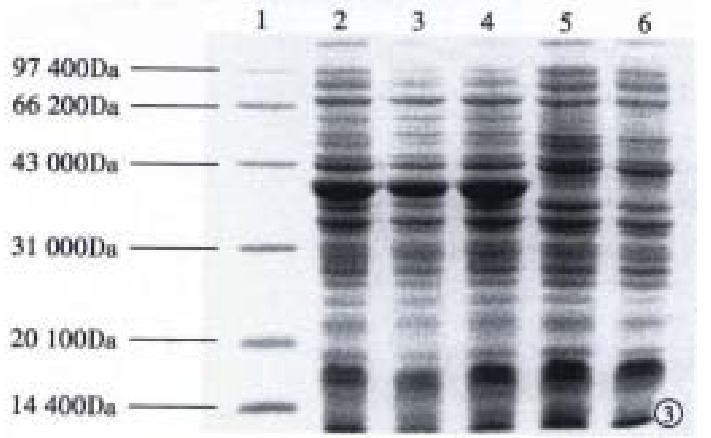
Analysis of FCagA expression by 12% SDS-PAGE.-1: Protein markers; 2, 3, 4: Heat induced pBV220/FCagA-DH5α; 5:pBV220/FCagA-DH5α; 6: Heat induced pBV220/ DH5α.
Figure 4.
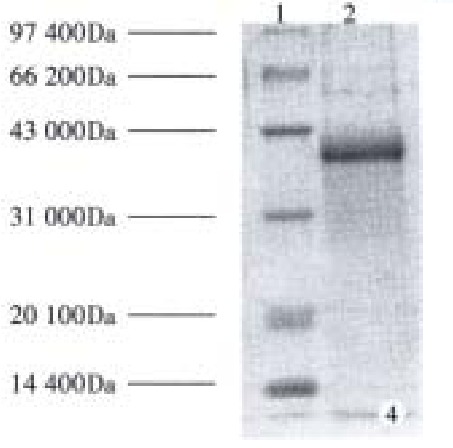
Determination of the purified FCagA by 12% SDS-PAGE. 1: Protein markers; 2: FCagA.
The antigenicity of FcagA
To identify the antigenicity of FCagA, DIGFA was performed with FCagA acting as antigen. The results showed that FCagA had the same antigenicity as CagA when 20 samples of anti-antibody positive serum were tested.
Specifications of the DIGFA kits
① Tests of specificity and sensitivity. Using the DIGFA kits, we detected the anti-CagA IgG in 262 cases. Comparing with ELISA, the specificity of DIGFA was 98.5%, the sensitivity was 96.8%, and the coincident rate was 97.7% (Table 1). ② Duplicate tests. From the 262 serum samples, we randomly selected 100 sera for the detection of anti-CagA IgG. The experiment was repeated 5 times and the same result was got: 42 serum positive, 58 serum negative. ③ Blocking tests. When 20 anti-CagA IgG positive sera were randomly chosen, a mixture of 10 µL serum and excess amount of FCagA (10 µg) would make the result negative, and a mixture of serum and BSA did not change the positive result, indicating that FCagA can specifically bind anti-CagA IgG. ④ Stability. The stability tests showed that the specifications of the kits were stable for at least 6 months at 4 °C.
Table 1.
Results of anti-CagA IgG in sera detected by DIGFA and ELISA
|
ELISA |
Total | |||
| Positive | Negative | |||
| DIGFA | Positive | 123 | 2 | 125 |
| Negative | 4 | 133 | 137 | |
| Total | 1 2 7 | 135 | 262 | |
DISCUSSION
Hp CagA is a bacterium surface protein with a MW of 120 kDa-128 kDa. A dominant characteristic of this protein is its high antigenicity[1]. The infection of CagA positive Hp results in local and systemic humoral immuno-reaction, and there is high concentration of anti-CagA in the patient’s blood and gastric juice[3,6]. This is the base of immuno-detection of anti-CagA antibody. Most recombinants of CagA previously expressed were at its carbonic end and the antigenicity of its amino end has not been reported. Since the 5’-end of CagA had no usable restriction endonuclease sites, we used PCR to amplify the 5’-end fragment of CagA, and cloned it into pBV220[12]. Subsequently the DH5α was transformed by pBV220/FCagA, and a 38 kDa recombinant was induced by heat when temperature reached 42 °C. The new protein accounted for 33% of the total bacterium proteins and could combine with anti-CagA IgG, indicating that the recombinant had the antigenicity of CagA. Purification of the soluble part of the recombinant protein made it possible to develop immuno-detecting method for CagA+Hp.
At present, the detecting methods of Hp infection can be divided into two categories: injury and non-injury. Since the injury methods are based on the gastrointestinal endoscope, the non-injury methods are preferred, among which the rapid urease test has low specificity, and the ELISA takes too much time. The same problems exist in the detection of CagA+Hp. Since the infection rate of CagA+Hp is high and its harmful effects on human being are great, it is necessary to develop rapid, simple, and non-injury approaches to detect the infection of CagA+Hp.
DIGFA is an immuno-method developed in recent years. In this method, the red immunogold is used as the marker and nitrocellulose as the substrate, which has the function of absorption, filtration, concentration, and capillary, and can let the antigen-antibody reaction take place rapidly. In this study, we established an anti-CagA assay and developed the test kits which have great applicable value and high sensitivity and specificity. This may contribute to the epidemic study of the infection of CagA+Hp in a large scale.
Footnotes
Edited by Wang XL
References
- 1.Covacci A, Censini S, Bugnoli M, Petracca R, Burroni D, Macchia G, Massone A, Papini E, Xiang Z, Figura N. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc Natl Acad Sci USA. 1993;90:5791–5795. doi: 10.1073/pnas.90.12.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ching CK, Wong BC, Kwok E, Ong L, Covacci A, Lam SK. Prevalence of CagA-bearing Helicobacter pylori strains detected by the anti-CagA assay in patients with peptic ulcer disease and in controls. Am J Gastroenterol. 1996;91:949–953. [PubMed] [Google Scholar]
- 3.Crabtree JE, Wyatt JI, Sobala GM, Miller G, Tompkins DS, Primrose JN, Morgan AG. Systemic and mucosal humoral responses to Helicobacter pylori in gastric cancer. Gut. 1993;34:1339–1343. doi: 10.1136/gut.34.10.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blaser MJ, Perez-Perez GI, Kleanthous H, Cover TL, Peek RM, Chyou PH, Stemmermann GN, Nomura A. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 1995;55:2111–2115. [PubMed] [Google Scholar]
- 5.Queiroz DMM, Mendes EN, Rocha GA, Soares TF, Lima Jr GF, Oliveira CA. H. pylori strains possessing cagA and vacuolating cytotoxin producers are associated to both types of gastric carcinoma. Gastroenterology. 1996;110:A236. [Google Scholar]
- 6.Crabtree JE, Taylor JD, Wyatt JI, Heatley RV, Shallcross TM, Tompkins DS, Rathbone BJ. Mucosal IgA recognition of Helicobacter pylori 120 kDa protein, peptic ulceration, and gastric pathology. Lancet. 1991;338:332–335. doi: 10.1016/0140-6736(91)90477-7. [DOI] [PubMed] [Google Scholar]
- 7.Klaamas K, Held M, Wadström T, Lipping A, Kurtenkov O. IgG immune response to Helicobacter pylori antigens in patients with gastric cancer as defined by ELISA and immunoblotting. Int J Cancer. 1996;67:1–5. doi: 10.1002/(SICI)1097-0215(19960703)67:1<1::AID-IJC1>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 8.Tummuru MK, Cover TL, Blaser MJ. Cloning and expression of a high-molecular-mass major antigen of Helicobacter pylori: evidence of linkage to cytotoxin production. Infect Immun. 1993;61:1799–1809. doi: 10.1128/iai.61.5.1799-1809.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han FC, Yan XJ, Zhang L, Hou Y, Su CZ, Zhang LX, Jiang M. Dot immunogold filtration assay for anti-Helicobacter pylori antibody. Shijie Huaren Xiaohua Zazhi. 1998;6:407–408. [Google Scholar]
- 10.De Roe C, Courtoy PJ, Baudhuin P. A model of protein-colloidal gold interactions. J Histochem Cytochem. 1987;35:1191–1198. doi: 10.1177/35.11.3655323. [DOI] [PubMed] [Google Scholar]
- 11.Wang BL, Scopsi L, Hartvig Nielsen M, Larsson LI. Simplified purification and testing of colloidal gold probes. Histochemistry. 1985;83:109–115. doi: 10.1007/BF00495139. [DOI] [PubMed] [Google Scholar]
- 12.Zhang ZQ, Yao LH, Hou YD. Construction a highly prokaryocytic expression vector with PRPL promoter and its aplication. Bingdu Xuebao. 1990;6:111–116. [Google Scholar]