

Abstract
The serine protease tissue-type plasminogen activator (t-PA) is involved in both vital physiological brain processes, such as synaptic plasticity, and pathophysiological conditions, such as neurodegeneration and ischemic stroke. Recent data suggest that epigenetic mechanisms play an important role in the regulation of t-PA in human endothelial cells. However, there are limited data on epigenetic regulation of t-PA in human brain-derived cells. We demonstrate that treatment of cultured human neurons and human astrocytes with the histone deacetylase inhibitors trichostatin A (TSA) and MS-275 resulted in a two- to threefold increase in t-PA mRNA and protein expression levels. Next, we performed a chromatin immunoprecipitation assay on treated astrocytes with antibodies directed against acetylated histones H3 and H4 (both markers of gene activation). Treatment with MS-275 and TSA for 24 hours resulted in a significant increase in H3 acetylation, which could explain the observed increase in t-PA gene activity after the inhibition of histone deacety-lation. Furthermore, DNA methylation analysis of cultured human neurons and astrocytes, as well as human postmortem brain tissue, revealed a stretch of unmethylated CpG dinucleotides in the proximal t-PA promoter, whereas more upstream CpGs were highly methylated. Taken together, these results implicate involvement of epigenetic mechanisms in the regulation of t-PA expression in the human brain.
Keywords: tissue-type plasminogen activator, brain tissue, histone acetylation, DNA methylation
Introduction
Tissue-type plasminogen activator (t-PA) is produced by endothelial cells (ECs) and is the main activator of the fibrinolytic system in the intravascular compartment. In the brain, t-PA is also expressed by neurons and glial cells.1 Under physiological conditions, t-PA promotes synaptic remodeling and neuronal plasticity.2 On the other hand, excessive release of t-PA into the extracellular space during pathological conditions, such as inflammatory and ischemic events, can potentiate excitotoxicity and neurodegeneration.3,4 Several physiological and pathological processes in the brain have been associated with epigenetic events, including synaptic plasticity, neurodegeneration, and ischemic stroke.5 Interestingly, histone deacetylase (HDAC) inhibitors have been shown to potentiate synaptic plasticity6 and show therapeutic efficiency in murine models of ischemic stroke.7
Studies on human ECs have demonstrated that the t-PA gene is under epigenetic control in this cell type.8 Epigenetic regulation is commonly cell-type specific. Thus, an increased knowledge regarding the epigenetic regulation of t-PA in the human brain is of importance. Here, we sought to investigate DNA methylation at the t-PA gene promoter and the effect of histone acetylation on t-PA expression in human brain-derived cells.
Materials and Methods
Cell culture and treatments
Human primary astrocytes (ScienCell Research Laboratories) were cultured in astrocyte growth medium supplemented with 2% fetal bovine serum, 1% astrocyte growth supplement, and 1% penicillin/streptomycin solution (ScienCell Research Laboratories). The cells were split and subcultured using trypsin-EDTA (0.25 mg/mL; ScienCell Research Laboratories) treatment. Human primary neurons (ScienCell Research Laboratories) were cultured on poly-l-lysine-coated flasks (ScienCell Research Laboratories) in neuronal medium supplemented with 1% neuronal growth supplement and 1% penicillin/streptomycin solution (ScienCell Research Laboratories). The cells were kept at 37 °C and 5% CO2 in a humidified environment. The medium was replaced in the first two days and every two to three days thereafter. To investigate the influence of histone modifications on t-PA gene expression, cultures of human neurons and astrocytes were treated with HDAC inhibitors trichostatin A (TSA, 1 μM; Sigma-Aldrich) or MS-275 (10 μM; Selleck Chemicals) for the indicated time periods. TSA inhibits class I and class II HDACs, whereas MS-275 specifically inhibits class I HDACs.9 HDAC inhibitors were prepared in dimethyl sulfoxide (DMSO) and diluted in astrocyte growth medium or neuronal medium. Control cultures were exposed to the maximum final concentration of DMSO (0.1%). All stimulations were performed in six separate cell culture wells (n = 6). For practical reasons, cells were derived from two different individuals. Following treatments, cell media and cellular extracts were collected and stored at −20 and −80 °C, respectively, until further analysis.
Human brain tissues
The study was conducted in compliance with the principles of the Declaration of Helsinki. Fresh frozen brain tissue samples derived from the cortex and the hippocampus (n = 10 per brain region) were obtained from the Neurological Foundation of New Zealand Human Brain Bank. All donated brains were collected with the full consent of the next of kin and in line with ethics approval from The University of Auckland Human Participants Ethics Committee (Ref 011654). There were no evident neurological injuries associated with the cause of death, and the assessment of all brains by a neuropathologist revealed no neurological abnormalities. Mean postmortem delay was 13 hours (range 7–21 hours).
Analysis of t-PA mRNA and protein expression
Total cellular RNA was isolated from cellular extracts using E.Z.N.A. Total RNA Isolation Kit (Omega Bio-tek) according to the manufacturer’s protocol. Isolated RNA was converted to cDNA using a GeneAmp RNA PCR Kit (Applied Biosystems). Expression levels of t-PA mRNA were determined using TaqMan Real-Time qPCR (Applied Biosystems). All samples were normalized relative to glyceraldehyde 3-phosphate dehydrogenase. The reactions were performed in a 384-well format using an ABI Prism 79100HT Sequence Detection System (Applied Biosystems) in a total volume of 10 μL. Thermocycling conditions were two minutes at 50 °C and 10 minutes at 95 °C, followed by 40 cycles for 15 seconds at 95 °C and one minute at 60 °C. Relative quantification of gene expression was analyzed as a treatment-to-control expression ratio using the comparative CT method. Each sample was analyzed in triplicate for both target and control genes. Probe and primer sequences are shown in Table 1.
Table 1.
Primer and probe nucleotide sequences used in this study.
| GENE EXPRESSION qPCR | ||
|---|---|---|
| GENE | PRIMER SEQUENCE | PROBE SEQUENCE |
| t-PA | Fw 5′-GGC CTT GTC TCC TTT CTA TTC G-3′ Rv 5′-AGC GGC TGG ATG GGT ACA G-3′ |
5′-TGA CAT GAG CCT CCT TCA GCC GCT-3′ |
| GAPDH | Fw 5′-CCA CAT CGC TCA GAC ACC AT-3′ Rv 5′-CCA GGC GCC CAA TAC G-3′ |
5′-AAG GTG AAG GTC GGA GTC AAC GGA TTT G-3′ |
| ChiP qPCR | ||
|---|---|---|
| REGION | PRIMER SEQUENCE | LOCATION* |
| t-PA promoter | Fw 5′-ACC CCC TGC CTG GAA ACT TA-3′ Rv 5′-GGT ACA GAA ACC CGA CCT ACC A-3′ |
−46 to +92 |
| BISULFITE SEQUENCING | ||||
|---|---|---|---|---|
| REGION | # | PRIMER SEQUENCE | LOCATION* | |
| t-PA promoter 1 | Outer | 1 | Fw 5′-TTT GAA AAG GTG TTA GTA AG-3′ Rv 5′-ACC ACT AAA AAA ACA AAA CC-3′ |
−721 to −382 |
| Inner | 2 | Fw 5′-TAA GGG AAA TGG TTT GTT TA-3′ Rv 5′-CTACRATAAAAAATACCCCCATA-3′ |
−705 to −416 | |
| t-PA promoter 2 | Outer | 3 | Fw 5′-TTT GGG TTT ATT TAA GGG GAT GT-3′ Rv 5′-AAA AAT TTT CTC TCC AAC CCT AAA C-3′ |
−577 to +156 |
| Inner | 4 | Fw 5′-GAG GTT ATT TAT TGT AGT TTT GTA TTT TAT-3′ Rv 5′-CAA CTC TAA ACT CCC CAC AAC TC-3′ |
−535 to +118 | |
| t-PA promoter 3 | Outer | 5 | Fw 5′-TTA GGA TTT TAA AGG AAG ATG ATT TTT AA-3′ Rv 5′-AAA AAA ACA AAC CCC AAA ATA CAA-3′ |
−176 to +222 |
| Inner | 6 | Fw 5′-AAA GGA AGA TGA TTT TTA AGG TTT TAT TT-3′ Rv 5′-AAA ATT TTC TCT CCA ACC CTA AAC T-3′ |
−166 to +155 | |
Notes:
Location with respect to the transcription start site of the t-PA gene. Top panel: Primers and TaqMan probes used in the real-time RT-PCR. mRNA of the t-PA gene was converted to complementary DNA and quantitatively amplified together with the reference gene GAPDH. Middle panel: Primes sequences used for SYBR Green real-time PCR quantification of chromatin immunoprecipitation (ChIP) samples. Bottom panel: Primer sequences used in the methylation analysis of the t-PA promoter. Three overlapping regions of the t-PA promoter (t-PA promoter 1, 2 and 3) were amplified in a nested PCR. The reaction was run in two stages with outer and an inner primers. Locations are given according to the NCBI Reference Sequences for t-PA. Fw denotes forward, Rv denotes reverse.
Antigen levels of t-PA in the cell culture media following treatment with HDAC inhibitors were determined by Enzyme-Linked Immunosorbent Assay (ELISA) (TriniLIZE t-PA; Trinity Biotech) according to the manufacturer’s protocol.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) was performed using the ChIP-IT Express Kit (Active Motif) as follows. Human astrocytes were cultured on 15-cm plates until ~80% confluence and stimulated with TSA (1 μM) or MS-275 (10 μM) for 24 hours. Control cultures were treated with the final maximum concentration of DMSO as described above. After treatments, chromatin was cross-linked by adding serum-free astrocyte growth medium supplemented with 1% formaldehyde to each plate and incubating for 10 minutes at room temperature. The cells were washed with ice-cold Phosphate-Buffered Saline (PBS), and the fixation reaction was stopped by the addition of 1 × glycine/PBS buffer for five minutes. The fixed cells were washed in PBS, and the plates were scraped using a rubber policeman and a cell-scraping solution of ice-cold PBS supplemented with 30 μL of 100 mM phenylmethanesulfonyl fluoride (PMSF). The harvested cells were pelleted by centrifugation, resuspended in ice-cold lysis buffer supplemented with PMSF and protein inhibitor cocktail (PIC), and incubated on ice for 30 minutes. The lysates were transferred to a homogenizer and dounced on ice. Following centrifugation, the pellets were resuspended in shearing buffer supplemented with PMSF and PIC and sonicated at 4 °C using a Bioruptor Pico (Diagenode). The sheared chromatin samples were centrifuged at 4 °C, and the supernatants containing the sheared chromatin were collected. At this point, 10 μL of each sample was removed and stored at 20 °C to serve as the input DNA control.
The ChIP reaction was performed with sheared chromatin, protein G magnet beads, ChIP buffer, PIC, and rabbit anti-acetyl-histone H3 (1 mg/mL) or anti-acetyl-histone H4 (Merck Millipore). No-antibody controls were also performed. All samples were incubated overnight on a rotator at 4 °C. The next day, beads were washed, resuspended in elution buffer, and incubated on a rotator for 10 minutes at room temperature. Reverse cross-linking buffer was added to each sample, and the captured DNA was eluted by magnetic separation of the beads.
In the next step, ChIP buffer 2 and 5 M NaCl was added to the input DNA samples. ChIP samples and input DNA samples were incubated at 95 °C for 15 minutes, followed by the addition of proteinase K and further incubation for one hour at 37 °C. Thereafter, samples were returned to room temperature, and proteinase K stop solution was added. Samples were cleaned using a Chromatin IP DNA Purification Kit (Active Motif), and the captured DNA was quantified using SYBR Green detection. Samples were analyzed in triplicates on a CFX384 Real-Time PCR Detection System (Bio-Rad). ChIP results are presented as the percentage of input fraction and corrected for background with no-antibody controls. Primer sequences are displayed in Table 1.
DNA methylation analysis of the t-PA promoter
Total DNA from cultured cells and brain tissues was isolated using the E.Z.N.A. Tissue DNA Kit (Omega Bio-tek) and modified with bisulfite using a MethylCode Bisulfite Conversion Kit (Invitrogen). Following bisulfite conversion, three overlapping regions of the t-PA promoter were amplified by nested Polymerase Chain Reaction (PCR). All primer sequences are shown in Table 1. Conditions for both the primary and the secondary reactions were 95 °C for two minutes, followed by 35 cycles at 94 °C for one minute, 58 °C for one minute, and 72 °C for two minutes, followed by 10 minutes at 72 °C. The sequencing of the PCR products was performed using the inner primers (primers 2, 4, and 6; Table 1), and the conditions for the reaction were 94 °C for three minutes, followed by 50 cycles at 96 °C for 30 seconds, 50 °C for 10 seconds, and 60 °C for three minutes. The methylation state of each individual CG dinucleotide (CpG) in the region of interest was analyzed subsequent to direct bisulfite sequencing, as described elsewhere.10
Statistical analysis
Differences in mRNA and protein expression levels and differences in histone acetylation levels between HDAC inhibitor-treated cells and control-treated cells were determined by the unpaired Student’s t-test. A P value of ≤0.05 was considered statistically significant. Values are presented as mean and standard error of the mean.
Results and Discussion
Following treatment with the HDAC inhibitors, TSA and MS-275, for 24 hours, we observed a two- to threefold increase in t-PA mRNA expression in astrocyte cultures compared to the control-treated cells (Fig. 1A). Similar results were obtained for neuronal cultures (Fig. 1B). t-PA antigen levels in astrocyte-conditioned media also increased ~1.5- to 2-fold in response to HDAC inhibitor treatment (Fig. 1A). Antigen levels of t-PA were below the detection limit in the culture media from neuronal cells. The observed increase in t-PA expression after HDAC inhibition suggests a role for histone modifications in t-PA gene regulation in brain-derived cells, either direct or indirect.
Figure 1.

Treatment with histone deacetylase (HDAC) inhibitors resulted in a significant increase in t-PA expression in cultured human astrocytes and neurons. (A) t-PA mRNA and protein expression levels in cultured human astrocytes following treatment with 1 μM trichostatin A (TSA) or 10 μM MS−275. (B) t-PA mRNA expression levels in cultured human neurons following treatment with TSA or MS-275. Cells were treated for 14 or 24 hours. Results are shown as fold induction compared with control-treated cells and presented as mean ± SEM. Each data point represents the average of three independent treatment series performed on two different occasions (n = 6). Cells were derived from two different individuals. Response to treatment was evaluated by the unpaired Student’s t-test: *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001.
Compared with astrocytes, induction of t-PA mRNA in neurons appeared to occur at a later time point. The reason for this delayed response could indicate a difference in the fine-tuning of t-PA regulation between the two cell types and should be examined further.
HDACs act not only on histones but also on a variety of other proteins, such as transcription factors, that may influence gene regulation. To determine whether treatment with HDAC inhibitors de facto increased t-PA promoter acetylation, we performed a ChIP assay on treated astrocytes with antibodies directed against acetylated histones H3 and H4 (both markers of gene activation). Treatment with MS-275 and TSA for 24 hours resulted in a significant increase in H3 acetylation (Fig. 2). This confirms that the treatment of astrocytes with HDAC inhibitors acts directly on histone H3 acetylation in the t-PA promoter, which could explain the observed increase in t-PA gene activity after the inhibition of histone deacetylation. In contrast, no significant increase in histone H4 acetylation was observed following treatment with MS-275 or TSA.
Figure 2.
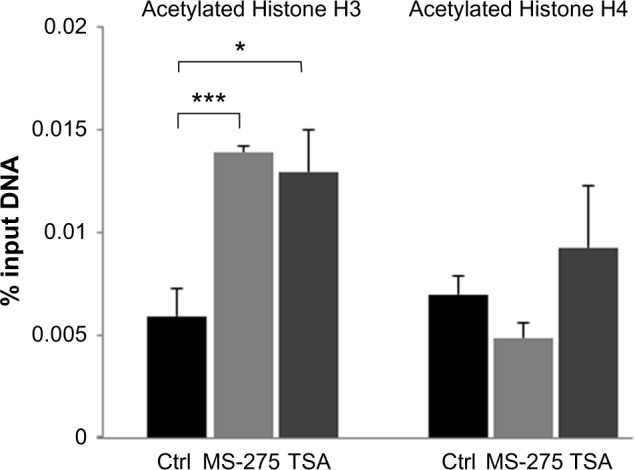
Treatment with histone deacetylase (HDAC) inhibitors resulted in a significant increase in acetylation of histone H3 but not histone H4 in the t-PA promoter. Results are presented as mean percentage of input DNA ± SEM. Each data point represents the average of two independent ChIP experiments performed in triplicates (n = 2).
In all human brain-derived cells and brain tissue samples examined, we observed a high degree of DNA methylation (50–100%) in the t-PA promoter CpG dinucleotides from position −618 to −366, relative to the transcription start site, whereas the CpG dinucleotides in the proximal promoter, from position −121 to +94, showed a minimal degree of methylation (0–20%; Fig. 3). Minor differences in the methylation state of the t-PA promoter were found between cultured human astrocytes and neurons, as well as hippocampal and cortical human brain tissues. The observed hypomethylation at the t-PA promoter region in human brain-derived cells and brain tissue is in line with previous results in human ECs, whereas human hepatoma cells and hepatocytes exhibit a considerably higher degree of DNA methylation in the same genetic region.8 Neurons, astrocytes, and ECs are cell types with a high basal expression of t-PA, whereas hepatoma cells and hepatocytes have a low basal expression.8 Thus, the expression level of t-PA appears inversely correlated with the degree of promoter methylation, which is expected for genes that are under epigenetic influence. However, the mechanistic significance for the regulation of the t-PA gene remains to be examined further.
Figure 3.

DNA methylation analysis revealed a stretch of unmethylated CpG dinucleotides in the proximal t-PA promoter, whereas more upstream CpGs were highly methylated. DNA methylation levels (%) are shown for human hippocampal tissue (n = 10), human cortical brain tissue (n = 10), cultured human astrocytes (n = 2), and cultured human neurons (n = 2). Results are presented as mean ± SEM.
Conclusion
Our study presents two novel findings considering epigenetic aspects of t-PA gene regulation in the human brain. First, we demonstrate that the expression of t-PA is induced in cultured human neurons and astrocytes after the inhibition of histone deacetylation and this is associated with an increase in histone H3 acetylation in cultured astrocytes. Second, our DNA methylation analysis of cultured human neurons, astrocytes, and human brain tissues revealed a stretch of unmethylated CpG dinucleotides in the proximal t-PA promoter, whereas more upstream CpGs were highly methylated.
Taken together, these results suggest that epigenetic mechanisms are of significance for the regulation of t-PA gene in the human brain. Thus, future studies on the putative impact of epigenetic modulation on t-PA gene expression in brain disorders are motivated.
Acknowledgments
The authors thank the Genomics Core Facility platform at the Sahlgrenska Academy, University of Gothenburg, for DNA sequencing and real-time qPCR support, the TATAA Biocenter, Gothenburg, for qPCR support, and the Neurological Foundation of New Zealand for providing human brain tissue samples.
Footnotes
ACADEMIC EDITOR: James Willey, Editor in Chief
PEER REVIEW: Four peer reviewers contributed to the peer review report. Reviewers’ reports totaled 794 words, excluding any confidential comments to the academic editor.
FUNDING: This study was supported by the Swedish Research Council (K2011-65X-14605-09-6), the Swedish state (ALFGBG-148861), the Swedish Heart and Lung Foundation (20100256), the Yngve Land, the Rune and Ulla Amlöv, the Edit Jacobsson, and the Swiss National Science Foundation (310030-133099). The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Conceived and designed the experiments: MO, KH, CJ. Analyzed the data: MO, KH, EKOK. Supplied the critical material: MO, KH, SD-G, MAC, RLMF, EKOK, CJ. Wrote the first draft of the article: MO, KH. Contributed to the writing of the article: CJ. Jointly developed the structure and arguments for the paper: MO, KH, CJ. Agreed with the article results and conclusions: MO, KH, SD-G, MAC, RLMF, EKOK, CJ. Made critical revisions: MO, KH, SD-G, MAC, RLMF, EKOK, CJ. All authors reviewed and approved the final article.
REFERENCES
- 1.Teesalu T, Kulla A, Simisker A, et al. Tissue plasminogen activator and neuroserpin are widely expressed in the human central nervous system. Thromb Haemost. 2004;92:358–68. doi: 10.1160/TH02-12-0310. [DOI] [PubMed] [Google Scholar]
- 2.Samson AL, Medcalf RL. Tissue-type plasminogen activator: a multifaceted modulator of neurotransmission and synaptic plasticity. Neuron. 2006;50:673–8. doi: 10.1016/j.neuron.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 3.Tsirka SE, Gualandris A, Amaral DG, et al. Excitotoxin-induced neuronal degeneration and seizure are mediated by tissue plasminogen activator. Nature. 1995;377:340–4. doi: 10.1038/377340a0. [DOI] [PubMed] [Google Scholar]
- 4.Benchenane K, Lopez-Atalaya JP, Fernandez-Monreal M, et al. Equivocal roles of tissue-type plasminogen activator in stroke-induced injury. Trends Neurosci. 2004;27:155–60. doi: 10.1016/j.tins.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 5.Graff J, Kim D, Dobbin MM, et al. Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiol Rev. 2011;91:603–49. doi: 10.1152/physrev.00012.2010. [DOI] [PubMed] [Google Scholar]
- 6.Vecsey CG, Hawk JD, Lattal KM, et al. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci. 2007;27:6128–40. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Langley B, Brochier C, Rivieccio MA. Targeting histone deacetylases as a multifaceted approach to treat the diverse outcomes of stroke. Stroke. 2009;40:2899–905. doi: 10.1161/STROKEAHA.108.540229. [DOI] [PubMed] [Google Scholar]
- 8.Dunoyer-Geindre S, Kruithof EK. Epigenetic control of tissue-type plasminogen activator synthesis in human endothelial cells. Cardiovasc Res. 2011;90:457–63. doi: 10.1093/cvr/cvr028. [DOI] [PubMed] [Google Scholar]
- 9.Hess-Stumpp H, Bracker TU, Henderson D, et al. MS-275, a potent orally available inhibitor of histone deacetylases – the development of an anticancer agent. Int J Biochem Cell Biol. 2007;39:1388–405. doi: 10.1016/j.biocel.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 10.Jiang M, Zhang Y, Fei J, et al. Rapid quantification of DNA methylation by measuring relative peak heights in direct bisulfite-PCR sequencing traces. Lab Invest. 2010;90:282–90. doi: 10.1038/labinvest.2009.132. [DOI] [PubMed] [Google Scholar]