

Abstract
Pediatric dysphagia—feeding and swallowing difficulties that begin at birth, last throughout childhood, and continue into maturity—is one of the most common, least understood complications in children with developmental disorders. We argue that a major cause of pediatric dysphagia is altered hindbrain patterning during pre-natal development. Such changes can compromise craniofacial structures including oropharyngeal muscles and skeletal elements as well as motor and sensory circuits necessary for normal feeding and swallowing. Animal models of developmental disorders that include pediatric dysphagia in their phenotypic spectrum can provide mechanistic insight into pathogenesis of feeding and swallowing difficulties. A fairly common human genetic developmental disorder, DiGeorge/22q11.2 Deletion Syndrome (22q11DS) includes a substantial incidence of pediatric dysphagia in its phenotypic spectrum. Infant mice carrying a parallel deletion to 22q11DS patients have feeding and swallowing difficulties. Altered hindbrain patterning, neural crest migration, craniofacial malformations, and changes in cranial nerve growth prefigure these difficulties. Thus, in addition to craniofacial and pharyngeal anomalies that arise independently of altered neural development, pediatric dysphagia may reflect disrupted hindbrain patterning and its impact on neural circuit development critical for feeding and swallowing. The mechanisms that disrupt hindbrain patterning and circuitry may provide a foundation to develop novel therapeutic approaches for improved clinical management of pediatric dysphagia.
Keywords: pediatric dysphagia, hindbrain patterning, oropharyngeal morphogenesis, brainstem circuitry, cranial nerves, 22q11.2 Deletion/DiGeorge Syndrome
INTRODUCTION
Approximately 25% of all infants and children, and as many as 80% with neurodevelopmental disorders have pediatric dysphagia (Figure 1A)—extreme difficulties with feeding and swallowing that slow weight gain, disrupt nutrition, cause acute choking that can lead to life threatening aspiration-based infections of the nasal sinuses, middle ears, and lungs [1–10]. Dysphagia-related symptoms likely compromise many aspects of postnatal development including sensory experience, motor activity, cognitive exploration, language acquisition and social engagement. Perhaps due to a general increase in the incidence of neurodevelopmental disorders or enhanced diagnostic awareness, current data suggest that the frequency of pediatric dysphagia is increasing [11]. Nevertheless, our current understanding of this serious complication for many infants and children is based primarily upon descriptions of clinical phenomena. The basic underlying biology remains unknown, in large measure because of a lack of valid animal models of human neurodevelopmental disorders that include early post-natal feeding and swallowing dysfunction in their phenotypic spectrum.
Figure 1.

A. Pediatric dysphagia is a frequent complication in early life. Top: Otherwise typically developing children with no diagnoses of known developmental or neurodevelopmental disorders. Current estimates suggest that as few as 25% and as many as 45% of typically developing children have dysphagia at some point during infancy or childhood (data from [5]). Middle: Incidence of pediatric dysphagia in all developmental disorders, including neurodevelopmental disorders suggesting that approximately 80% of children with developmental disorders have pediatric dysphagia (data from [5]). Bottom: Frequency of pediatric dysphagia in children with DiGeorge/22q11.2 Deletion Syndrome (22q11DS). Available estimates [13] indicate that all of these children have dysphagia at birth, and 57% continue to have dysphagic symptoms from 4 years of age onward. B. A summary of clinically significant morphogenetic anomalies seen in infants with 22q11DS. As these infants grow, they will also encounter behavioral difficulties that identify 22q11DS as a neurodevelopmental disorder. C. The minimal critical deletion on human Chromosome 22, q11.2 that causes 22q11DS, and the parallel deletion in LgDel mice that model 22q11DS. In humans, low copy repeats (LCR) flank the deleted region, providing a potential mechanism for deletion during meiotic recombination. In mice, there are no LCRs that flank the orthologous region to 22q11.2 on mmChromosome 16. The deletion was engineered by placing lox-p sites at the 3′ and 5′ genes in the orthologous region followed by Cre recombination to recover heterozygous deleted mice [62]. Figure adapted from (Meechan et al, 2015).
22q11.2 Deletion Syndrome (22q11DS), also known as DiGeorge Syndrome has become a “model” genetic disorder [12] for understanding the relationship between complex behavioral disruptions and additional phenotypes that are often coincident in a broad range of neurodevelopmental disorders (Figure 1B,C). One of the most clinically relevant 22q11DS phenotypes, aside from cognitive and social impairment, is disrupted feeding and swallowing that defines pediatric dysphagia [13]. All 22q11DS patients have neonatal feeding and swallowing impairments, and nearly 60% continue to experience complications from 4 years of age onward [13]. In some patients, these difficulties likely result from craniofacial dysmorphology including cleft palate and other anomalies that often require surgical intervention. Unfortunately, current surgical treatments are only effective in approximately 25% of patients [14, 15], suggesting mechanisms beyond oropharyngeal mechanics may contribute to pathology. Indeed, many cases of pediatric dysphagia in 22q11DS patients are not accompanied by overt craniofacial dysmorphology that requires surgical intervention [13, 16]. Nevertheless, these patients have the same nutritional and respiratory complications. Apparently, pediatric dysphagia can arise due to disruptions in developmental mechanisms other than those responsible for oropharyngeal morphogenesis.
The lack of foundational knowledge of pediatric dysphagia pathogenesis in 22q11DS or any other developmental disorder makes it difficult to predict clinical course, and design effective new therapies. Current therapies—often based upon approaches used for dysphagic adults—focus on oral motor interventions like non-nutritive sucking or oral stimulation [17, 18], modified feeding schedules, altered food consistency [19–22], and peripheral neuromuscular stimulation [11, 23, 24]. These approaches, however, are only marginally effective [11, 25], and most have not been adequately evaluated in controlled clinical trials [26]. Moreover, while possibly helpful, these approaches do not define or ameliorate underlying pathology. In this review, we will address likely developmental biological foundations of pediatric dysphagia. We argue that key mechanisms for understanding the disorder, especially in neuro-developmental disorders, reflect early hindbrain patterning and its consequences for neural and oropharyngeal development. We summarize our use of 22q11DS mouse models to discover craniofacial and related neuro-developmental perturbations that cause pediatric dysphagia. We discuss how understanding coordination of early hindbrain patterning for differentiation of brainstem motor and cranial sensory neural circuitry as well as oropharyngeal structures necessary for feeding and swallowing may lead to novel therapeutic interventions to improve the quality of life of affected children.
Pediatric dysphagia: the face, the hindbrain, and neurodevelopmental disorders
The ability to feed is one of the earliest—and most essential—complex behaviors to emerge in all animals. It is not surprising that disruption of this behavior has significant, and in some cases, life threatening consequences. In most vertebrates, this essential behavior begins in utero with swallowing of amniotic fluid and becomes essential for survival within hours of birth. Thus, the developmental mechanisms critical for feeding and swallowing must operate during embryonic and fetal development to prepare the newborn for these essential behaviors. Similarly, pathogenic mechanisms must also operate primarily during embryonic and fetal development. The high incidence of dysphagia in children with neurodevelopmental disorders suggests that the developing nervous system, in addition to the oropharyngeal periphery, must be a pathogenic target. Of course, developmental disorders that do not primarily target the nervous system include pediatric dysphagia in their clinical spectrum. For example, Pompe Disease, a lysomal glycogen storage disease that primarily compromises cardiac and skeletal muscle includes pediatric dysphagia as a significant complication [27–29]. It remains uncertain, therefore, whether dysphagia in infants and children is fundamentally a cranial musculo-skeletal disorder, or a disorder of neural circuits that control feeding and swallowing behavior. Thus, understanding the relationship between neural and craniofacial development, especially in neurodevelopmental disorders, is likely to provide new insight into causes of pediatric dysphagia.
The fundamental relationship between craniofacial anomalies and neurodevelopmental disorders has been recognized for nearly half a century [30]. Remarkably, the relationship between peripheral oropharyngeal structures, brainstem neural circuits, and disrupted feeding and swallowing in newborns and young children remain undefined. Clinical manifestations for pediatric dysphagia have been well documented [24, 31, 32]. Due to a lack of research focused on pediatric dysphagia pathology, even the primary targets for pathologic changes are not clear. Dysphagia might be due to developmental disruption of the facial and oropharyngeal cartilages, bones, and muscles (which we will collectively call the “oropharyngeal apparatus”; Figure 2A) that are critical for feeding and swallowing [33], independent of a major contribution from the developing nervous system. Alternately, it may arise from disruption of hindbrain neurons or cranial sensory neurons that innervate these structures (Figure 2B). Since normal feeding and swallowing depends critically upon the coordinated activity of hindbrain motor and cranial sensory neural circuits that regulate the movements of the oropharyngeal apparatus (Figure 2C), it seems likely that all three entities are either structurally or functionally impaired.
Figure 2.

A summary of oropharyngeal structures, cranial nerves, and neural circuits necessary for normal feeding and swallowing. A. The key oropharyngeal skeletal (gray/white), muscular (pink) and other soft tissue (dark pink) structures necessary for feeding and swallowing around birth (left) and in adulthood (right). Note that the soft palate (velum; stipple) must be elevated and the epiglottis must be lowered to prevent aspiration into the nasopharynx and trachea, respectively. The relationship between these structures, the mouth and the esophagus (blue) changes between birth and adulthood (after [33]). B. Multiple cranial nerves are essential for optimal feeding and swallowing. These nerves are clearly defined in the embryo (left) and in the adult (right). Cranial Nerve (CN) V (trigeminal) provides much of the innervation for the lips, tongue and palate. CN VII (facial) innervates primarily the tongue. CN IX (glossopharyngeal) innervates primarily the tongue and pharynx/esophagus, and CN X innervates the larynx and the pharynx. Finally, CN XII innervates the muscles of the tongue. C. The basic organization of the neural circuit for feeding and swallowing reflects sensory input from peripheral cranial sensory ganglia whose axons run in the cranial nerves summarized in panel B, as well as cranial motor neurons in the brainstem whose activity is modulated by multiple inhibitory interneurons, and whose axons also travel to peripheral muscles via the cranial nerves summarized in panel B.
Hindbrain neural circuits are implicated in pediatric dysphagia by two sets of observations. First, most children with pediatric dysphagia, including those with 22q11DS, do not have severe craniofacial malformations and there is no clear indication that surgical repair can effectively eliminate symptoms [34–37]. Second, studies of another key behavior that, like feeding and swallowing, depends upon the integrity of the hindbrain from birth onward—control of eye movements—demonstrate the important contribution of hindbrain neural circuitry to disrupted craniofacial function. Until the mid-2000s, it was thought that most eye movement deficits in infants and children could be corrected by surgical adjustment of defective extra-ocular muscles [38]. In the last decade, however, multiple genetic studies have shown that hindbrain patterning, neuronal differentiation, axon growth and circuit formation underlie several severe disorders [39–42]. Similar to morphogenesis of structures necessary for optimal feeding and swallowing, eye movements depend on hindbrain neural crest migration and differentiation necessary for differentiation of ocular structures [43], as well as hindbrain motor and cranial sensory circuit development [44, 45]. The assessment of human genetic eye movement disorders in mouse models has led to a new emphasis on neural as well as oculo-muscular mechanisms in the clinical management of perinatal oculo-motor disturbances [46, 47]. These insights may provide alternatives to surgical interventions that under- or over-correct disrupted eye movements, leading to further complications [48, 49]. These insights have transformed clinical diagnosis and treatment of eye movement disorders [40]. To gain parallel insight into developmental origins of pediatric dysphagia, it is imperative to assess potential contributions of neural circuitry as well as the oropharyngeal apparatus. Developmental disruptions in hindbrain patterning may contribute to feeding and swallowing pathogenesis—similar to that now established for ocular motor disruptions—and provide a new foundation for more precise diagnoses, and effective therapeutic interventions.
The mechanics and neural circuitry of normal feeding and swallowing
Normal feeding and swallowing relies upon the coordination of oral, lingual, palatal, laryngeal, and esophageal musculo-skeletal structures ([33]; Figure 2). The biomechanical apparatus that ingests and propels food from the mouth to the stomach is controlled by motor commands and sensory feedback from cranial nerves. These include the maxillary and mandibular branches of the trigeminal nerve (Cranial Nerve V) for jaw closing, the facial nerve (CN VII) for jaw opening and movements of relevant facial muscles (e.g., the lips), the glossopharyngeal nerve (CN IX) and the vagus nerve (CN X) for pharyngeal and laryngeal muscles, and the hypoglossal nerve (CN XII) for tongue muscles (Figure 2C). The activity of hindbrain motor neurons that contribute to each cranial nerve is modulated by local interneurons as well as sensory input from ganglion neurons associated with each nerve. Descending forebrain inputs also regulate these circuits; however, the details are beyond the scope of this review. Together, three neuronal types: peripheral cranial sensory neurons, hindbrain motor neurons and interneurons (Figure 3) constitute neural circuits for sequential control of distinct phases of feeding and swallowing [50, 51], analogous to the distributed brainstem circuitry that regulates coordinated eye movements [52].
Figure 3.

Hindbrain patterning and differentiation in the mouse. Embryos between Embryonic day E9.5 and E10.5 are shown. A. Rhombomeres, repeating segments that begin at the anterior aspect of the spinal cord (r7) and end at the region of the hindbrain that will give rise to the cerebellum (r1), are sites of differential gene expression. This information is maintained by migratory neural crest in the branchial arches (ba). B. Neural crest migration from the hindbrain at E10.5, visualized by Wnt1:Cre-dependent recombination of a tdTomato reporter, generates the cranial ganglia essential for feeding and swallowing (gCN V, VII, IX, and X). C. Cranial ganglion cells express a variety of transcription factors (Onecut1, or Oc1 is shown here) as cranial sensory neurons differentiate and extend axons into the peripheral structures that will mediate feeding and swallowing. D. Cranial motor neurons and their progenitors express multiple transcriptional regulators (Nkx6.1 and Isl1 are shown here) that identify subsets of these cells during mid-gestation. TuJ1 = anti neuron-specific β-tubulin (Tubb3).
The neural circuit for feeding and swallowing has been mostly characterized in adults [53]. Four phases of feeding [33, 53]—(1) ingestion/transport; (2) chewing and processing; (3) oropharyngeal accumulation/transport; (4) swallowing—rely upon sequential activation of motor and sensory components of CN V, CN VII, CN IX, CN X, and CN XII (Figure 2C). By synchronous activation and inhibition of cranial motor neurons, the network generates voluntary ingesting, chewing, and swallowing movements as well as reflexes that ensure safe and efficient feeding [2, 54]. Patterned muscular contractions reflect rhythmic, sequential hindbrain sensory relay and motor neuron activity [55, 56]. The network depends upon appropriate excitation and inhibition of motor neurons reflexively activated for food ingestion, transport, and chewing (CN V, VII, XII), oropharyngeal accumulation, transport, and swallowing (CN IX, X). GABAergic internuclear and premotor interneurons inhibit motor neurons to ensure unidirectional, sequential movement of food, preventing regurgitation and aspiration. There is little information about how this neural circuit develops, how it functions during early postnatal life, or fails to function in pediatric dysphagia [50]. It is unclear whether differences in early effects of inhibitory neurotransmitters seen in several other brain regions [57] influence initial excitatory/inhibitory balance, whether there is a critical period for adaptive change to adjust to peripheral variations [58], or whether early experience has a long lasting impact on feeding and swallowing [59, 60].
The anatomical and physiological details of feeding and swallowing suggest that very early disruption of embryonic patterning that could affect both the peripheral and central elements required for optimal food intake. Assessing such early developmental changes in human patients is all but impossible—feeding and swallowing deficits are diagnosed after birth, long after pathogenic insults likely occur. Thus, to assess whether pediatric dysphagia may be a disorder of early hindbrain patterning or additional mechanisms that contribute to craniofacial and oropharyngeal development, one must identify valid animal models of dysphagia. Using such models it should be possible to explore causal associations between early patterning changes, oropharyngeal and neural circuit anomalies, and biologically significant impairment of perinatal feeding and swallowing.
Dysphagia, animal models and 22q11DS
Valid animal models of any developmental disorder must meet at least three criteria: First, the model must approximate clinical phenomena seen in patients. Second, the model must have a relationship to either causal genetic mutations or end stage pathology in patients. Third, the model must provide opportunities to test mechanistic hypotheses that could not easily be tested in patients. Unlike analysis of developmental disruption of eye movement control, which moved forward based upon human genetic observations of single gene mutations associated with eye movement disorders subsequently reproduced in mutant mice (see above), there has been no focused genetic analysis of pediatric dysphagia in human patients. Instead there are a small number of developmental disorders including 22q11DS with a fairly high incidence of dysphagia as part of a broader phenotypic spectrum [5, 6, 61]. Therefore, we set out to determine whether mice that model 22q11DS genetically—particularly the LgDel mouse that has a heterozygous 28 gene deletion that parallels the minimal critical deletion in human 22q11DS patients (see Figure 1C) and the Tbx1 mouse, with a mutation of a candidate gene for 22q11DS phenotypes [12, 62]—accurately model pediatric dysphagia.
Our first challenge was to assess whether LgDel or Tbx1+/− mutant mice have any of the essential features associated with pediatric dysphagia. An initial clinical sign of dysphagia is delayed or diminished weight gain from birth onward [63]. LgDel mice, like children with 22q11DS [64], fail to gain weight from birth onward compared to their typically developing counterparts (Figure 4A). Tbx1+/− neonates, on the other hand, do not show this phenotype. In addition, like many 22q11DS patients, LgDel mice have signs of food aspiration-related naso-sinus, middle ear, and respiratory inflammation and infection (Figure 4B). There are protein inclusions in the nasal sinuses, middle ear and lungs that are composed primarily of murine milk protein, consistent with aspiration during nursing. These protein inclusions are accompanied by leukocyte infiltration including neutrophils and macrophages at all three sites, increased frequency of mucus producing goblet cells in the middle ear, accumulations of red blood cells in the lungs—all indicators of infection and significant inflammatory responses [65–67]. These infections were seen at high frequency (4/5) in LgDel P7 pups, but not in wild type (WT) controls. Finally, we developed a behavioral assay of acute feeding and swallowing in mouse pups that parallels the barium swallow test [68] used to assess dysphagia in children. We adapted a previously published mouse pup nursing protocol that uses appropriately sized nipples to maximize the effectiveness of hand feeding [69]. Fluorescent microspheres were added to formula and immediately following one nursing session, pups were sacrificed and analyzed for distribution of the fluorescent beads. The accumulation of fluorescent milk in the stomachs of WT controls confirmed the effectiveness of the general approach. Using this test, we found that LgDel pups acutely aspirate milk, while typically developing littermates do not (Figure 4C). Based upon these observations, the LgDel, but not the Tbx1+/− mouse, apparently provides a robust model in which we can rigorously define the behavioral, pathological, and perinatal developmental dimensions of dysphagia.
Figure 4.

Evidence of dysphagia in LgDel neo-natal mice. A. Left: Growth curves for 22q11DS patients from birth through 3 years of age (a time when all 22q11DS patients have dysphagic syndromes) in females (pink) and males (blue). Female and male 22q11DS patients have equivalent slowing of weight gain. Right: Growth curves for LgDel female (pink) and male (blue) mice from birth through 30 days of age (identified individually and weighed daily) show similar diminished weight gain in the 22q11DS model mice. B. Aspiration infections in the nasopharynx of Postnatal day 7 (P7) LgDel mouse. Left: There are protein accumulations that are infiltrated with leukocytes (*asterisks). Right: These accumulations are composed primarily of murine milk protein (red inset), and are coincident with accumulations of neutrophils (blue inset). C. Acute aspiration of milk (labeled with fluorescent green microspheres) into the nasopharynx (NP) of LgDel (right) but not Wild Type (WT) P7 mouse pups. To = tongue. Figure adapted from (Karpinski et al., 2014, Meechan et al., 2015).
How does 22q11 deletion disrupt feeding and swallowing?
Our confirmation of the LgDel mouse as a model of pediatric dysphagia, based upon parallel features to clinical manifestations of the disorder makes it possible to ask fundamental mechanistic questions about etiology that are more or less impossible to ask in patients. One key question is whether pediatric dysphagia is primarily a peripheral craniofacial development disorder that leads to disrupted oropharyngeal biomechanics, or a neural circuit disorder accompanied by subsequent craniofacial malformations and dysfunction? This distinction is significant, particularly for neurodevelopmental disorders, because neural circuits that control feeding and swallowing may be altered in the context of broader disruptions of brain development that also impact cognition, social interaction and communication. General pathogenic mechanisms may disrupt all neural circuit development in these disorders, with variable consequences for distinct behaviors. Furthermore, clinical management of oropharyngeal dysmorphology versus neural circuit changes that lead to dysphagia would likely be distinct. Accordingly, without determining if defects are peripheral versus central in origin, there can be no rational foundation for refining diagnostic criteria to improve clinical management, identifying useful therapeutic targets, or optimizing pre-natal health for at-risk fetuses to minimize later dysphagia complications. Thus, we analyzed fundamental aspects of craniofacial differentiation as well as central and peripheral nervous system development in the LgDel mouse, and identified three key developmental disruptions that prefigure these postnatal dysphagic symptoms: branchial arch-related craniofacial malformations, disrupted early cranial nerve growth, and altered anterior-posterior hindbrain patterning.
Oropharyngeal dysmorphogenesis in 22q11 DS, Tbx1 mutants, and the LgDel mouse
Despite clinical acknowledgement of mild to severe craniofacial anomalies in 22q11DS, there are only a small number of rigorous analyses of key craniofacial phenotypes in patients. The consensus of recent studies [70, 71] indicates cranial skeletal changes in the skull, nasal processes, maxilla, and teeth—all sites of cranial neural crest contributions. 22q11DS patients also have variable palatal defects ranging from overt cleft, submucusoal cleft or a shorter palate [72]. These defects result in the diagnostic “velo-pharyngeal insufficiency” that characterizes the syndrome [73]. Velo-pharyngeal insufficiency complicates feeding and swallowing by allowing aspiration from the mouth into the nasal sinuses [13, 16]; it is also associated with sleep apnea [74, 75], as well as speech and language difficulties including hyper-nasality at later stages due to altered airflow during articulation [76–78]. It remains uncertain, however, whether this phenotype, a likely a critical contributor to dysphagia in 22q11DS infants and children, reflects oropharyngeal structure, or altered palatal control due to hypotonia. In addition, laryngotracheoesophageal anomalies are often present in 22q11DS patients [72], which can further disrupt feeding and swallowing. Speech difficulties in 22q11DS have also been associated with velo-pharyngeal insufficiency in 22q11DS [79]. Surgical interventions to correct velo-pharyngeal insufficiency are variably successful [80–82]. 22q11DS mouse models provide an opportunity to assess the relative developmental contributions of peripheral oropharyngeal versus neural disruption of mouth, tongue and palate movement in dysphagia. Such information will provide a useful new dimension for assessing dysphagia-related dysfunction, and may serve as the foundation for selecting clinical treatments, and predicting outcomes for dysphagic 22q11DS patients.
Previous analysis of craniofacial anomalies in 22q11DS mouse models has focused primarily on Tbx1 mutants, in which cardiovascular anomalies are accompanied, at variable penetrance, by craniofacial dysmorphology [83–90]. Tbx1 null mutants exhibit a variety of craniofacial phenotypes including micrognathia, cleft palate and missing or abnormal cranial bones [91, 92] as well as defects in development of mastication and facial muscles derived from branchial arches 1 and 2 [87, 93]. The hyoid bone that anchors the tongue and muscles of mastication is missing in Tbx1 null mutants [94]. Nevertheless, the tongue muscles themselves, derived from the anterior somites rather than the cranial mesenchyme, appear normal in Tbx1 null mice [93]. In addition, Tbx1 is required for cranial tendon development [95]. Finally, there are non-neural as well as neural ear anomalies in Tbx1 mutant embryos [96–99], raising the possibility that pharyngeal mesoderm, neural crest as well as cranial placode morphogenetic mechanisms may be compromised. Most phenotypes reported have been in Tbx1−/− embryos; therefore, it is difficult to assess the relevance of these data to phenotypes caused by diminished Tbx1 dosage in the context of broader 22q11 gene deletion. There are a limited number of reports in Tbx1−/− mice of altered neural crest migration and signaling interactions, including for neural crest that contributes to aortic arch arteries [100] as well as branchial arches [101], even though Tbx1 itself is expressed primarily in pharyngeal and cranial epithelia, rather than presumed neural crest-derived mesenchyme in craniofacial and aortic arch primordia [102]. Thus, while heterozygous Tbx1 loss of function in the context of broader 22q11 deletion may contribute to anomalies relevant for dysphagia, these anomalies have not been assessed in mice carrying deletions parallel to those in 22q11DS patients.
We evaluated craniofacial malformations in the developing and mature LgDel mouse [9], and found at two distinct variably penetrant phenotypes. At mid-gestation (E14.5) there is an apparent delay in palatal shelf elevation that occurs at relatively low penetrance (Figure 5A). Young adult LgDel mice have smaller mandibles with altered morphometric features (Figure 5B). Since these structures depend in part upon neural crest contributions, it is possible that these phenotypes ultimately reflect altered neural crest differentiation due to 22q11 gene deletion. Indeed, in a qPCR expression analysis of 22q11-deleted genes in WT branchial arch BA1A/BA1B/BA2 (Figure 5C), we found robust expression of 19 22q11 genes in these structures [9]. This is in agreement with our previous PCR and in situ hybridization observations localizing multiple 22q11 genes (20/28 murine orthologues) to craniofacial primordia [103–105]. For two of these genes, Prodh2 [105] and Ranbp1 [104], detailed in situ hybridization analyses show robust expression at sites where cranial neural crest cells are found. Similar analyses remain to be completed for the additional 18 murine 22q11 orthologues expressed in craniofacial structures that include neural crest contributions. Mechanistic details of cranial neural crest migration and differentiation have not yet been assessed in LgDel or other broadly deleted 22q11DS mouse models; accordingly, the relevance of Tbx1−/− phenotypes remains uncertain. Resolution of whether the neural crest is developmentally intact upon departure from the hindbrain, and altered peripherally by disrupted interactions with placodal ectoderm, cranial mesoderm, or visceral endoderm due to broader 22q11 deletion will be key to determining peripheral cranial versus early central neural morphogenetic contributions to the pathogenesis of pediatric dysphagia.
Figure 5.

Craniofacial anomalies in developing and mature LgDel mice are prefigured by expression of multiple 22q11 genes in the craniofacial primordia. A. Failure of palatal elevation in the E14.5 LgDel mouse (right). The palatal shelves (arrows) have not completed the characteristic upward growth, as is seen in the WT E14.5 embryo (+/+; left). B. The mandible of the young adult LgDel mouse (P30) is smaller and morphometrically anomalous (measurements between cardinal points identified in circles at left are provided in the histogram at right). C. Multiple 22q11 genes are expressed at substantial levels in WT craniofacial primordia. qPCR measurements of these genes for branchial arch 1b (ba1b; mandible) and branchial arch 2 (ba2; hyoid) are shown here; similar values are seen for ba1a [9].
Multiple origins: mesodermal and neural crest contributions to dysphagia?
The development of the oropharyngeal apparatus and the neural circuits that control its movements for optimal feeding and swallowing depends upon a shared mechanism: patterning the embryonic hindbrain into domains along the anterior-posterior axis called rhombomeres [106, 107] (Figure 3A). For the oropharyngeal apparatus, however, development also engages pharyngeal mesoderm, including that from cardiac regions, as well as the hindbrain-derived neural crest [108, 109]. For the peripheral cranial sensory innervation, both ectodermal placodes and the neural crest are involved [110, 111], and for motor innervation, motor neuron progenitors are specified based upon their position in the hindbrain [112]. Thus, although hindbrain pattering is a common regulator of key events that generate muscular, skeletal and neural components necessary for feeding and swallowing, it alone does not account for normal development. Furthermore, although neural components are essential for controlling the behavior of feeding and swallowing, optimal myogenesis and skeletogenesis, due to mesodermal patterning and differentiation independent of the nervous system, is also required. Indeed in 22q11DS, it is clear that both neural crest [113] and pharyngeal mesoderm are compromised [87, 114]. This dual disruption may contribute to the origins of perinatal dysphagia in 22q11DS and other neurodevelopmental disorders.
Mesoderm, particularly from posterior pharyngeal regions and cranial neural crest cells, specified at distinct rhombomeric locations (Figure 3B,C) contribute to the mesenchyme of the branchial arches [109, 115, 116]. The branchial arches in turn differentiate into the facial and oropharyngeal skeletal elements crucial for feeding and swallowing. Branchial arches consist of pharyngeal ectoderm, endoderm, and mesenchyme composed of cells from both the mesoderm and hindbrain neural crest [95]. The branchial arch mesenchyme differentiates into facial and oropharyngeal skeletal and muscular elements crucial for feeding and swallowing. In parallel, the hindbrain cranial motor neurons and their associated interneurons arise from distinct rhombomeres (Figure 3D) and the growth of their axons into the periphery is constrained by their positions. Accordingly, neural crest-derived oropharyngeal target structures generally have the same rhombomeric origin as the nerve that innervates them [117–119]. Development of branchial arches into skeletal, connective and muscle derivatives of the oropharyngeal apparatus requires complex interactions of these different cell types. The anterior posterior origin of hindbrain specifies neural crest cells and determines which branchial arches they will contribute to. These arches associate with specific cranial nerves and muscles. For example, brachial arch 1 (maxillo-mandibular arch) will contribute to the jaw, muscles of mastication and will be innervated by CN V; brachial arch 2 (hyoid arch) will contribute to bones and muscles associated with the face and innervated by CN VII; brachial arch 3 will contribute to the hyoid bone and the Stylopharyngeus muscle and is innervated by CN IX. More posterior arches, comprised of mesodermal as well as neural crest cells, contribute to connective tissues and muscles associated with the palate and more posterior structures. Thus, there may be multiple pathogenic disruptions of mesoderm and neural crest, as well as ectodermal placodes, leading to the oro-pharnygeal and cranial neural dysfunction underlying dysphagia.
Cranial sensory and motor nerve disruption in LgDel embryos
Appropriate control of all cranial functions, including feeding and swallowing, depends upon the coordination of cranial sensory information from the periphery with ongoing cranial motor neuron activity in the brainstem. The basic organization of this peripheral/central coordination is established during early hindbrain patterning and differentiation [112, 120]. Thus, by evaluating the developing cranial nerves in mid-gestation embryos, one can recognize disruption of these fundamental processes. We found that several of the cranial nerves that control feeding and swallowing have aberrant growth patterns early in development. LgDel embryos have significant, frequent defects in CN V and CN IX/X, with a lower penetrance of defects in CN VII (Figure 6A). These observations suggest that aberrant development of hindbrain motor and/or sensory neurons, both of which contribute to cranial nerves, could underlie neural circuit dysfunction leading to aberrant feeding and swallowing. It is still not possible, however, to discount the contribution of disrupted differentiation of the oropharyngeal periphery to this potential pathogenic foundation for dysphagia. The rhombomeric origins of the neural crest from which these nerves arise, is shared by branchial arch skeletal elements, which may be disrupted in parallel. Moreover, the periphery may be compromised by 22q11 gene-dependent, hindbrain independent mechanisms. Cranial nerve phenotypes in LgDel would then be a compensatory response by otherwise unaffected populations of central and peripheral neurons and their axons.
Figure 6.

Development of cranial nerves that are critical for feeding and swallowing is disrupted in LgDel mice. A. A comparison of cranial nerve differentiation at E10.5 in WT and LgDel mice. Left: a dorsal view of the hindbrain from an E10.5 WT (left) and E10.5 LgDel embryo (right) in which the cranial nerves have been labeled in the whole specimen using a neurofilament antibody. The arrows indicate the diminished distance between gCN V and gCN VII in the LgDel. The asterisks indicate the diminished presence of presumed motor axons exiting into CN V and CN VII in the LgDel. Finally, gCN IX and X are distinct entities in the WT, but are fused and appear to be shifted anteriorly (possibly due to diminished size of the anterior regions), in the LgDel. Right: High magnification lateral views compare WT cranial nerves with those compromised in the LgDel mouse. Both CN V and CN VII appear smaller and dysmorphic. The most consistent changes (statistically significant based on blind scoring by 4 independent observers; [9]) are: diminished thickness and lack of branching of all three divisions of CN V (ophthalmic-op; maxillary-mx; mandibular-md; arrows and bracket; upper panels); lack of terminal bifurcated branching in CN VII (arrows, middle panels), and fusions or anomalous axon branches that join CN IX and X (arrows, lower panels). B. Distinct changes in cranial nerve development are seen in Tbx1+/− E10.5 embryos. Left: The distance between CN V and VII, seen in a dorsal view of the neurofilament labeled E10.5 hindbrain in the Tbx1+/− embryo is not diminished (arrows), as is the case in the LgDel (shown for comparison in the right panel), and the size and branching of CN V and VII are comparable to WT. In contrast, we found fusions and anomalous axon fascicles between CN IX and X, and these approximate the phenotypes seen in the LgDel embryo. We note, however, that CN IX and X appear smaller in the LgDel than in the Tbx1+/− mouse. Right: WT CN IX and X (top) are separated (arrows) and their axons extend in an orderly fashion both centrally and peripherally. Tbx1+/− CN IX and X are fused (arrows), as in the LgDel (compare with lower right panel in A). These fusions in the Tbx1+/− embryo happen at the same frequency as those seen in the LgDel [9], supporting a key role for heterozygous loss of function of Tbx1+/− for this CN phenotype.
To begin to distinguish the relative contributions of central and peripheral mechanisms and their consequences for the oropharyngeal periphery or cranial neural circuits to dysphagia pathogenesis, we returned to a comparison of Tbx1 and LgDel 22q11DS models. Tbx1 is particularly useful for this comparison, since the gene is not expressed in the developing brain [121], suggesting that mutant phenotypes are most likely the consequence of peripheral changes. We confirmed previous observations [90] that suggested that Tbx1 mutant mice had partially penetrant CN IX/X dysmorphic phenotypes—primarily ganglion fusions or aberrant axon fascicles, as well as diminished peripheral axon growth (Figure 6B). We did not find, however, any evidence for CN V or VII phenotypes in these mice. This distinction between Tbx1 and LgDel mice indicates that changes caused by 22q11 gene deletion beyond Tbx1 are likely focused on anterior cranial nerves, particularly CN V. The absence of signs of peri-natal dysphagia in Tbx1+/− mouse pups that are seen in LgDel (see above) reinforces the interpretation that anterior cranial nerve dysmorphology due to 22q11 deletion beyond Tbx1 is an essential contributor to dysphagia, whereas posterior cranial nerve dysmorphology due primarily to Tbx1 diminished dosage is not.
Anterior-posterior hindbrain patterning is disrupted in the LgDel mouse
The available evidence in 22q11DS animal models points to disrupted hindbrain patterning and its consequences—particularly anterior hindbrain patterning based upon comparison of LgDel (CN V) and Tbx1 (CN IX/X) dependent phenotypes—as a likely pathogenic mechanism that leads to pediatric dysphagia. This evidence raises another central question: which mechanisms that normally regulate hindbrain patterning are modified by 22q11 deletion leading to dysphagia-related pathology? It is well known that anterior-posterior patterning in the hindbrain is sensitive to retinoic acid (RA) signaling [122–125]. RA, an essential morphogenetic signal, is the biologically active derivative of dietary Vitamin A. Rigorous regulation of RA levels is crucial for normal development of multiple tissues and organs. Indeed, disruption of RA levels, or transcription of RA-regulated genes can lead to changes in anterior-posterior identity of hindbrain neurons and cranial nerves [126, 127]. Changes in RA signaling result in phenotypes similar to those in 22q11DS mouse models or patients [128–140]. In 22q11DS mouse models, diminished 22q11 gene dosage alters expression of both RA synthetic and degradation enzymes, and disrupts RA signaling in the heart, face and forebrain [103, 141–144]. Thus, we asked if there were parallel local changes in RA signaling, patterning, and subsequent differentiation in the developing LgDel or Tbx1+/− hindbrain.
To assess the relationship between CN developmental anomalies, hindbrain patterning, RA signaling and dysphagia in 22q11DS mouse models we first quantified expression of RA-regulated genes in microdissected embryonic day (E) 9.5 hindbrain, a time when patterning mechanisms have established rhombomeric domains. Excess RA signaling can result in phenotypes similar to those seen in the LgDel [145]; therefore, disrupted RA signaling is a likely candidate for hindbrain anomalies in LgDel embryos. We found that multiple RA-regulated genes were increased in expression level in the LgDel hindbrain (Figure 7A). These data led us to evaluate whether increased expression was due to locally enhanced transcription of RA-regulated genes in the hindbrain with maintenance of expression patterns, or changes in patterning that might alter anterior-posterior rhombomere identities and subsequent differentiation. We selected an RA-regulated gene that showed substantial up-regulation and had a distinctive posterior expression pattern, Cyp26b1, and performed in situ hybridization (ISH) analysis in E9.5 WT, LgDel, and Tbx1+/− mice [9]. Cyp26b1, normally maximally expressed in rhombomeres 5 and 6, with minimal expression in r3 and r2, expands into r2 and is increased in expression in r3 and r4 in LgDel embryos (Figure 7B). We did not see parallel changes in Tbx1+/− embryos. All of these changes are consistent with a posteriorizing shift in RA-dependent hindbrain patterning in the LgDel embryo, potentially leading to selective disruption of anterior cranial nerves, particularly CN V, that regulate distinct aspects of feeding and swallowing.
Figure 7.

Retinoic acid (RA) signaling is altered in the LgDel hindbrain, leading to changes in anterior-posterior patterning. A. qPCR analysis, in microdissected E9.5 hindbrains from WT and LgDel embryos, of a subset of highly RA-regulated genes known to be expressed at substantial levels in the E9.5 hindbrain, as well as two control genes (Shh, and Cdc45, a 22q11 gene). Expression levels for all but one of these genes (Cyp26a1) are significantly increased in the E9.5 hindbrain. B. Disrupted pattern of RA-regulated gene expression in the E9.5 hindbrain of LgDel, but not Tbx1+/− embryos. We used in situ hybridization (ISH) for Cyp26b1, which is highly retinoid regulated in WT, and has a distinct posterior-hindbrain expression pattern: enhanced in r5 and r6 with diminished expression in r3 and r4 and little to no expression in r2 (left). There is a dramatic anterior shift of expression, in parallel with the increased expression seen in our qPCR measurements (see panel A), in the LgDel embryo (middle). There is now substantial Cyp26b1 expression in r3 and r4, and expression continues into r2. This shift is consistent with a “posteriorizing” influence of altered RA signaling on hindbrain differentiation in the LgDel. A similar expansion of Cyb26b1 labeling is not seen in the Tbx1+/− hindbrain (right). C. Lowering RA signaling levels in LgDel mice by approximately 25% [103] using a heterozygous null allele of the RA synthetic enzyme Raldh2, results in rescue of the CN V phenotypes in LgDel, but not CN IX and X phenotypes. The CN V rescue in LgDel was remarkably statistically robust (far right, top); however there is no statistical evidence for rescue of the CN IX/X phenotype (far right, bottom). D. ISH analysis of Cyp26b1 expression in Raldh2+/−;LgDel compound embryos shows that the expression levels and pattern of this RA-regulated gene are now comparable to WT. qPCR analysis (not shown; Karpinski et al, 2014) is consistent with this rescue; RA dependent genes are restored in their expression to levels indistinguishable from WT. Figure adapted from (Karpinski et al., 2014).
To begin to assess the contribution of RA signaling to disrupted hindbrain patterning following 22q11 gene deletion more thoroughly, we asked whether we could restore developmental phenotypes in CN V or other dysmorphic CNs in the LgDel to the WT state by manipulating RA signaling levels. Since RA signaling is increased in the LgDel hindbrain, at least based upon our qPCR and ISH analysis of RA-regulated genes, we chose to genetically diminish RA synthesis in WT and LgDel embryo [103, 146, 147] and assess the consequences for anterior versus posterior cranial nerve patterning, as well as levels and patterns of RA-dependent genes. We bred into WT and LgDel mice one inactivated allele of Raldh2, the key RA synthetic enzyme for hindbrain patterning [103, 136]. This heterozygous Raldh2+/− mutation diminishes RA signaling, assessed using an RARE reporter mouse, by approximately 25% [103]. When we scored CN V versus VII/VIII versus IX/X phenotypes, we found that the frequency of CN V phenotypes diminishes significantly—so that there is a lower frequency of anomalies than those seen, occasionally in WT embryos—and the morphology of CN V was returned to WT appearance (Figure 7C). We did not see a parallel change in CN IX/X phenotypes, consistent with selective rescue of anterior versus posterior rhombomeres and cranial nerves. We have not yet analyzed the consequences of this rescue of cranial nerve phenotypes in post-natal LgDel:Raldh2+/− mice ( which are viable) based upon diminished weight gain or milk aspiration (see Figure 4). Nevertheless, the current data demonstrate that the 22q11DS cranial nerve developmental phenotypes result from the combined effects of disrupted RA signaling for anterior cranial nerves and disrupted Tbx1 function for posterior cranial nerves. Our comparison of LgDel and Tbx1 phenotypes relevant for dysphagia suggests that disrupted anterior patterning in the LgDel, independent of Tbx1, may be an essential determinant for the developmental origins of pediatric dysphagia.
Easier to swallow? Hindbrain patterning and new therapies for pediatric dysphagia
Our new understanding of potentially pathogenic changes in RA-dependent hindbrain patterning and subsequent disruption of brainstem circuits that prefigure pediatric dysphagic phenotypes in 22q11DS mouse models provides a foundation for improving diagnosis and treatment of the dysphagia in 22q11DS, and more generally in neurodevelopmental disorders. Three broad questions must be answered to fully assess the clinical utility of basic insights into hindbrain pathogenesis of pediatric dysphagia:
1. Can insight into developmental disruption improve diagnosis?
There is currently no framework for assessing genetic risk for pediatric dysphagia, including risk in the context of genetic neurodevelopmental syndromes like 22q11DS. Analysis of the role of individual 22q11 genes beyond Tbx1 in disrupting hindbrain patterning or differentiation can identify novel genes, their upstream regulators, and downstream targets that contribute to dysphagia risk in 22q11DS, and potentially other neurodevelopmental disorders. By identifying additional hindbrain, cranial sensory, and oropharyngeal gene expression changes, particularly those regulated by RA, in the developing LgDel hindbrain, it should be possible to identify broader gene networks that compromise mechanisms necessary for optimal development of circuits and structures for feeding and swallowing. Some of these genes no doubt will emerge as candidate genes for pediatric dysphagia, independent of established genetic syndromes, or modifiers in the context of such syndromes. Accordingly, a new understanding of genetic networks that regulate hindbrain patterning and differentiation critical for feeding and swallowing, made possible by the use of animal models and a variety of gene discovery approaches, will provide a strong foundation for a new clinical genetics of pediatric dysphagia. This insight will aid in improved outcomes for dysphagia by enhancing diagnostic precision and providing a new spectrum of targets for intervention.
2. What is the contribution of neural circuit dysfunction to pediatric dysphagia pathology?
Currently, there is no evidence for whether the neural circuits that innervate oropharyngeal structures critical for feeding and swallowing are altered in pediatric dysphagia. Since early rhombomere patterning and cranial nerve outgrowth are disrupted, at least in the LgDel 22q11DS model, it is likely that there are functional disruptions in segmental hindbrain circuits; however, this is yet to be demonstrated experimentally. Disrupted neural circuits, either those that coordinate motor control based on concerted activity of segmentally distributed brainstem motor neurons, or appropriate proprioceptive or nociceptive feedback from cranial sensory neurons, can clearly lead to difficulty in each phase of feeding and swallowing. Identification of circuit anomalies at the cell biological level based upon assessment of dendritic and axonal differentiation and the frequency/distribution of synaptic inputs can identify essential processes that are disrupted by earlier hindbrain changes. Physiological assessment of circuit function relevant for feeding and swallowing will provide an outline of how behavioral control can be compromised. These circuit features include membrane properties of relevant motor, sensory or interneurons, excitatory/inhibitory balance, conductance of channels, and activation of neurotransmitter receptors or other regulators of synaptic function. These receptors, channels or synaptic regulators may prove to be useful targets for therapeutic intervention to improve circuit function, and thus feeding and swallowing. Modeling pediatric dysphagia in LgDel and other 22q11DS model mice will provide a new opportunity to analyze the physiology and pharmacology of hindbrain motor and sensory circuit anomalies and their relationship to feeding and swallowing behaviors.
3. Are there feasible strategies to prevent pediatric dysphagia?
Appropriate levels of maternal micronutrients protect the fetus from stress and facilitate morphogenesis; altered levels disrupt specific developmental mechanisms. Recent clinical experience demonstrates that controlling fetal micronutrient exposure via the maternal diet dramatically improves outcomes for at-risk pregnancies [148]. Two micronutrients, retinoic acid (RA) and folic acid (FA), have been implicated in developmental processes potentially disrupted in pediatric dysphagia: hindbrain patterning and craniofacial differentiation. Studies in animal models demonstrate that FA levels influence dysphagia-related craniofacial development [149, 150]. Of particular interest, FA supplementation in mice prevents RA-induced neural tube, palate, heart, and thymus malformations—all 22q11DS phenotypes [151–154]. Since RA signaling in the hindbrain of the LgDel mouse is associated with dysphagic phenotypes, FA may prevent some of these defects. Interestingly, similar developmental anomalies result from exposure to too much or too little RA itself [145], which is metabolized from maternal sources of Vitamin A during critical stages of development [155]. We have found that LgDel embryos are sensitized to RA exposures that are apparently benign for WT embryos [103]. Thus, small changes in RA levels, due to variation in maternal Vitamin A intake, may significantly alter hindbrain and cranial nerve development in the 22q11DS embryo. Investigating whether maternal micronutrient-based therapies—including rigorous control of Vitamin A/RA levels—can prevent specific feeding and swallowing deficits by correcting disrupted developmental mechanisms could result in dietary strategies to prevent pediatric dysphagia in at-risk pregnancies.
Summary
Understanding the pathology, developmental origins, and prevention of pediatric dysphagia remains one of the major challenges for improving treatment, quality of life, and attainment of developmental milestones for infants and children with neurodevelopmental disorders. Pediatric dysphagia likely arises from a combination of disrupted oropharyngeal musculo-skeletal as well as cranial neural circuit development. We identified the first functionally validated genetic model of pediatric dysphagia. The LgDel 22q11DS model has remarkably parallel perinatal feeding and swallowing difficulties to those seen in infants and children with 22q11DS. We used developmental biological, genetic, behavioral, and pharmacological approaches to define pre-natal causes, and likely consequences of disrupted hindbrain patterning that lead to disrupted feeding and swallowing in this model. Our data indicate that the pathogenic mechanism is likely focused on alterations of the developing hindbrain and related neural crest, rather than disrupted peripheral mechanisms that compromise pharyngeal mesendoderm, cranial ectoderm, or visceral endoderm (Figure 8). Using this insight, made possible only by the availability of a valid animal model, it is now possible to identify genetic networks, disrupted morphogenetic mechanisms and altered neural circuits that account for specific aspects of feeding and swallowing difficulty in pediatric dysphagia from birth onward. These new data will facilitate design of pre- and peri-natal interventions that might ameliorate dysphagic complications. Thus, by understanding the early developmental biology that makes it “hard to swallow” in neurodevelopmental disorders, it should be possible to define a new basis for the clinical management of pediatric dysphagia and improve outcomes for the children whose early lives are complicated by this disorder.
Figure 8.
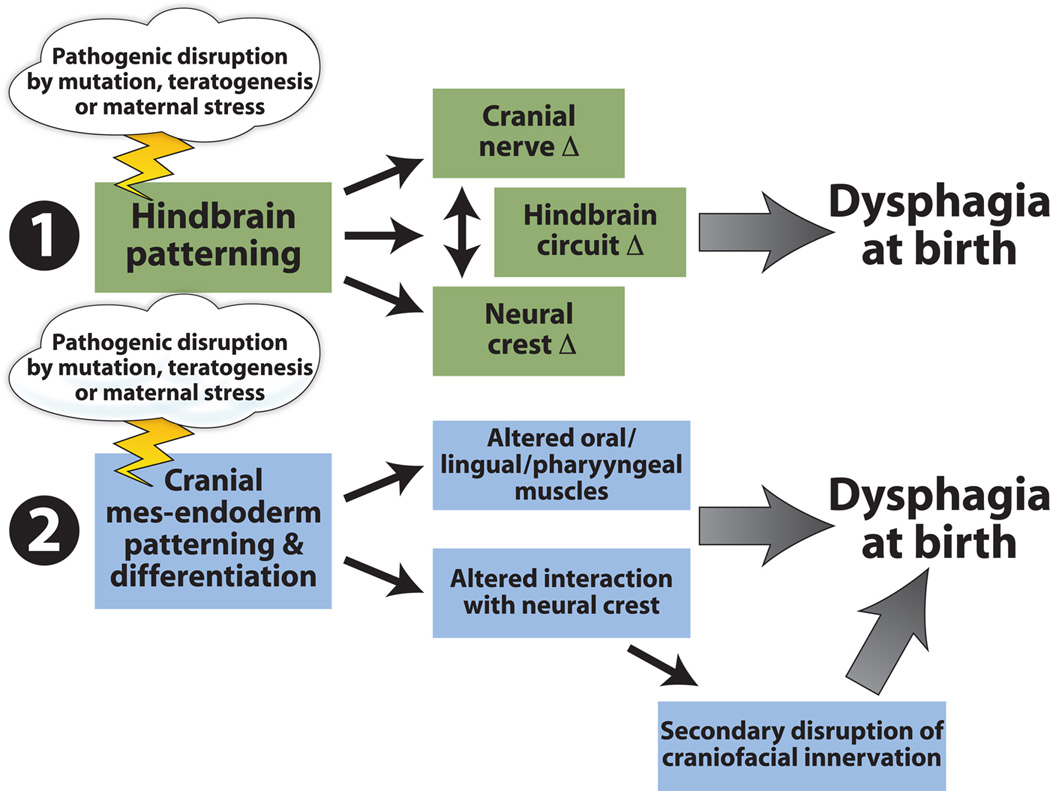
Multiple developmental disruptions may underlie pediatric dysphagia. One possibility, supported by some of the observations in the LgDel mouse model of 22q11DS summarized in this review, is that feeding and swallowing dysfunction arises due to disruptions of hindbrain patterning. These disruptions then compromise hindbrain motor neuron differentiation as well as neural crest specification that is key for cranial nerve growth, appropriate sensory and motor innervation as well as morphogenesis of oropharyngeal structures critical for optimal feeding and swallowing. A second possibility is that pediatric dysphagia is primarily a disorder of disrupted craniofacial/oropharyngeal development due to altered patterning and differentiation of cranial mesendoderm and ectodermal placodes—independent of any significant, primary involvement of the central nervous system or neuroectodermal derivatives. These changes would lead primarily to oropharyngeal morphogenetic anomalies that would alter feeding and swallowing mechanics, with only secondary changes in motor or sensory innervation and circuit function—some of which might be compensatory for the peripheral dysmorphology. We note that these two possible general mechanisms are not mutually exclusive. Indeed mutations, teratogens, or maternal stress might alter both in parallel, modifying normal central/peripheral interactions leading to pathogenesis that results in pediatric dysphagia.
HIGHLIGHTS.
Pediatric dysphagia is very common in children with developmental disorders
We assessed this problem in mouse models of DiGeorge/22q11.2 Deletion Syndrome
Mouse models of 22q11 Deletion Syndrome have multiple phenotypes that are parallel to key clinical features of pediatric dysphagia.
We present evidence that a primary cause is altered hindbrain patterning
This results in craniofacial defects and changes in growth of cranial nerves
Mechanisms that disrupt hindbrain patterning may idenfity new targets for effective therapies
ACKNOWLEDGEMENTS
We thank Matthew Fralish and Elizabeth Paronett for their assistance with multiple phases of the work summarized here. Imaging was performed in the GWU Center for Microscopic Imaging and Analysis, supported by HD040677.
Grant support:
NIH R01 DE022065 (SAM)
DC011534 (A-S.L.), HD083157 (A-S.L. and SAM)
REFERENCES
- 1.Tutor JD, Gosa MM. Dysphagia and aspiration in children. Pediatr Pulmonol. 2012;47(4):321–337. doi: 10.1002/ppul.21576. [DOI] [PubMed] [Google Scholar]
- 2.Miller CK. Updates on pediatric feeding and swallowing problems. Curr Opin Otolaryngol Head Neck Surg. 2009;17(3):194–199. doi: 10.1097/MOO.0b013e32832b3117. [DOI] [PubMed] [Google Scholar]
- 3.Arvedson JC. Assessment of pediatric dysphagia and feeding disorders: clinical and instrumental approaches. Dev Disabil Res Rev. 2008;14(2):118–127. doi: 10.1002/ddrr.17. [DOI] [PubMed] [Google Scholar]
- 4.Cooper-Brown L, et al. Feeding and swallowing dysfunction in genetic syndromes. Dev Disabil Res Rev. 2008;14(2):147–157. doi: 10.1002/ddrr.19. [DOI] [PubMed] [Google Scholar]
- 5.Lefton-Greif MA. Pediatric dysphagia. Phys Med Rehabil Clin N Am. 2008;19(4):837–851. doi: 10.1016/j.pmr.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 6.Prasse JE, Kikano GE. An overview of pediatric dysphagia. Clin Pediatr (Phila) 2009;48(3):247–251. doi: 10.1177/0009922808327323. [DOI] [PubMed] [Google Scholar]
- 7.Bingham PM. Deprivation and dysphagia in premature infants. J Child Neurol. 2009;24(6):743–749. doi: 10.1177/0883073808329530. [DOI] [PubMed] [Google Scholar]
- 8.Arvedson J, et al. Silent aspiration prominent in children with dysphagia. Int J Pediatr Otorhinolaryngol. 1994;28(2–3):173–181. doi: 10.1016/0165-5876(94)90009-4. [DOI] [PubMed] [Google Scholar]
- 9.Karpinski BA, et al. Dysphagia and disrupted cranial nerve development in a mouse model of DiGeorge (22q11) deletion syndrome. Dis Model Mech. 2014;7(2):245–257. doi: 10.1242/dmm.012484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Durvasula VS, O'Neill AC, Richter GT. Oropharyngeal Dysphagia in children: mechanism, source, and management. Otolaryngol Clin North Am. 2014;47(5):691–720. doi: 10.1016/j.otc.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 11.Kakodkar K, Schroeder JW., Jr Pediatric dysphagia. Pediatr Clin North Am. 2013;60(4):969–977. doi: 10.1016/j.pcl.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 12.Meechan DW, Jr, et al. Modeling a model: Mouse genetics, 22q11.2 Deletion Syndrome, and disorders of cortical circuit development. Prog Neurobiol. 2015;130:1–28. doi: 10.1016/j.pneurobio.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eicher PS, et al. Dysphagia in children with a 22q11.2 deletion: unusual pattern found on modified barium swallow. J Pediatr. 2000;137(2):158–164. doi: 10.1067/mpd.2000.105356. [DOI] [PubMed] [Google Scholar]
- 14.Brandao GR, et al. Speech outcomes and velopharyngeal function after surgical treatment of velopharyngeal insufficiency in individuals with signs of velocardiofacial syndrome. J Craniofac Surg. 2011;22(5):1736–1742. doi: 10.1097/SCS.0b013e31822e624f. [DOI] [PubMed] [Google Scholar]
- 15.Widdershoven JC, et al. Outcome of velopharyngoplasty in patients with velocardiofacial syndrome. Arch Otolaryngol Head Neck Surg. 2008;134(11):1159–1164. doi: 10.1001/archotol.134.11.1159. [DOI] [PubMed] [Google Scholar]
- 16.Rommel N, et al. Videomanometric evaluation of pharyngo-oesophageal dysmotility in children with velocardiofacial syndrome. J Pediatr Gastroenterol Nutr. 2008;46(1):87–91. doi: 10.1097/01.mpg.0000304460.07423.68. [DOI] [PubMed] [Google Scholar]
- 17.Arvedson J, et al. Evidence-based systematic review: effects of oral motor interventions on feeding and swallowing in preterm infants. Am J Speech Lang Pathol. 2010;19(4):321–340. doi: 10.1044/1058-0360(2010/09-0067). [DOI] [PubMed] [Google Scholar]
- 18.Pinelli J, Symington. A., Jr Non-nutritive sucking for promoting physiologic stability and nutrition in preterm infants. Cochrane Database Syst Rev. 2005;(4):CD001071. doi: 10.1002/14651858.CD001071.pub2. [DOI] [PubMed] [Google Scholar]
- 19.Vandenplas Y. Thickened infant formula does what it has to do: decrease regurgitation. Pediatrics. 2009;123(3):e549–e550. doi: 10.1542/peds.2008-3815. author reply e550. [DOI] [PubMed] [Google Scholar]
- 20.Strowd L, et al. Dysphagia dietary guidelines and the rheology of nutritional feeds and barium test feeds. Chest. 2008;133(6):1397–1401. doi: 10.1378/chest.08-0255. [DOI] [PubMed] [Google Scholar]
- 21.Dion S, et al. Use of Thickened Liquids to Manage Feeding Difficulties in Infants: A Pilot Survey of Practice Patterns in Canadian Pediatric Centers. Dysphagia. 2015 doi: 10.1007/s00455-015-9625-2. [DOI] [PubMed] [Google Scholar]
- 22.Stuart S, Motz JM. Viscosity in infant dysphagia management: comparison of viscosity of thickened liquids used in assessment and thickened liquids used in treatment. Dysphagia. 2009;24(4):412–422. doi: 10.1007/s00455-009-9219-y. [DOI] [PubMed] [Google Scholar]
- 23.Huckabee ML, Doeltgen S. Emerging modalities in dysphagia rehabilitation: neuromuscular electrical stimulation. N Z Med J. 2007;120(1263):U2744. [PubMed] [Google Scholar]
- 24.Sharp WG, et al. Pediatric feeding disorders: a quantitative synthesis of treatment outcomes. Clin Child Fam Psychol Rev. 2010;13(4):348–365. doi: 10.1007/s10567-010-0079-7. [DOI] [PubMed] [Google Scholar]
- 25.Christiaanse ME, et al. Neuromuscular electrical stimulation is no more effective than usual care for the treatment of primary dysphagia in children. Pediatr Pulmonol. 2011;46(6):559–565. doi: 10.1002/ppul.21400. [DOI] [PubMed] [Google Scholar]
- 26.Morgan AT, Dodrill P, Ward EC. Interventions for oropharyngeal dysphagia in children with neurological impairment. Cochrane Database Syst Rev. 2012;10:CD009456. doi: 10.1002/14651858.CD009456.pub2. [DOI] [PubMed] [Google Scholar]
- 27.Jones HN, et al. Oropharyngeal dysphagia in infants and children with infantile Pompe disease. Dysphagia. 2010;25(4):277–283. doi: 10.1007/s00455-009-9252-x. [DOI] [PubMed] [Google Scholar]
- 28.van Gelder CM, et al. Facial-muscle weakness, speech disorders and dysphagia are common in patients with classic infantile Pompe disease treated with enzyme therapy. J Inherit Metab Dis. 2012;35(3):505–511. doi: 10.1007/s10545-011-9404-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hobson-Webb LD, Jones HN, Kishnani PS. Oropharyngeal dysphagia may occur in late-onset Pompe disease, implicating bulbar muscle involvement. Neuromuscul Disord. 2013;23(4):319–323. doi: 10.1016/j.nmd.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 30.Demyer W, Zeman W, Palmer CG. The Face Predicts the Brain: Diagnostic Significance of Median Facial Anomalies for Holoprosencephaly (Arhinencephaly) Pediatrics. 1964;34:256–263. [PubMed] [Google Scholar]
- 31.Arvedson JC. Management of pediatric dysphagia. Otolaryngol Clin North Am. 1998;31(3):453–476. doi: 10.1016/s0030-6665(05)70064-5. [DOI] [PubMed] [Google Scholar]
- 32.Udall JN., Jr Infant feeding: initiation, problems, approaches. Curr Probl Pediatr Adolesc Health Care. 2007;37(10):374–399. doi: 10.1016/j.cppeds.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 33.Matsuo K, Palmer JB. Anatomy and physiology of feeding and swallowing: normal and abnormal. Phys Med Rehabil Clin N Am. 2008;19(4):691–707. doi: 10.1016/j.pmr.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jabbour J, et al. Pediatric Vocal Fold Immobility: Natural History and the Need for Long-Term Follow-up. JAMA Otolaryngol Head Neck Surg. 2014 doi: 10.1001/jamaoto.2014.81. [DOI] [PubMed] [Google Scholar]
- 35.Dobbie AM, White DR. Laryngomalacia. Pediatr Clin North Am. 2013;60(4):893–902. doi: 10.1016/j.pcl.2013.04.013. [DOI] [PubMed] [Google Scholar]
- 36.Karnak I, et al. Achalasia in childhood: surgical treatment and outcome. Eur J Pediatr Surg. 2001;11(4):223–229. doi: 10.1055/s-2001-17154. [DOI] [PubMed] [Google Scholar]
- 37.Khazanchi A, Katz PO. Strategies for treating severe refractory dysphagia. Gastrointest Endosc Clin N Am. 2001;11(2):371–386. [PubMed] [Google Scholar]
- 38.Buck D, et al. Surgical intervention in childhood intermittent exotropia: current practice and clinical outcomes from an observational cohort study. Br J Ophthalmol. 2012;96(10):1291–1295. doi: 10.1136/bjophthalmol-2012-301981. [DOI] [PubMed] [Google Scholar]
- 39.Oystreck DT, Engle EC, Bosley TM. Recent progress in understanding congenital cranial dysinnervation disorders. J Neuroophthalmol. 2011;31(1):69–77. doi: 10.1097/WNO.0b013e31820d0756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bosley TM, Abu-Amero KK, Oystreck DT. Congenital cranial dysinnervation disorders: a concept in evolution. Curr Opin Ophthalmol. 2013;24(5):398–406. doi: 10.1097/ICU.0b013e3283645ad6. [DOI] [PubMed] [Google Scholar]
- 41.Rucker JC, et al. Characterization of ocular motor deficits in congenital facial weakness: Moebius and related syndromes. Brain. 2014;137(Pt 4):1068–1079. doi: 10.1093/brain/awu021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Engle EC. The genetic basis of complex strabismus. Pediatr Res. 2006;59(3):343–348. doi: 10.1203/01.pdr.0000200797.91630.08. [DOI] [PubMed] [Google Scholar]
- 43.Creuzet S, Vincent C, Couly G. Neural crest derivatives in ocular and periocular structures. Int J Dev Biol. 2005;49(2–3):161–171. doi: 10.1387/ijdb.041937sc. [DOI] [PubMed] [Google Scholar]
- 44.Tischfield MA, et al. Homozygous HOXA1 mutations disrupt human brainstem, inner ear, cardiovascular and cognitive development. Nat Genet. 2005;37(10):1035–1037. doi: 10.1038/ng1636. [DOI] [PubMed] [Google Scholar]
- 45.Engle EC. Oculomotility disorders arising from disruptions in brainstem motor neuron development. Arch Neurol. 2007;64(5):633–637. doi: 10.1001/archneur.64.5.633. [DOI] [PubMed] [Google Scholar]
- 46.Cederquist GY, et al. An inherited TUBB2B mutation alters a kinesin-binding site and causes polymicrogyria, CFEOM and axon dysinnervation. Hum Mol Genet. 2012;21(26):5484–5499. doi: 10.1093/hmg/dds393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miyake N, et al. Human CHN1 mutations hyperactivate alpha2-chimaerin and cause Duane's retraction syndrome. Science. 2008;321(5890):839–843. doi: 10.1126/science.1156121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clarke WN, Noel LP. Surgical results in intermittent exotropia. Can J Ophthalmol. 1981;16(2):66–69. [PubMed] [Google Scholar]
- 49.Coffey B, et al. Treatment options in intermittent exotropia: a critical appraisal. Optom Vis Sci. 1992;69(5):386–404. doi: 10.1097/00006324-199205000-00008. [DOI] [PubMed] [Google Scholar]
- 50.Barlow SM. Central pattern generation involved in oral and respiratory control for feeding in the term infant. Curr Opin Otolaryngol Head Neck Surg. 2009;17(3):187–193. doi: 10.1097/MOO.0b013e32832b312a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bieger D, Neuhuber W. Neural circuits and mediators regulating swallowing in the brainstem. GI Motility online. 2006 [Google Scholar]
- 52.Buttner-Ennever JA, Horn AK. Anatomical substrates of oculomotor control. Curr Opin Neurobiol. 1997;7(6):872–879. doi: 10.1016/s0959-4388(97)80149-3. [DOI] [PubMed] [Google Scholar]
- 53.Mistry S, Hamdy S. Neural control of feeding and swallowing. Phys Med Rehabil Clin N Am. 2008;19(4):709–728. doi: 10.1016/j.pmr.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 54.Miller AJ. Oral and pharyngeal reflexes in the mammalian nervous system: their diverse range in complexity and the pivotal role of the tongue. Crit Rev Oral Biol Med. 2002;13(5):409–425. doi: 10.1177/154411130201300505. [DOI] [PubMed] [Google Scholar]
- 55.Schwartz MW. Brain pathways controlling food intake and body weight. Exp Biol Med (Maywood) 2001;226(11):978–981. doi: 10.1177/153537020122601103. [DOI] [PubMed] [Google Scholar]
- 56.Lang IM. Brain stem control of the phases of swallowing. Dysphagia. 2009;24(3):333–348. doi: 10.1007/s00455-009-9211-6. [DOI] [PubMed] [Google Scholar]
- 57.Ben-Ari Y, et al. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. 2007;87(4):1215–1284. doi: 10.1152/physrev.00017.2006. [DOI] [PubMed] [Google Scholar]
- 58.Takesian AE, Hensch TK. Balancing plasticity/stability across brain development. Prog Brain Res. 2013;207:3–34. doi: 10.1016/B978-0-444-63327-9.00001-1. [DOI] [PubMed] [Google Scholar]
- 59.Als H, et al. Early experience alters brain function and structure. Pediatrics. 2004;113(4):846–857. doi: 10.1542/peds.113.4.846. [DOI] [PubMed] [Google Scholar]
- 60.Blauw-Hospers CH, Hadders-Algra M. A systematic review of the effects of early intervention on motor development. Dev Med Child Neurol. 2005;47(6):421–432. doi: 10.1017/s0012162205000824. [DOI] [PubMed] [Google Scholar]
- 61.Benfer KA, Weir KA, Boyd RN. Clinimetrics of measures of oropharyngeal dysphagia for preschool children with cerebral palsy and neurodevelopmental disabilities: a systematic review. Dev Med Child Neurol. 2012;54(9):784–795. doi: 10.1111/j.1469-8749.2012.04302.x. [DOI] [PubMed] [Google Scholar]
- 62.Merscher S, et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 2001;104(4):619–629. doi: 10.1016/s0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- 63.Arvedson JC. Behavioral issues and implications with pediatric feeding disorders. Semin Speech Lang. 1997;18(1):51–69. doi: 10.1055/s-2008-1064062. quiz 69–70. [DOI] [PubMed] [Google Scholar]
- 64.Tarquinio DC, et al. Growth charts for 22q11 deletion syndrome. Am J Med Genet A. 2012;158A(11):2672–2681. doi: 10.1002/ajmg.a.35485. [DOI] [PubMed] [Google Scholar]
- 65.Caye-Thomasen P, Tos M. Eustachian tube goblet cell density during and after acute otitis media caused by Streptococcus pneumoniae: a morphometric analysis. Otol Neurotol. 2003;24(3):365–370. doi: 10.1097/00129492-200305000-00003. [DOI] [PubMed] [Google Scholar]
- 66.Wang Q, et al. CD44 deficiency leads to enhanced neutrophil migration and lung injury in Escherichia coli pneumonia in mice. Am J Pathol. 2002;161(6):2219–2228. doi: 10.1016/S0002-9440(10)64498-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang H, et al. Histological and immunological observations of bacterial and allergic chronic rhinosinusitis in the mouse. Am J Rhinol. 2008;22(4):343–348. doi: 10.2500/ajr.2008.22.3184. [DOI] [PubMed] [Google Scholar]
- 68.Hiorns MP, Ryan MM. Current practice in paediatric videofluoroscopy. Pediatr Radiol. 2006;36(9):911–919. doi: 10.1007/s00247-006-0124-3. [DOI] [PubMed] [Google Scholar]
- 69.Hoshiba J. Method for hand-feeding mouse pups with nursing bottles. Contemp Top Lab Anim Sci. 2004;43(3):50–53. [PubMed] [Google Scholar]
- 70.Wang K, et al. Utilization of three-dimensional computed tomography for craniofacial phenotypic analysis in children with velocardiofacial syndrome. J Craniofac Surg. 2009;20(6):2013–2019. doi: 10.1097/SCS.0b013e3181bd2e34. [DOI] [PubMed] [Google Scholar]
- 71.Dalben Gda S, Richieri-Costa A, Taveira LA. Craniofacial morphology in patients with velocardiofacial syndrome. Cleft Palate Craniofac J. 2010;47(3):241–246. doi: 10.1597/08-278.1. [DOI] [PubMed] [Google Scholar]
- 72.McDonald-McGinn DM, Emanuel BS, Zackai EH. 22q11.2 Deletion Syndrome. In: Pagon RA, et al., editors. GeneReviews(R) Seattle WA: 1993. [Google Scholar]
- 73.Vantrappen G, et al. The velo-cardio-facial syndrome: the otorhinolaryngeal manifestations and implications. Int J Pediatr Otorhinolaryngol. 1998;45(2):133–141. doi: 10.1016/s0165-5876(98)00067-6. [DOI] [PubMed] [Google Scholar]
- 74.Crockett DJ, et al. Obstructive sleep apnea syndrome in children with 22q11.2 deletion syndrome after operative intervention for velopharyngeal insufficiency. Front Pediatr. 2014;2:84. doi: 10.3389/fped.2014.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kennedy WP, et al. 22q11.2 Deletion syndrome and obstructive sleep apnea. Int J Pediatr Otorhinolaryngol. 2014;78(8):1360–1364. doi: 10.1016/j.ijporl.2014.05.031. [DOI] [PubMed] [Google Scholar]
- 76.Rommel N, et al. Retrospective analysis of feeding and speech disorders in 50 patients with velo-cardio-facial syndrome. Genet Couns. 1999;10(1):71–78. [PubMed] [Google Scholar]
- 77.Persson C, et al. Speech and hearing in adults with 22q11.2 deletion syndrome. Am J Med Genet A. 2012;158A(12):3071–3079. doi: 10.1002/ajmg.a.35589. [DOI] [PubMed] [Google Scholar]
- 78.Baylis AL, Watson PJ, Moller KT. Structural and functional causes of hypernasality in velocardiofacial syndrome. A pilot study. Folia Phoniatr Logop. 2009;61(2):93–96. doi: 10.1159/000209252. [DOI] [PubMed] [Google Scholar]
- 79.Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet. 2007;370(9596):1443–1452. doi: 10.1016/S0140-6736(07)61601-8. [DOI] [PubMed] [Google Scholar]
- 80.Milczuk HA, Smith DS, Brockman JH. Surgical outcomes for velopharyngeal insufficiency in velocardiofacial syndrome and nonsyndromic patients. Cleft Palate Craniofac J. 2007;44(4):412–417. doi: 10.1597/05-136.1. [DOI] [PubMed] [Google Scholar]
- 81.Losken A, et al. Surgical correction of velopharyngeal insufficiency in children with velocardiofacial syndrome. Plast Reconstr Surg. 2006;117(5):1493–1498. doi: 10.1097/01.prs.0000206377.14083.ce. [DOI] [PubMed] [Google Scholar]
- 82.Kirschner RE, Baylis AL. Surgical considerations in 22Q11.2 deletion syndrome. Clin Plast Surg. 2014;41(2):271–282. doi: 10.1016/j.cps.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 83.Dastjerdi A, et al. Tbx1 regulation of myogenic differentiation in the limb and cranial mesoderm. Dev Dyn. 2007;236(2):353–363. doi: 10.1002/dvdy.21010. [DOI] [PubMed] [Google Scholar]
- 84.van Bueren KL, et al. Hes1 expression is reduced in Tbx1 null cells and is required for the development of structures affected in 22q11 deletion syndrome. Dev Biol. 2010;340(2):369–380. doi: 10.1016/j.ydbio.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Scambler PJ. 22q11 deletion syndrome: a role for TBX1 in pharyngeal and cardiovascular development. Pediatr Cardiol. 2010;31(3):378–390. doi: 10.1007/s00246-009-9613-0. [DOI] [PubMed] [Google Scholar]
- 86.Okubo T, et al. Ripply3, a Tbx1 repressor, is required for development of the pharyngeal apparatus and its derivatives in mice. Development. 2011;138(2):339–348. doi: 10.1242/dev.054056. [DOI] [PubMed] [Google Scholar]
- 87.Kong P, et al. Tbx1 is required autonomously for cell survival and fate in the pharyngeal core mesoderm to form the muscles of mastication. Hum Mol Genet. 2014;23(16):4215–4231. doi: 10.1093/hmg/ddu140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liao J, et al. Full spectrum of malformations in velo-cardio-facial syndrome/DiGeorge syndrome mouse models by altering Tbx1 dosage. Hum Mol Genet. 2004;13(15):1577–1585. doi: 10.1093/hmg/ddh176. [DOI] [PubMed] [Google Scholar]
- 89.Arnold JS, et al. Inactivation of Tbx1 in the pharyngeal endoderm results in 22q11DS malformations. Development. 2006;133(5):977–987. doi: 10.1242/dev.02264. [DOI] [PubMed] [Google Scholar]
- 90.Vitelli F, et al. Tbx1 mutation causes multiple cardiovascular defects and disrupts neural crest and cranial nerve migratory pathways. Hum Mol Genet. 2002;11(8):915–922. doi: 10.1093/hmg/11.8.915. [DOI] [PubMed] [Google Scholar]
- 91.Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. 2001;27(3):286–291. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- 92.Lindsay EA, et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410(6824):97–101. doi: 10.1038/35065105. [DOI] [PubMed] [Google Scholar]
- 93.Kelly RG, Jerome-Majewska LA, Papaioannou VE. The del22q11.2 candidate gene Tbx1 regulates branchiomeric myogenesis. Hum Mol Genet. 2004;13(22):2829–2840. doi: 10.1093/hmg/ddh304. [DOI] [PubMed] [Google Scholar]
- 94.Funato N, et al. Loss of Tbx1 induces bone phenotypes similar to cleidocranial dysplasia. Hum Mol Genet. 2015;24(2):424–435. doi: 10.1093/hmg/ddu458. [DOI] [PubMed] [Google Scholar]
- 95.Grenier J, et al. Relationship between neural crest cells and cranial mesoderm during head muscle development. PLoS One. 2009;4(2):e4381. doi: 10.1371/journal.pone.0004381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Arnold JS, et al. Tissue-specific roles of Tbx1 in the development of the outer, middle and inner ear, defective in 22q11DS patients. Hum Mol Genet. 2006;15(10):1629–1639. doi: 10.1093/hmg/ddl084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Moraes F, et al. Tbx1 is required for proper neural crest migration and to stabilize spatial patterns during middle and inner ear development. Mech Dev. 2005;122(2):199–212. doi: 10.1016/j.mod.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 98.Vitelli F, et al. TBX1 is required for inner ear morphogenesis. Hum Mol Genet. 2003;12(16):2041–2048. doi: 10.1093/hmg/ddg216. [DOI] [PubMed] [Google Scholar]
- 99.Raft S, et al. Suppression of neural fate and control of inner ear morphogenesis by Tbx1. Development. 2004;131(8):1801–1812. doi: 10.1242/dev.01067. [DOI] [PubMed] [Google Scholar]
- 100.Calmont A, et al. Tbx1 controls cardiac neural crest cell migration during arch artery development by regulating Gbx2 expression in the pharyngeal ectoderm. Development. 2009;136(18):3173–3183. doi: 10.1242/dev.028902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Aggarwal VS, et al. Mesodermal Tbx1 is required for patterning the proximal mandible in mice. Dev Biol. 2010;344(2):669–681. doi: 10.1016/j.ydbio.2010.05.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zoupa M, et al. Tbx1 is expressed at multiple sites of epithelial-mesenchymal interaction during early development of the facial complex. Int J Dev Biol. 2006;50(5):504–510. doi: 10.1387/ijdb.052116mz. [DOI] [PubMed] [Google Scholar]
- 103.Maynard TM, et al. 22q11 Gene dosage establishes an adaptive range for sonic hedgehog and retinoic acid signaling during early development. Hum Mol Genet. 2013;22(2):300–312. doi: 10.1093/hmg/dds429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Maynard TM, et al. RanBP1, a velocardiofacial/DiGeorge syndrome candidate gene, is expressed at sites of mesenchymal/epithelial induction. Mech Dev. 2002;111(1–2):177–180. doi: 10.1016/s0925-4773(01)00616-5. [DOI] [PubMed] [Google Scholar]
- 105.Meechan DW, et al. Gene dosage in the developing and adult brain in a mouse model of 22q11 deletion syndrome. Mol Cell Neurosci. 2006;33(4):412–428. doi: 10.1016/j.mcn.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 106.Guthrie S. Patterning the hindbrain. Curr Opin Neurobiol. 1996;6(1):41–48. doi: 10.1016/s0959-4388(96)80007-9. [DOI] [PubMed] [Google Scholar]
- 107.Narita Y, Rijli. FM. Hox genes in neural patterning and circuit formation in the mouse hindbrain. Curr Top Dev Biol. 2009;88:139–167. doi: 10.1016/S0070-2153(09)88005-8. [DOI] [PubMed] [Google Scholar]
- 108.Rinon A, et al. Cranial neural crest cells regulate head muscle patterning and differentiation during vertebrate embryogenesis. Development. 2007;134(17):3065–3075. doi: 10.1242/dev.002501. [DOI] [PubMed] [Google Scholar]
- 109.Tzahor E, Evans SM. Pharyngeal mesoderm development during embryogenesis: implications for both heart and head myogenesis. Cardiovasc Res. 2011;91(2):196–202. doi: 10.1093/cvr/cvr116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Steventon B, Mayor R, Streit A. Neural crest and placode interaction during the development of the cranial sensory system. Dev Biol. 2014;389(1):28–38. doi: 10.1016/j.ydbio.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Saint-Jeannet JP, Moody SA. Establishing the pre-placodal region and breaking it into placodes with distinct identities. Dev Biol. 2014;389(1):13–27. doi: 10.1016/j.ydbio.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Guthrie S. Patterning and axon guidance of cranial motor neurons. Nat Rev Neurosci. 2007;8(11):859–871. doi: 10.1038/nrn2254. [DOI] [PubMed] [Google Scholar]
- 113.Walker MB, Trainor PA. Craniofacial malformations: intrinsic vs extrinsic neural crest cell defects in Treacher Collins and 22q11 deletion syndromes. Clin Genet. 2006;69(6):471–479. doi: 10.1111/j.0009-9163.2006.00615.x. [DOI] [PubMed] [Google Scholar]
- 114.Wurdak H, Ittner LM, Sommer L. DiGeorge syndrome and pharyngeal apparatus development. Bioessays. 2006;28(11):1078–1086. doi: 10.1002/bies.20484. [DOI] [PubMed] [Google Scholar]
- 115.Trainor PA. Specification and patterning of neural crest cells during craniofacial development. Brain Behav Evol. 2005;66(4):266–280. doi: 10.1159/000088130. [DOI] [PubMed] [Google Scholar]
- 116.Creuzet S, Couly G, Le Douarin NM. Patterning the neural crest derivatives during development of the vertebrate head: insights from avian studies. J Anat. 2005;207(5):447–459. doi: 10.1111/j.1469-7580.2005.00485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wilkinson DG. Molecular mechanisms of segmental patterning in the vertebrate hindbrain and neural crest. Bioessays. 1993;15(8):499–505. doi: 10.1002/bies.950150802. [DOI] [PubMed] [Google Scholar]
- 118.Carpenter EM, et al. Loss of Hox-A1 (Hox-1.6) function results in the reorganization of the murine hindbrain. Development. 1993;118(4):1063–1075. doi: 10.1242/dev.118.4.1063. [DOI] [PubMed] [Google Scholar]
- 119.Briscoe J, Wilkinson DG. Establishing neuronal circuitry: Hox genes make the connection. Genes Dev. 2004;18(14):1643–1648. doi: 10.1101/gad.1227004. [DOI] [PubMed] [Google Scholar]
- 120.Cordes SP. Molecular genetics of cranial nerve development in mouse. Nat Rev Neurosci. 2001;2(9):611–623. doi: 10.1038/35090039. [DOI] [PubMed] [Google Scholar]
- 121.Maynard TM, et al. A comprehensive analysis of 22q11 gene expression in the developing and adult brain. Proc Natl Acad Sci U S A. 2003;100(24):14433–1448. doi: 10.1073/pnas.2235651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gavalas A. ArRAnging the hindbrain. Trends Neurosci. 2002;25(2):61–64. doi: 10.1016/s0166-2236(02)02067-2. [DOI] [PubMed] [Google Scholar]
- 123.Maden M. Heads or tails? Retinoic acid will decide. Bioessays. 1999;21(10):809–812. doi: 10.1002/(SICI)1521-1878(199910)21:10<809::AID-BIES2>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 124.Glover JC, Renaud JS, Rijli FM. Retinoic acid and hindbrain patterning. J Neurobiol. 2006;66(7):705–725. doi: 10.1002/neu.20272. [DOI] [PubMed] [Google Scholar]
- 125.Mark M, Ghyselinck NB, Chambon P. Retinoic acid signalling in the development of branchial arches. Curr Opin Genet Dev. 2004;14(5):591–598. doi: 10.1016/j.gde.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 126.Niederreither K, et al. Retinoic acid synthesis and hindbrain patterning in the mouse embryo. Development. 2000;127(1):75–85. doi: 10.1242/dev.127.1.75. [DOI] [PubMed] [Google Scholar]
- 127.Karpinski BA, et al. A Cellular and Molecular Mosaic Establishes Growth and Differentiation States for Cranial Sensory Neurons. Dev Biol. 2015 doi: 10.1016/j.ydbio.2016.03.015. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dupe V, et al. Key roles of retinoic acid receptors alpha and beta in the patterning of the caudal hindbrain, pharyngeal arches and otocyst in the mouse. Development. 1999;126(22):5051–5059. doi: 10.1242/dev.126.22.5051. [DOI] [PubMed] [Google Scholar]
- 129.Lammer EJ, et al. Retinoic acid embryopathy. N Engl J Med. 1985;313(14):837–841. doi: 10.1056/NEJM198510033131401. [DOI] [PubMed] [Google Scholar]
- 130.Wilson JG, Roth CB, Warkany J. An analysis of the syndrome of malformations induced by maternal vitamin A deficiency. Effects of restoration of vitamin A at various times during gestation. Am J Anat. 1953;92(2):189–217. doi: 10.1002/aja.1000920202. [DOI] [PubMed] [Google Scholar]
- 131.Vermot J, et al. Decreased embryonic retinoic acid synthesis results in a DiGeorge syndrome phenotype in newborn mice. Proc Natl Acad Sci U S A. 2003;100(4):1763–1768. doi: 10.1073/pnas.0437920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Corcoran J. What are the molecular mechanisms of neural tube defects? Bioessays. 1998;20(1):6–8. doi: 10.1002/(SICI)1521-1878(199801)20:1<6::AID-BIES3>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 133.Diez-Pardo JA, et al. Neural tube defects: an experimental model in the foetal rat. Eur J Pediatr Surg. 1995;5(4):198–202. doi: 10.1055/s-2008-1066204. [DOI] [PubMed] [Google Scholar]
- 134.Quemelo PR, Lourenco CM, Peres LC. Teratogenic effect of retinoic acid in swiss mice. Acta Cir Bras. 2007;22(6):451–456. doi: 10.1590/s0102-86502007000600007. [DOI] [PubMed] [Google Scholar]
- 135.Ryckebusch L, et al. Retinoic acid deficiency alters second heart field formation. Proc Natl Acad Sci U S A. 2008;105(8):2913–2918. doi: 10.1073/pnas.0712344105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Niederreither K, et al. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat Genet. 1999;21(4):444–448. doi: 10.1038/7788. [DOI] [PubMed] [Google Scholar]
- 137.Wendling O, et al. Retinoid signaling is essential for patterning the endoderm of the third and fourth pharyngeal arches. Development. 2000;127(8):1553–1562. doi: 10.1242/dev.127.8.1553. [DOI] [PubMed] [Google Scholar]
- 138.Quinlan R, et al. Deficits in the posterior pharyngeal endoderm in the absence of retinoids. Dev Dyn. 2002;225(1):54–60. doi: 10.1002/dvdy.10137. [DOI] [PubMed] [Google Scholar]
- 139.Matt N, et al. Retinoic acid-induced developmental defects are mediated by RARbeta/RXR heterodimers in the pharyngeal endoderm. Development. 2003;130(10):2083–2093. doi: 10.1242/dev.00428. [DOI] [PubMed] [Google Scholar]
- 140.Mulder GB, Manley N, Maggio-Price L. Retinoic acid-induced thymic abnormalities in the mouse are associated with altered pharyngeal morphology, thymocyte maturation defects, and altered expression of Hoxa3 and Pax1. Teratology. 1998;58(6):263–275. doi: 10.1002/(SICI)1096-9926(199812)58:6<263::AID-TERA8>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 141.Ryckebusch L, et al. Decreased levels of embryonic retinoic acid synthesis accelerate recovery from arterial growth delay in a mouse model of DiGeorge syndrome. Circulation Research. 2010;106(4):686–694. doi: 10.1161/CIRCRESAHA.109.205732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Guris DL, et al. Dose-dependent interaction of Tbx1 and Crkl and locally aberrant RA signaling in a model of del22q11 syndrome. Dev Cell. 2006;10(1):81–92. doi: 10.1016/j.devcel.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 143.Roberts C, et al. Cyp26 genes a1, b1 and c1 are down-regulated in Tbx1 null mice and inhibition of Cyp26 enzyme function produces a phenocopy of DiGeorge Syndrome in the chick. Human Molecular Genetics. 2006;15(23):3394–3410. doi: 10.1093/hmg/ddl416. [DOI] [PubMed] [Google Scholar]
- 144.Ivins S, et al. Microarray analysis detects differentially expressed genes in the pharyngeal region of mice lacking Tbx1. Dev Biol. 2005;285(2):554–569. doi: 10.1016/j.ydbio.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 145.Mulder GB, et al. Effects of excess vitamin A on development of cranial neural crest-derived structures: a neonatal and embryologic study. Teratology. 2000;62(4):214–226. doi: 10.1002/1096-9926(200010)62:4<214::AID-TERA7>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 146.Anchan RM, et al. Disruption of local retinoid-mediated gene expression accompanies abnormal development in the mammalian olfactory pathway. J Comp Neurol. 1997;379(2):171–184. [PubMed] [Google Scholar]
- 147.Haskell GT, LaMantia AS. Retinoic acid signaling identifies a distinct precursor population in the developing and adult forebrain. J Neurosci. 2005;25(33):7636–7647. doi: 10.1523/JNEUROSCI.0485-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhu H, Kartiko S, Finnell RH. Importance of gene-environment interactions in the etiology of selected birth defects. Clin Genet. 2009;75(5):409–423. doi: 10.1111/j.1399-0004.2009.01174.x. [DOI] [PubMed] [Google Scholar]
- 149.Piedrahita JA, et al. Mice lacking the folic acid-binding protein Folbp1 are defective in early embryonic development. Nat Genet. 1999;23(2):228–232. doi: 10.1038/13861. [DOI] [PubMed] [Google Scholar]
- 150.Zhu H, et al. Cardiovascular abnormalities in Folr1 knockout mice and folate rescue. Birth Defects Res A Clin Mol Teratol. 2007;79(4):257–268. doi: 10.1002/bdra.20347. [DOI] [PubMed] [Google Scholar]
- 151.Reynolds PR, Schaalje GB, Seegmiller RE. Combination therapy with folic acid and methionine in the prevention of retinoic acid-induced cleft palate in mice. Birth Defects Res A Clin Mol Teratol. 2003;67(3):168–173. doi: 10.1002/bdra.10036. [DOI] [PubMed] [Google Scholar]
- 152.Yao Z, et al. Folic acid rescue of ATRA-induced cleft palate by restoring the TGF-beta signal and inhibiting apoptosis. Journal of Oral Pathology and Medicine. 2011;40(5):433–439. doi: 10.1111/j.1600-0714.2010.00994.x. [DOI] [PubMed] [Google Scholar]
- 153.Cipollone D, et al. Folic acid and methionine in the prevention of teratogen-induced congenital defects in mice. Cardiovasc Pathol. 2009;18(2):100–109. doi: 10.1016/j.carpath.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 154.Zhang Z, et al. Prevention of retinoic acid-induced early craniofacial abnormalities by folinic acid and expression of endothelin-1/dHAND in the branchial arches in mouse. Br J Nutr. 2006;96(3):418–425. [PubMed] [Google Scholar]
- 155.White JC, et al. Defects in embryonic hindbrain development and fetal resorption resulting from vitamin A deficiency in the rat are prevented by feeding pharmacological levels of all-trans-retinoic acid. Proc Natl Acad Sci U S A. 1998;95(23):13459–13464. doi: 10.1073/pnas.95.23.13459. [DOI] [PMC free article] [PubMed] [Google Scholar]