

Abstract
The Forkhead Box M1 (FOXM1) transcription factor plays important roles in tumorigenesis and tumor metastasis in multiple human carcinomas. However, the underlying mechanisms for FOXM1 function remain to be classified. In the present study, we employed quantitative proteomic approach to search new downstream targets of FOXM1 in breast cancer MDA-MB-231 cells. A total of 4125 proteins were identified and quantified by label-free quantitation, of which 318 proteins were significantly changed (with P-value <0.05) between FOXM1 knockdown cells and control cells. Among them, three proteins ACSL4, CGGBP1 and PGRMC2 were significantly downregulated with FOXM1 reduction by western blot analysis. Further functional assays revealed that knockdown of the three proteins in MDA-MB-231 cells attenuated the ability of cell migration, consistent with the phenotype of FOXM1 knockdown. These results suggest that new potential downstream effectors of FOXM1 were identified by proteomic approach, and may provide new potential therapeutic targets in breast cancer.
Keywords: FOXM1, quantitative proteomic analysis, breast cancer, cell migration
Introduction
Forkhead box protein M1 (FOXM1), a member of Forkhead transcription factor superfamily, is overexpressed in a large variety of human cancers [1] and is a key regulator of cancer pathogenesis [2,3]. There is growing evidence that FOXM1 is required for tumor cell proliferation, differentiation, longevity, transformation and metastasis. Besides, FOXM1 overexpression also confers the potential of drug resistance to tumor cells. Studies to date have demonstrated that FOXM1 expression correlates positively with tumor grade, stage, size or incidence of metastasis in most human tissues, including breast, liver, stomach, pancreas, etc. In breast cancer, FOXM1 has been reported to be frequently upregulated and knockdown of it leads to the inhibition of cell migration. Although some metastasis-related genes such as Slug, uPAR, MMP-2 have been identified as downstream effectors of FOXM1 [4-6], the exact mechanism and more downstream targets of FOXM1 on cell migration of breast cancer remains to be further elucidated.
Quantitative proteomic approaches, such as isotopecoded affinity tags (ICAT), stable isotope labeling with amino acids in cell culture and label-free quantitation (LFQ) methods, have been widely used to quantify the proteins from cells, tissues, and body fluids under different conditions [7]. Label-free protein quantitation methods are becoming promising and popular in shotgun datasets, for which can be applied without introducing isotopes for quantitation. Recently, this approach has been moved to a new stage with the supportive software development [8,9]. It is encouraging that a number of studies have identified novel targets or partners of specific proteins with this technology [10-12].
In the present study, label-free quantitative proteomic approaches were performed to identify and quantify those proteins differentially expressed betweenFOXM1 knockdown breast cancer cells (MDA-MB-231/Lv-shFOXM1) and control cells (MDA-MB-231/Lv-shNC). A total of 4125 proteins were identified and quantified by label-free quantitation, of which 318 proteins were significantly changed (with P-value <0.05) between the two groups. Among them, ACSL4, CGGBP1 and PGRMC2 were identified as potential downstream targets of FOXM1 by Western blot. Knockdown any of the three genes attenuated the ability of cell migration in MDA-MB-231 cells through transwell chamble assays. Thus, our data provided new insights into FOXM1 function in cell migration by the potential new downstream target genes.
Materials and methods
Cell culture and cell line
The human breast carcinoma cell line MDA-MB-231 were obtained from the American Type Culture Collection (Manassas, VA) and cultured in Dulbecco’s Modified Eagle Medium (DMEM, Corning), supplemented with 10% fetal bovine serum (FBS) and 100 units/ml penicillin/streptomycin in a humidified incubator in an atmosphere of 5% CO2 at 37°C.
RNA interference and cell transfection
The small interfering RNAs targeting FoxM1 were designed as described previously [13]. The sequence was as follow: siFoxM1 (sense 5-CUCUUCUCCCUCAGAUAUAdTdT-3). The irrelevant nucleotides not targeting any annotated human genes were used as negative control: siNC (sense 5-UUCUCCGAACGUGUCACGUdTdT-3). For ACSL4, CGGBP1, and PGRMC2 gene silencing, the siRNAs sequences specific against these genes respectively were: siACSL4 (sense 5-GGAUAUUCUUCUCCGCUUAdTdT-3), siCGGBP1 (sense 5-CCAUGACUGUCUGAUACGUUUdTdT-3) and siPGRMC2 (sense 5-UUCUCCGAACGUGUCACGUdTdT-3). All of siRNAs were chemically synthesized by GenePharma, Shanghai. Cell transfection with siRNAs was conducted using Lipofectamine 2000 (Invitrogen, USA) in accordance with the manufacturer’s instructions. The lentivirus knocking down FOXM1 or control (LV-shFOXM1, LV-shNC) were packaged and purchased from GenePharma, Shanghai using above corresponding sequences.
Quantitative real-time PCR analysis
Total RNA from MDA-MB-231 cells was isolated with RNAiso Plus reagent (TaKaRa, Japan), and the total RNA was reverse transcribed using the PrimeScriptTM RT Reagent Kit with gDNA Eraser (TaKaRa, Japan). Quantitative real-time reverse transcription-PCR (qRT-PCR) was performed with ABI 7500 Real Time System and SYBER green reagent (TaKaRa, Japan). The mRNA level of each sample was normalized to β-actin prior to comparative analysis using 2-ΔCt method. The following primer pairs were used to amplify and measure the amount of FOXM1 and β-actin: FOXM1-F: 5-GGGCGCACGGCGGAAGATGAA-3, FOXM1-R: 5-CCACTCTTCCAAGGGAGGGCTC-3 and actin-F: 5-CCTGGCACCCAGCACAATG-3, actin-R: 5-GGGCCGGACTCGTCATACT-3. Each sample was run in triplicate for the target gene and the internal control gene.
Western blot analysis
Standard Western blotting was performed using whole-cell protein lysates and protein concentration of cell lysates was measured. Samples were boiled for 5 minutes, subjected to electrophoresis in SDS-PAGE (10% w/v) and transferred onto a polyvinylidenedifluoride (PVDF) membrane. The membrane was blocked in 5% blocking buffer (5% nonfat milk and 0.1% Tween-20 in PBS) for 2 h at room temperature, and then incubated with the primary antibody in PBST (0.1% Tween-20 in PBS) overnight at 4°C. Incubation with the secondary antibody was performed for 1 h at room temperature. Primary antibodies were used against FOXM1 and b-actin from Santa Cruz Biotechnology, USA, against RASGAP, CGGBP1, PGRMC2 ACSL4, AMPK, GOLGA2, LENG8, C12orf11 and MAPK3 from Proteintech, Wuhan, China. The detection of proteins was achieved by using the Odyssey Infared Imaging System (Li-COR, USA).
Cell migration assay
Cells were performed by using the transwell chamber (BD Bioscience) with the vendor’s protocol. Briefly, cells were pretreated with siRNAs for 48 h and were trypsinized and resuspended into serum-free DMEM medium. Cells were then added to the upper chambers at 3×104 cells per well. DMEM containing 5% FBS was added to the lower chambers as attractant. After 48 h, the migrated cells on the membrane’s undersurface were stained with Crystal Violet Staining Solution and counted in 5 fields per well. All experiments were conducted in triplicate and repeated twice.
Mass spectrometry
The LC-MS/MS was performed as Zhou previous described [14,15] with some modifications. Briefly, samples were acidified with formic acid to a final concentration of 1% v/v and loaded on a 75 μm×150 mm fused silica precolumn packed in house with 3-μm ReproSil-Pur C18 beads (120 A ; Dr. Maisch GmbH, Ammerbuch, Germany) using an Easy nano-UPLC 1000 (Thermo Electron, Waltham, MA). The peptides were eluted using a gradient (5-80% ACN with 0.1% formic acid) at a flow rate of 300 nL/min over 200 min period into an nano-ESI Velos Pro-Orbitrap Elite mass spectrometer (Thermo Electron, Waltham, MA). MS/MS spectra were acquired in a data-dependent acquisition mode that automatically selected and fragmented the ten most intense peaks from each MS spectrum generated by high-energy collisional dissociation (HCD).
Data analysis
The MS data were analyzed using the software MaxQuant [16,17] (http://maxquant.org/, version 1.3.0.5). Carbamidomethyl (C) was set as a fixed modification, and oxidation (M, +15.99492 Da) was set as a variable modification. Proteins were identified by searching MS and MS/MS data of peptides against a decoy version of the International Protein Index (IPI) human database (version 3.87, 91464 protein sequences; European Bioinformatics Institute). Trypsin/P was selected as the digestive enzyme with two potential missed cleavages. The false discovery rate (FDR) for peptides and protein groups was rigorously controlled to be <1% by the Andromeda search engine [18]. FDR was calculated by the number of hits from the reverse database divided by the number of forward hits [18,19]. Label-free quantification was carried out in MaxQuant using intensity determination and normalization algorithm [16]. The peptide peak intensities were calculated by summing up the intensities of different isotopic peaks in an isotope pattern of this peptide, only peptides that undoubtedly belong to a protein were used for further analysis [16]. The maximum ratio information from peptide signals across samples was extracted for protein label-free quantitation. The “LFQ intensity” of each protein in different samples was calculated as the best estimate satisfying all the pair-wise peptide comparisons, and this LFQ intensity was almost at the same scale of the summed-up peptide intensities [16]. The peptide intensity and protein LFQ intensity lists of the two cell lines were further processed using Perseus software (version 1.2.0.16). The LFQ intensity values were logarithmized (Log2), and the missing value was imputed with random numbers from a normal distribution (Width = 0.3, Shift = 1.8).
Statistical analysis was performed with SPSS version 17.0. A one-way Student’s t-test was used to compare differences between the two groups of the qRT-PCR data. And a two-tailed Student’s t-test to determine the significance of the Cell migration assay results. A P value of less than 0.05 was considered to indicate a significant difference, “*” indicates P<0.05; “**” indicates P<0.01.
Results
Construction and identification of FOXM1 knockdown stable cell line
To search FOXM1-regulated downstream targets in breast cancer, two stable cell lines MDA-MB-231/LV-shFoxM1, MDA-MB-231/LV-shNC were generated by infecting FoxM1 knockdown lentivirus and negative control (NC) lentivirus. As shown in Figure 1A, both of the lentivirus infection efficiency reached nearly 100% via the green fluorescent protein (GFP) assay. The qRT-PCR and western blot data demonstrated that FoxM1 mRNA and protein levels were successfully decreased in the MDA-MB-231/LV-shFoxM1 cells compared with control LV-shNC cells (Figure 1B). Then, the two cell lines were used to compare the proteomes for differentially expressed proteins. The procedures for protein process from the two cell lines using centrifugal proteomic reactor, nano-LC-MS/MS analysis of the resulting peptides, database searching, and LFQ intensity calculation by Maxquant and bioinformatic analysis are illustrated in Figure 1C.
Figure 1.
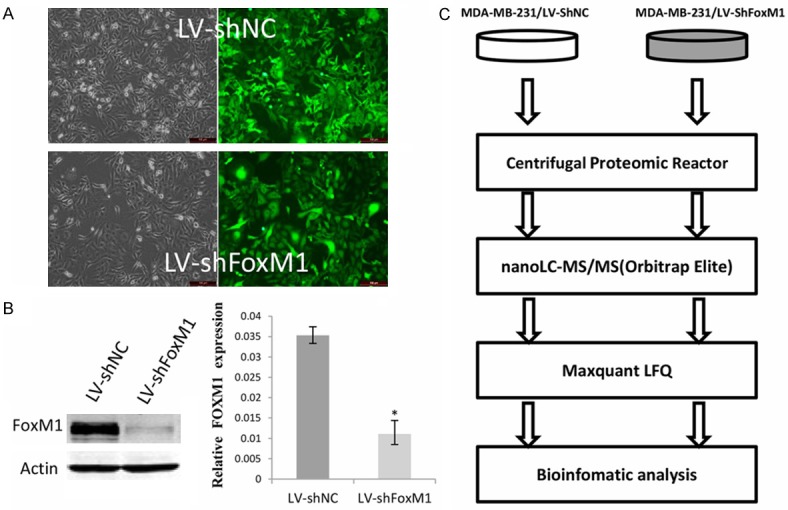
Identification of FOXM1 stable knock down cell line and flow chart of the proteomic study. A. Infection efficiency of MDA-MB-231 cells by LV-shNC and LV-shFoxM1 lentivirus. Left, image under light microscope; right, image under fluorescence microscope showing the expression of green fluorescent protein-positive cells. B. The expression level of FOXM1 in MDA-MB-231/LV-shNC cell and MDA-MB-231/LV-shFoxM1 by Western blot and qRT-PCR analysis. *Statistically different at P<0.05. C. Flow chart of the proteomic study of MDA-MB-231/LV-shFoxM1 and MDA-MB-231/LV-shNC cells. The resulting peptides were analyzed by high resolution nano-LC-MS/MS and quantified with the label-free algorithm in MaxQuant software.
Label-free quantitative proteomic analysis of FOXM1 knockdown stable cell lines
Tryptic peptides from two stable cell lines were analyzed by LC-MS/MS on a linear ion trap Orbitrap mass spectrometer [20]. 39686 unique peptides were identified and a total number of 4125 unique protein groups were quantified. Next, we estimated the quantitative capacity of our dataset. As shown in Figure 2A, a box plot analysis was applied to compare the LFQ intensity average of all the 8 individual samples (each cohort 4 replicates). The LFQ intensity average is at the same level across the samples which indicate that the results of LFQ analysis have no biases towards different samples. As shown in Figure 2B, relative label-free quantitation was highly reproducible between biological replicates inside each cohort or between the LC-MS/MS runs from different cohorts, and correlation between normalized LFQ intensities was higher than 0.93. To globally discern the proteomic changes, we generated a heat map plot of a hierarchical clustering analysis (HCA) of the total 4125 protein intensities of the 8 samples (Figure 2C).
Figure 2.
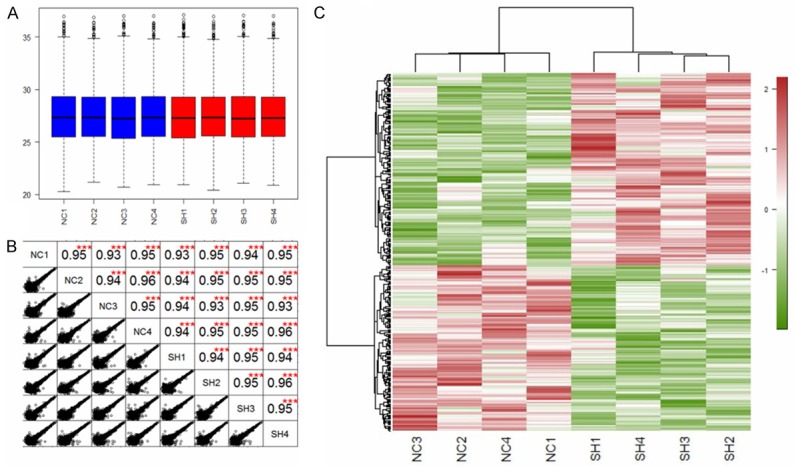
Box plots of the protein LFQ intensity (log2) for each sample (A). A scatter plot and correlation between two samples (B), and hierarchical clustering analysis of the 4125 proteins identified by quantitative proteomics (C). The protein samples were collected from 4 replicates of LV-shFoxM1 (SH1, SH2, SH3 and SH4) and 4 replicates of LV-shNC (NC1, NC2, NC3 and NC4).
In order to identify differentially expressed proteins between the LV-shFoxM1 and /LV-shNC cells, a t-test was performed. Of the 4125 unique protein groups, 318 proteins showed statistically significant differences between these two subsets (P value <0.05). Out of which 171 proteins were down-regulated and 147 proteins were up-regulated. And 69 proteins changed significantly (P value <0.05 and fold change over 1.5 as the cutoff). Selected 20 proteins that were significantly changed with knockdown FoxM1 are listed in Table 1.
Table 1.
Selected proteins with altered abundance in MDA-MB-231/LV-shFoxM1 versus LV-shNC
| Accession number (gi) | Protein Name | Abbreviation | Molecular weight (Kda) | LV-shNC/LV-shFoxM1 Ratio | p value |
|---|---|---|---|---|---|
| IPI00220356 | Isoform 2 of RasGTPase-activating protein 1 | RASGAP/RASA1 | 100 | 0.2 | 1.71e-02 |
| IPI00795467 | Uncharacterized protein | CGGBP1 | 12 | 0.32 | 1.05e-03 |
| IPI01015100 | Membrane-associated progesterone receptor component 2 | PGRMC2 | 24 | 0.08 | 2.97e-02 |
| IPI00219897 | Isoform Short of Long-chain-fatty-acid--CoA ligase 4 | ACSL4 | 74 | 0.81 | 3.23e-04 |
| IPI00792482 | Isoform 1 of 5’-AMP-activated protein kinase catalytic subunit alpha-1 | AMPK/PRKAA1 | 64 | 0.26 | 2.92e-02 |
| IPI00943357 | Isoform 1 of Golgin subfamily A member 2 | GOLGA2 | 113 | 0.63 | 1.72e-03 |
| IPI00550986 | Cell cycle regulator Mat89Bb homolog | C12orf11 | 80 | 0.36 | 1.21e-02 |
| IPI00852904 | Leukocyte receptor cluster (LRC) member 8 | LENG8 | 79 | 6.33 | 2.42e-02 |
| IPI00018195 | Mitogen-activated protein kinase3 | MAPK3 | 43 | 3.18 | 3.31e-02 |
| IPI00001541 | Mitochondrial import innermembrane translocase subunit Tim9 | TIMM9 | 10 | 0.34 | 1.69e-02 |
| IPI00816836 | Solute carrier family 2 (Facilitated glucose transporter), member 3 variant | SLC2A3 | 32 | 0.36 | 1.66e-02 |
| IPI01010298 | Uncharacterized protein | EPDR1 | 38 | 0.38 | 4.37e-02 |
| IPI00854856 | Isoform 4 of Shootin-1 | KIAA1598 | 63 | 0.46 | 1.36e-02 |
| IPI00410034 | Isoform 1 of Sodium-coupled neutral amino acid transporter 2 | SLC38A2 | 56 | 0.64 | 9.02e-03 |
| IPI00010187 | Elongation of very long chain fatty acids protein 1 | ELOVL1 | 33 | 0.65 | 1.72e-02 |
| IPI00005809 | Serum deprivation-response protein | SDPR | 47 | 1.54 | 2.02e-03 |
| IPI00301107 | Isoform 1 of Importin-11 | IPO11 | 113 | 1.55 | 2.07e-02 |
| IPI00797249 | L-xylulosereductase isoform 2 | DCXR | 26 | 1.57 | 9.74e-04 |
| IPI00019551 | Pleckstrin homology-like domainfamily A member 2 | PHLDA2 | 17 | 1.74 | 6.46e-04 |
| IPI00397526 | Isoform 1 of Myosin-10 | MYH10 | 229 | 1.81 | 6.76e-03 |
Validation of identified candidates via quantitative mass spectrometry analysis using western blot
Several of the proteins identified to be decreased and a few of proteins identified to be increased in FOXM1 knockdown cells were further quantified and validated by Western blot. As shown in Figure 3, RASGAP, CGGBP1, PGRMC2, ACSL4, AMPK, GOLGA2, C12orf11 and LENG8 were downregulated with FOXM1 reduction, whereas MAPK3 was observed to be opposite. The majority of western bolt results were in line with quantitative mass spectrometry data, indicating the proteomic data was reliable and reproducible.
Figure 3.
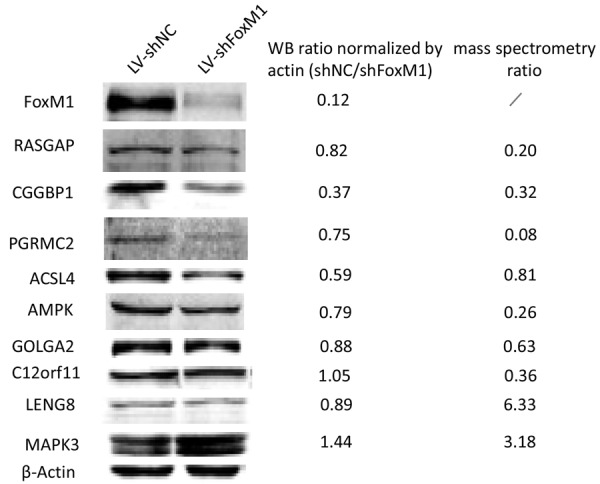
Verification of the expression of the proteins identified. Validation of the proteomic results of selected proteins as indicated in immunoblots of protein extracts from the breast cancer cells analyzed. Antibodies displaying a single predominant band at the expected molecular weights were accepted. β-actin was used as the loading control.
Functional investigation of ACSL4, CGGBP1 and PGRMC2 on cell migration
Based on the good agreement between mass spectrometry result and western blot analysis for CGGBP1, PGRMC2 and ACSL4, the three proteins were selected for further functional analysis using the aggressive phenotype of MDA-MB-231 cells. For this goal, specific small interference RNA, siCGGBP1, siPGRMC2 and siACSL4 or negative control siNC were synthesized and transfected into MDA-MB-231 cells, respectively. Figure 4A showed the knockdown efficiency of each gene and western blot data indicated the three genes were successfully downregulated by RNA inference. As the same time, transwell migration assays indicated that CGGBP1, PGRMC2 or ACSL4 knockdown led to a significant decrease in cell migration of MDA-MB-231 cells (Figure 4B and 4C), demonstrating that CGGBP1, PGRMC2 and ACSL4 have positively effects on cell migration of breast cancer cells as FOXM1 does.
Figure 4.
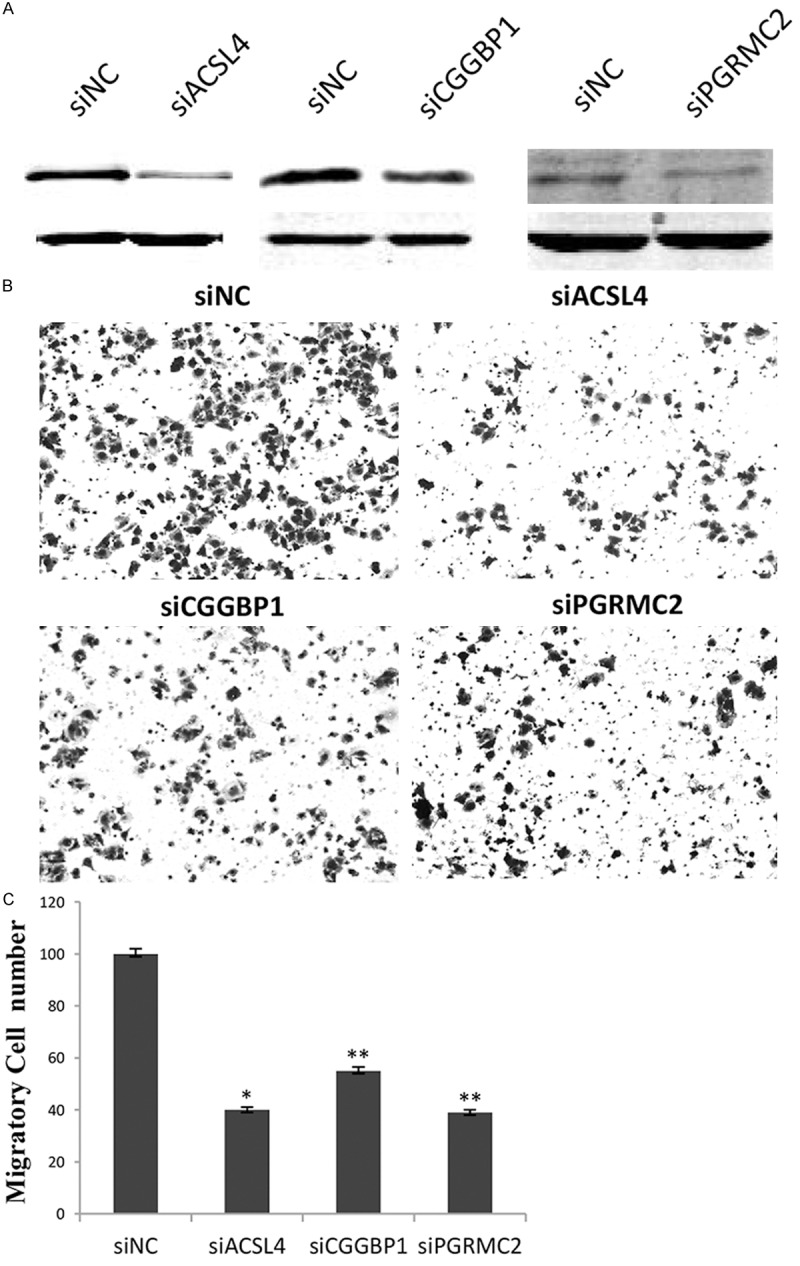
Knockdown of ACSL4, CGGBP1 and PGRMC2 attenuates the ability of cell migration. A. Specific small inference RNA against ACSL4, CGGBP1 or PGRMC2 was respectively transfected into MDA-MB-231 cells. The correspondingprotein levels were detected by western blot analysis. B. Transwell chamber assays were performed to investigatethe change of migratory ability of breast cancer cells,where those with siNC were used as a control. The experiments were repeated three times, and the histograms C representmean numbers of the migrated cells in five selected fields from triplicate tests (mean ± SD). *indecates P<0.05, **indecates P<0.01.
Discussion
FOXM1 transcriptionally modulates a series of gene expression involved in cell growth, proliferation, differentiation, longevity, transformation and migration. To understand the mechanism underlying the various role of FOXM1, it is required to determine FOXM1 regulated downstream targets. In the present study, we employed quantitative label-free mass spectrometry to explore FOXM1 regulated protein expression for the first time. Although researchers have reported FOXM1 targets by cDNA microarray before, it is well known the two approaches may come out different profiling [21].
A total of 318 proteins were finally identified and quantified by label-free quantitation (with P-value <0.05), of which 69 proteins changed significantly (P value <0.05 and fold change over 1.5 as the cutoff) between the two groups: MDA-MB-231/LV-shNC and MDA-MB-231/shFoxM1 cells. The MDA-MB-231 cell represents an aggressive cell model of breast cancer. Breast cancer is ranked as the fifth cause of death among all cancers [22]. The majority of primary tumors that remain confined to the breast are amenable to surgical resection, and 5-year survival rates for patients with non-metastatic disease are -98%. Once the tumor has metastasized to a distant site, despite research efforts and the development of new therapies, 5-year survival decreases to -26% [23]. Finding new molecular determinants associated with breast cancer metastasis are essential for the future development of successful therapeutic strategies. Therefore, of all potential downstream targets of FOXM1, we focus on the role on cell migration, for which is thought as early stage of tumor metastasis.
Among these differentially expressed proteins, RASGAP, CGGBP1, PGRMC2, ACSL4, AMPK, GOLGA2, C12orf11, LENG8 and MAPK were further confirmed by western blotting, supporting the reliability of the mass spectrometry-based LFQ analysis. Subsequently, we selected three genes for the further functional analysis. ACSL4, the acyl-CoA synthetase 4, esterifies mainly arachidonic acid into acyl-CoA and is increased in breast, colon and hepatocellular carcinoma [24,25]. CGGBP1 is a repetitive DNA-binding transcription regulator with target sites at CpG-rich sequences, which implies it may act as a possible mediator of CpG methy-lation [26,27]. PGRMC2, a component of progesterone receptor membrane, belongs to the heme-binding protein family and may serve as a receptor for progesterone [28]. Knocking down any of three genes by RNAi technique has an inhibitory effect on cell migration, implicating the three genes play a role in tumor metastasis. Thus, we might provide three new potential therapeutic targets against cell metastasis in breast cancer.
In conclusion, a label-free quantitative proteomic strategy for the first time was applied to compare protein expression between FOXM1 knockdown cells and control cells. A total of 318 unique proteins were identified and quantified in our study which provides to date the largest quantitative proteomic data regulated by FOXM1 in breast cancer. ACSL4, CGGBP1 and PGRMC2, the three irrelevant proteins involved in distinct biological process were confirmed by western blot and involvement of cell migration by transwell chamble assay, suggesting they may act as downstream effectors of FOXM1 in cancer metastasis. Undoubtedly, FOXM1 regulated targets might exist in other unidentified proteins within our proteomic profiling, which should be deserved to further study in future.
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (81172022 and 81272917) and Outstanding Leaders Training Program of Pudong Health Bureau of Shanghai (PWRl2013-02), Shanghai Natural Science Foundation of China (13ZR1434000) and Specialized Research Fund for the Doctoral Program of Higher Education (20130072120060), the Science and Technology Development Foundation from Pudong New District (PKJ2010-Y06), and Key Disciplines Group Construction Project of Pudong Health Bureau of Shanghai (PWZxq2014-04).
Disclosure of conflict of interest
None.
References
- 1.Park HJ, Gusarova G, Wang Z, Carr JR, Li J, Kim KH, Qiu J, Park YD, Williamson PR, Hay N, Tyner AL, Lau LF, Costa RH, Raychaudhuri P. Deregulation of FoxM1b leads to tumour metastasis. EMBO Mol Med. 2011;3:21–34. doi: 10.1002/emmm.201000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anders L, Ke N, Hydbring P, Choi YJ, Widlund HR, Chick JM, Zhai H, Vidal M, Gygi SP, Braun P, Sicinski P. A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell. 2011;20:620–634. doi: 10.1016/j.ccr.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalin TV, Ustiyan V, Kalinichenko VV. Multiple faces of FoxM1 transcription factor: lessons from transgenic mouse models. Cell Cycle. 2011;10:396–405. doi: 10.4161/cc.10.3.14709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahmad A, Wang Z, Kong D, Ali S, Li Y, Banerjee S, Ali R, Sarkar FH. FoxM1 down-regulation leads to inhibition of proliferation, migration and invasion of breast cancer cells through the modulation of extra-cellular matrix degrading factors. Breast Cancer Res Treat. 2010;122:337–346. doi: 10.1007/s10549-009-0572-1. [DOI] [PubMed] [Google Scholar]
- 5.Huang C, Xie D, Cui J, Li Q, Gao Y, Xie K. FOXM1c promotes pancreatic cancer epithelial-to-mesenchymal transition and metastasis via upregulation of expression of the urokinase plasminogen activator system. Clin Cancer Res. 2014;20:1477–1488. doi: 10.1158/1078-0432.CCR-13-2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang C, Chen H, Tan G, Gao W, Cheng L, Jiang X, Yu L, Tan Y. FOXM1 promotes the epithelial to mesenchymal transition by stimulating the transcription of Slug in human breast cancer. Cancer Lett. 2013;340:104–112. doi: 10.1016/j.canlet.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 7.Ong SE, Mann M. Mass spectrometry-based proteomics turns quantitative. Nat Chem Biol. 2005;1:252–262. doi: 10.1038/nchembio736. [DOI] [PubMed] [Google Scholar]
- 8.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p. p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 9.Jagtap P, Bandhakavi S, Higgins L, McGowan T, Sa R, Stone MD, Chilton J, Arriaga EA, Seymour SL, Griffin TJ. Workflow for analysis of high mass accuracy salivary data set using MaxQuant and ProteinPilot search algorithm. Proteomics. 2012;12:1726–1730. doi: 10.1002/pmic.201100097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adeola HA, Soares NC, Paccez JD, Kaestner L, Blackburn JM, Zerbini LF. Discovery of novel candidate urinary protein biomarkers for prostate cancer in a multiethnic cohort of South African patients via label-free mass spectrometry. Proteomics. Clin Appl. 2015;9:597–609. doi: 10.1002/prca.201400197. [DOI] [PubMed] [Google Scholar]
- 11.Tsai TH, Song E, Zhu R, Di Poto C, Wang M, Luo Y, Varghese RS, Tadesse MG, Ziada DH, Desai CS, Shetty K, Mechref Y, Ressom HW. LC-MS/MS-based serum proteomics for identification of candidate biomarkers for hepatocellular carcinoma. Proteomics. 2015;15:2369–2381. doi: 10.1002/pmic.201400364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang H, Hanash S. Mass spectrometry based proteomics for absolute quantification of proteins from tumor cells. Methods. 2015;81:34–40. doi: 10.1016/j.ymeth.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Q, Zhang N, Jia Z, Le X, Dai B, Wei D, Huang S, Tan D, Xie K. Critical role and regulation of transcription factor FoxM1 in human gastric cancer angiogenesis and progression. Cancer Res. 2009;69:3501–3509. doi: 10.1158/0008-5472.CAN-08-3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou H, Hou W, Denis NJ, Zhou H, Vasilescu J, Zou H, Figeys D. Glycoproteomic reactor for human plasma. J Proteome Res. 2009;8:556–566. doi: 10.1021/pr800734r. [DOI] [PubMed] [Google Scholar]
- 15.Zhou H, Wang F, Wang Y, Ning Z, Hou W, Wright TG, Sundaram M, Zhong S, Yao Z, Figeys D. Improved recovery and identification of membrane proteins from rat hepatic cells using a centrifugal proteomic reactor. Mol Cell Proteomics. 2011;10:O111.008425. doi: 10.1074/mcp.O111.008425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cox J, Matic I, Hilger M, Nagaraj N, Selbach M, Olsen JV, Mann M. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat Protoc. 2009;4:698–705. doi: 10.1038/nprot.2009.36. [DOI] [PubMed] [Google Scholar]
- 18.Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- 19.Choi H, Nesvizhskii AI. False discovery rates and related statistical concepts in mass spectrometry-based proteomics. J Proteome Res. 2008;7:47–50. doi: 10.1021/pr700747q. [DOI] [PubMed] [Google Scholar]
- 20.Li J, Tan Z, Li M, Xia T, Liu P, Yu W. Proteomic analysis of endometrium in fertile women during the prereceptive and receptive phases after luteinizing hormone surge. Fertil Steril. 2011;95:1161–1163. doi: 10.1016/j.fertnstert.2010.09.033. [DOI] [PubMed] [Google Scholar]
- 21.Xia L, Huang W, Tian D, Chen Z, Zhang L, Li Y, Hu H, Liu J, Chen Z, Tang G, Dou J, Sha S, Xu B, Liu C, Ma J, Zhang S, Li M, Fan D, Nie Y, Wu K. ACP5, a direct transcriptional target of FoxM1, promotes tumor metastasis and indicates poor prognosis in hepatocellular carcinoma. Oncogene. 2014;33:1395–1406. doi: 10.1038/onc.2013.90. [DOI] [PubMed] [Google Scholar]
- 22.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 [Internet] Lyon, France: International Agency for Research on Cancer; GLOBOCAN 2012 v1.0 2013. [Google Scholar]
- 23.Xie Y, Cui D, Kong Y. FoxM1 influences embryo implantation and is regulated by 17 beta-estradiol and progesterone in mouse uteri and endometrium cells. Int J Clin Exp Pathol. 2014;7:6585–6595. [PMC free article] [PubMed] [Google Scholar]
- 24.Orlando UD, Garona J, Ripoll GV, Maloberti PM, Solano ÁR, Avagnina A, Gomez DE, Alonso DF, Podestá EJ. The functional interaction between Acyl-CoA synthetase 4, 5-lipooxygenase and cyclooxygenase-2 controls tumor growth: a novel therapeutic target. PLoS One. 2012;7:e40794. doi: 10.1371/journal.pone.0040794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu X, Li Y, Wang J, Wen X, Marcus MT, Daniels G, Zhang DY, Ye F, Wang LH, Du X, Adams S, Singh B, Zavadil J, Lee P, Monaco ME. Long chain fatty Acyl-CoA synthetase 4 is a biomarker for and mediator of hormone resistance in human breast cancer. PLoS One. 2013;8:e77060. doi: 10.1371/journal.pone.0077060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu Y, Liao Z, Chen C, Qin N, Zheng J, Tian D, Li Y, Zhu S, Luo J, Xu L. [Over-expressed microRNA-7 inhibits the growth of human lung cancer cells via suppressing CGGBP1 expression] . Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2014;30:125–130. [PubMed] [Google Scholar]
- 27.Singh U, Roswall P, Uhrbom L, Westermark B. CGGBP1 regulates cell cycle in cancer cells. BMC Mol Biol. 2011;12:28. doi: 10.1186/1471-2199-12-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Causey MW, Huston LJ, Harold DM, Charaba CJ, Ippolito DL, Hoffer ZS, Brown TA, Stallings JD. Transcriptional analysis of novel hormone receptors PGRMC1 and PGRMC2 as potential biomarkers of breast adenocarcinoma staging. J Surg Res. 2011;171:615–622. doi: 10.1016/j.jss.2010.04.034. [DOI] [PubMed] [Google Scholar]