
Fig. 3.
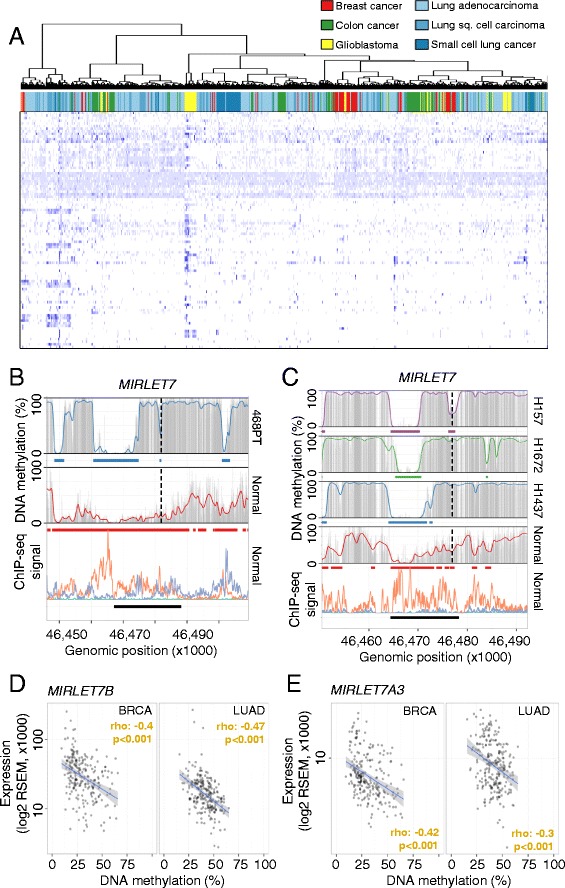
Cancer type-specific alterations of DNA methylation signatures at super-enhancer loci. a Hierarchical clustering of common hypomethylated super-enhancer regions in normal tissues (rows, <25 % average DNA methylation) in 714 cancer samples (columns). Average CpG methylation levels in common regions were clustered using Canberra distances and the Ward cluster method. DNA methylation levels are color-coded from 0 % (light blue) to 100 % (dark blue) and the different cancer types are color-coded. b, c DNA methylation profiles of the super-enhancer regions associated with MIRLET7 in normal tissues and cell lines derived from breast (b) and lung cancer (c). Smoothed (colored line), raw (gray bars) CpG methylation levels, hypomethylated regions (colored bars) and super-enhancers (black bars) are indicated. The enhancer-related histone marks (bottom panel) H3K27ac (orange) and H3K4me1 (purple) are displayed as ChIP-seq signal intensities [11]. Transcription start sites are indicated (broken line). d, e Association of DNA methylation levels (TCGA, HumanMethylation450 BeadChip, averaged probe levels within the super-enhancer) and gene expression (TCGA, RNA-seq, absolute expression values) related to the MIRLET7 super-enhancer and targeted microRNAs MIRLET7B (d) and MIRLET7A3 (e) in breast (n = 201) and lung (n = 216) cancer samples. Significances of a Spearman’s correlation test are indicated. RSEM RNA-Sequencing by Expectation Maximization