

Abstract
Genome-wide association studies (GWAS) of lung cancer in Asian never-smoking women have previously identified six susceptibility loci associated with lung cancer risk. To further discover new susceptibility loci, we imputed data from four GWAS of Asian non-smoking female lung cancer (6877 cases and 6277 controls) using the 1000 Genomes Project (Phase 1 Release 3) data as the reference and genotyped additional samples (5878 cases and 7046 controls) for possible replication. In our meta-analysis, three new loci achieved genome-wide significance, marked by single nucleotide polymorphism (SNP) rs7741164 at 6p21.1 (per-allele odds ratio (OR) = 1.17; P = 5.8 × 10−13), rs72658409 at 9p21.3 (per-allele OR = 0.77; P = 1.41 × 10−10) and rs11610143 at 12q13.13 (per-allele OR = 0.89; P = 4.96 × 10−9). These findings identified new genetic susceptibility alleles for lung cancer in never-smoking women in Asia and merit follow-up to understand their biological underpinnings.
Introduction
Lung cancer is the leading cause of cancer mortality among adults worldwide, accounting for more than 1.59 million deaths each year (1). The incidence rates of lung cancer among never-smoking females in some parts of East Asia are among the highest in the world (2). Previous studies have attributed the excess lung cancer risk to environmental risk factors such as exposure to environmental tobacco smoke (ETS) and household air pollution (3,4), but in light of the emerging evidence of genetic susceptibility to many cancers, including lung cancer, the opportunity to conduct studies in never-smoking females should lead to new insights into lung carcinogenesis, particularly as it relates to primary carcinogenesis and not tobacco driven lung cancer, most commonly observed in Europe and the USA.
To further understand the genetic etiology of lung cancer among Asian never-smoking women, we established the Female Lung Cancer Consortium in Asia (FLCCA), which consists of 18 studies in Mainland China, Hong Kong, Taiwan, South Korea, Singapore and Japan, with a total of 6609 cases and 7457 controls. We then conducted a large-scale multistage genome-wide association study (GWAS) of lung cancer restricted to never-smoking females and reported six susceptibility loci in our study population including 3q28, 5p15.33, 6p21.32, 6q22.2, 10q25.2 and 17q24.3 (5). In addition, there have been three other GWAS over the past several years for lung cancer in Asia among men and women and smokers and non-smokers, which reported additional susceptibility loci which were not confirmed in our never-smoking study in Asian women (6–8).
To discover additional lung susceptibility alleles among never-smoking Asian females, we imputed the four published GWAS data sets with a total of 6877 cases and 6277 controls (5–8), and genotyped an additional 5878 cases and 7046 controls for possible replication. We identified three new susceptibility loci that achieved genome-wide significance for lung cancer risk.
Results
Study overview
We imputed four previously reported GWAS scans individually and then combined the association test statistics for a total of 7 564 751 SNPs. We conducted a fixed-effects meta-analysis for a total of 6877 cases and 6277 controls (see ‘Materials and Methods’ section and Supplementary Material, Table S1). The genomic control factor λ = 1.03 showed that there was very little evidence of systematic inflation from population stratification for the meta-analysis of the four GWAS scans in the discovery stage (Supplementary Material, Fig. S1). We followed up 13 loci that were associated with lung cancer risk at P < 5 × 10−5 (Supplementary Material, Table S2) by genotyping the most promising SNPs in an additional set of 5878 cases and 7046 controls from 12 different centers including Mainland China (n = 7), Japan (n = 4) and Taiwan (n = 1). The final meta-analysis combining both discovery and replication stages included a total of 12 755 cases and 13 323 controls (Supplementary Material, Tables S1 and S2).
New lung cancer susceptibility loci reaching genome-wide significance
We identified three new risk loci in our population of never-smoking Asian females that were associated with lung cancer risk: rs7741164 (P = 5.80 × 10−13) at 6p21.1, rs72658409 (P = 1.41 × 10−10) at 9p21.3 and rs11610143 (P = 4.96 × 10−9) at 12q13.13, with P-values exceeding the threshold for genome-wide significance. No significant heterogeneity was observed across the four GWAS scans as well as replication studies for these three SNPs (Table 1). The SNP marker rs7741164 (G>A) maps to the intron of FOXP4-AS1 and is ∼20 kb upstream of the FOXP4 gene on 6p21.1 (Fig. 1A). The SNP marker rs72658409 (C>T) maps 40 kb downstream of CDKN2B-AS1 and 150 kb upstream of CDKN2B, a well-known tumor suppressor gene on 9p21 (Fig. 1B). The SNP marker rs11610143 (C>G) resides in an intron of ACVR1B, a gene implicated in an inflammation pathway (9) (Fig. 1C).
Table 1.
Three SNPs associated with lung cancer risk above genome-wide significance level
| Cytoband | SNP | Loc | Nearest gene(s) | Stage | Control | Case | Ref allele | Effect allele | EAF | OR | 95% CI | P | Phet |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 6p21.1 | rs7741164a | 41493412 | DQ141194 | Discovery | 6277 | 6877 | G | A | 0.306 | 1.18 | (1.10–1.26) | 2.05E−06 | |
| Replication | 7019 | 5842 | G | A | 0.316 | 1.17 | (1.10–1.24) | 5.88E−08 | |||||
| Combined | 13 296 | 12 719 | 1.17 | (1.12–1.22) | 5.80E−13 | 2.17E−01 | |||||||
| 9p21.3 | rs72658409 | 22160087 | NA | Discovery | 6277 | 6877 | C | T | 0.070 | 0.75 | (0.67–0.83) | 1.37E−07 | |
| Replication | 6684 | 5494 | C | T | 0.065 | 0.80 | (0.72–0.90) | 1.69E−04 | |||||
| Combined | 12 961 | 12 371 | 0.77 | (0.72–0.84) | 1.41E−10 | 2.79E−01 | |||||||
| 12q13.13 | rs11610143 | 52349071 | ACVR1B | Discovery | 6277 | 6877 | C | G | 0.320 | 0.88 | (0.83–0.93) | 2.21E−06 | |
| Replication | 6564 | 5006 | C | G | 0.340 | 0.90 | (0.85–0.95) | 4.78E−04 | |||||
| Combined | 12 841 | 11 883 | 0.89 | (0.85–0.92) | 4.96E−09 | 7.16E−01 |
LOC, location; Ref, reference; EAF, effect allele frequency in controls; OR, odds ratio; CI, confidence interval.
ars7741164 was imputed with low quality.
Figure 1.
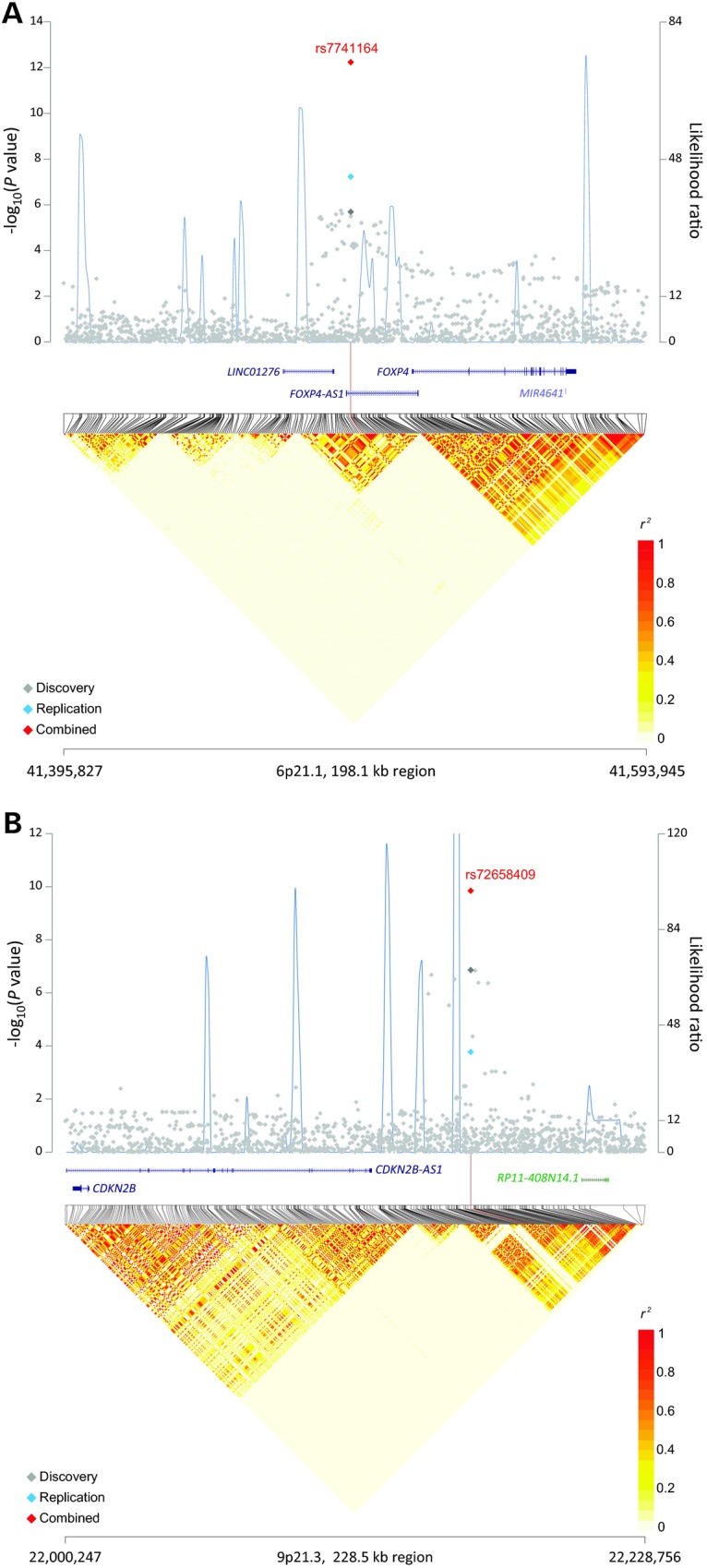
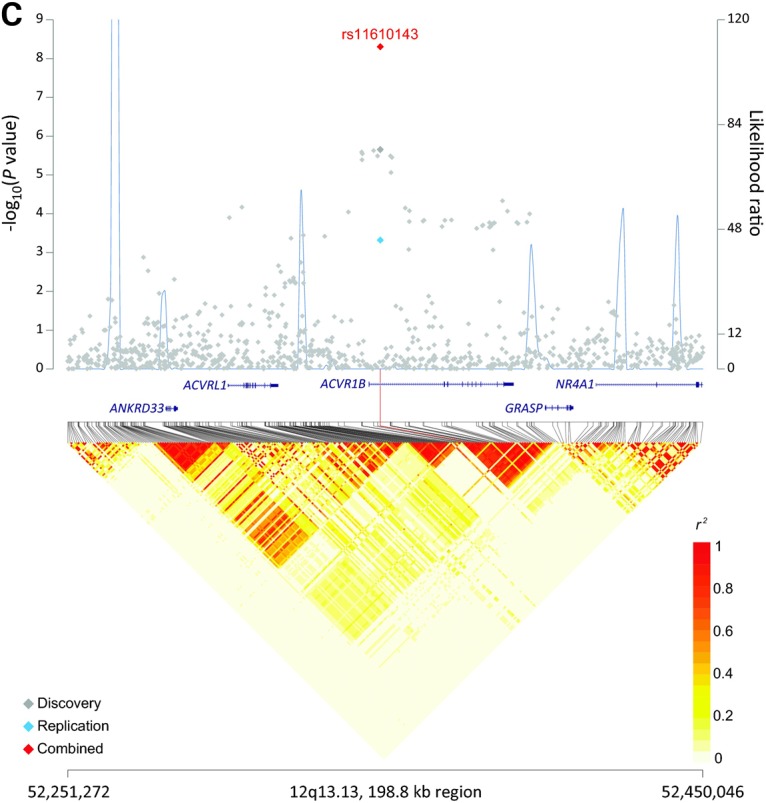
Association results, recombination hotspots and LD plot for three newly identified regions associated with lung cancer risk in never-smoking Asian women. (A–C) Top, association P values from meta-analysis of four imputed GWAS scans included for discovery stage (gray diamond) were plotted on a negative log scale (left y-axis) against genomic coordinates (hg19). For each region, meta-analysis result of replication sets (blue diamond), and overall combined meta-analysis (red diamond) for the index SNP are also shown. Overlaid (blue line) are likelihood ratio statistics (right y-axis) for recombination hotspots inferred from the 1000 Genomes Project phase 1 Asian populations (100 random samples). Bottom, Linkage disequilibrium heat map based on r2 using the 1000 Genomes Project phase 1 Asian data (n = 286). Shown are results for (A) 6p21.1 (chr6:41395827-41593945); (B) 9p21.3 (chr9:22000247-22228756) and (C) 12p13.13 (chr12:52251272-52450046).
In addition, we found suggestive evidence of association at rs3794742 in the intron of SYNGR2 at 17q25.3 (P = 4.3 × 10−7) with risk of lung cancer in this population (Supplementary Material, Table S2). SYNGR2 belongs to the synaptogyrin gene family, plays a role in membrane traffic regulation in non-neuronal cells in vivo and is associated with neuronitis disease (10). In an analysis of the ENCODE data set, rs3794742 and SNPs in high LD are implicated in a rich set of putative functional elements including promoter/enhancer histone marks, transcription factor bindings, motif changes and DNAse peak (Supplementary Material, Table S3). Still, further studies are needed to confirm this locus and then laboratory studies are needed to explain the biological basis of the susceptibility allele.
In silico bioinformatics analyses
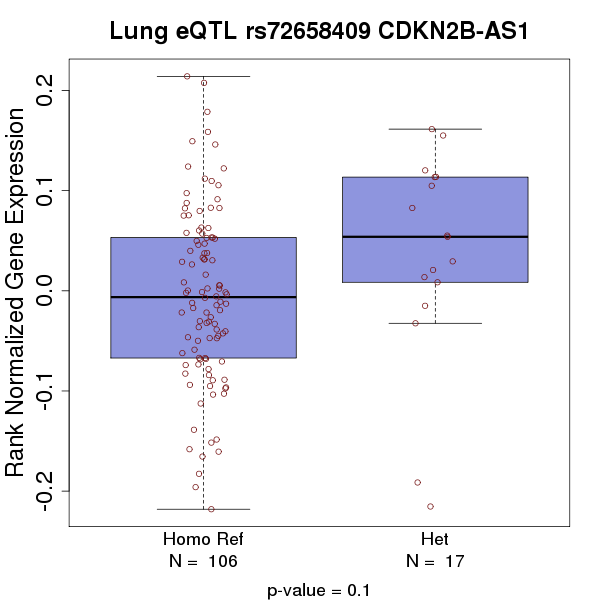
HaploReg data (11) (Supplementary Material, Table S3) showed that the minor allele (A) of rs7741164 is present in a substantially higher proportion of Asians [minor allele frequency (MAF) = 0.33] compared with Europeans (MAF = 0.03). rs72658409 influences both promoter histone marks of blood monocytes and enhancer histone marks for cells derived from nine organs including lung fibroblasts. Genotype-Tissue Expression (GTEx) (12) (see URLs) data showed that genotypes for rs72658409 are suggestively associated with expression level of CDKN2B gene (P = 0.04) but not CDKN2B-AS1 (P = 0.1) in normal lung tissue samples (n = 123) (Supplementary Material, Figs S2a and b). rs11610143 resides in a conserved region inferred by both GERP(13) and SiPhy (14), and it influences promoter histone marks in 8 organs including lung carcinoma and enhancer histone marks in 18 organs including fetal lung. Additionally, rs11610143 has a RegulomeDB (15) score of 4 with minimal evidence supporting transcription factor binding site (Supplementary Material, Table S3).
Technical validation of imputed SNPs
In order to check the quality of imputation, we performed TaqMan genotyping on a subset of GWAS samples (details in ‘Materials and Methods’ section). The squared correlation (r2) for the allelic dosage between the imputed genotypes and the genotypes measured by TaqMan were 0.21 (n = 2930), 0.979 (n = 606) and 0.997 (n = 674) for rs7741164, rs72658409 and rs11610143, respectively. Since the technical validation of rs7741164 showed that the correlation between the imputed and measured genotypes was moderately low, we attempted to impute the same region including rs7741164 based on an alternative imputation approach (details in ‘Materials and Methods’ section) and the r2 improved to 0.33. Nevertheless, the P value based on the replication stage alone was 5.4 × 10−8. Furthermore, a total of 2930 samples scanned at NCI as part of the discovery stage (∼30% of total) were genotyped with an optimized TaqMan assay for rs7741164. For this subset of discovery samples, the association result was P = 1.12 × 10−4 when using the TaqMan genotypes versus P = 2.45 × 10−3 when using imputed genotypes. When combining all the samples with TaqMan genotype data available from the discovery and replication stages (7293 cases and 8498 controls), the association result was P = 3.05 × 10−10 (Supplementary Material, Table S4). Consequently, our finding is likely to be stable despite the described imputation issue.
Discussion
Our first finding SNP rs7741164 maps to an intron of FOXP4-AS1 on 6p21.1. Other genetic variants at 6p21.1 have been shown by multiple GWAS to be associated with multiple cancers. For instance, rs2494938 was associated with lung, non-cardia gastric and esophageal squamous cell carcinoma (ESCC) in Han Chinese (16), rs10484761 was associated with ESCC in a GWAS in Chinese (17) and rs1983891 in the intron of FOXP4 was associated with prostate cancer in Japanese (18). The pair-wise linkage disequilibrium (LD) among all four SNPs including our novel finding of rs7741164 is low (r2 < 0.02 in 1000 Genomes Project data Asian population). The nearest plausible candidate gene, FOXP4-AS1, is a non-coding RNA gene (ncRNA) that belongs to the antisense RNA class. Thus far, ncRNAs have demonstrated key molecular functions such as the ability to regulate the expression of nearby protein-coding genes and modulate carcinogenesis pathways (19–21). As such, it is possible that FOXP4-AS1 acts by regulating the expression of key genes to influence lung cancer risk.
Our second finding SNP rs72658409 maps to an intergenic region on 9p21. Variants in the 9p21 region have been associated with risk for a number of cancers, including glioma (rs4977756 (22,23); rs1412829 (24)), melanoma (rs7023329 (25,26)), breast cancer (rs1011970 (27,28)), nasopharyngeal cancer (rs1412829 (29)), childhood acute lymphoblastic leukemia (rs3731217 (30)), chronic lymphocytic leukemia (rs1679013 (31)), basal cell carcinoma (rs2151280 (32)) and lung squamous cell carcinoma (rs1333040 (5)). This region also harbors highly penetrant mutations that explain a substantial fraction of hereditary melanoma (33). However, the LD is low (r2 < 0.02 in 1000 Genomes Project data Asian population) between our newly identified SNP rs72658409 and each of the SNPs listed above as well as other SNPs in this region reported to be associated with multiple cancers (34). Therefore, our new finding represents a new independent locus and illustrates the complex genetic architecture of 9p21. Notably, somatic 9p21 deletions have been frequently observed in human cancers, including lung cancer, lymphoid leukemia and esophageal cancer (35), and it is important to investigate how the germline susceptibility alleles inform such somatic alterations in different cancer sites including lung. Further functional validation studies are warranted for this complex locus in order to understand its role in lung carcinogenesis as well as associations between other independent SNPs and other cancers.
Our third finding SNP rs11610143 maps to an intron of ACVR1B on 12q13.13. A distinct intronic SNP, rs12809597, in ACVR1B was previously reported in a population of European descent to be associated with risk of lung cancer in never smokers (754 cases and 819 controls; OR = 0.72; P = 0.0002), especially among women (OR = 0.72; P = 0.0013) and/or those with exposure to ETS (OR = 0.67; P = 7.8 × 10−5) (36). However, this SNP is monomorphic in Eastern Asian populations and therefore its association with lung cancer observed in populations of European descent cannot be directly assessed in our data set. Furthermore, the LD between our novel SNP rs11610143 and rs12809597 is very low (r2 < 0.003 in 1000 Genomes Project data CEU population; BSD (between marker distance) is 7.2 kb). Therefore, the SNP we identified in our GWAS possibly tags an independent causal variant in this locus and underscores the importance of fine-mapping and pursuing further studies of this susceptibility allele.
In summary, the meta-analysis of four imputed GWAS of lung cancer among never-smoking women in Asian with further replication in independent case/control sets from similar populations has yielded three new risk loci for lung cancer at 6p21.1, 9p21.3 and 12q13.13. More than 80% of cases in our study were adenocarcinoma, and the effect sizes of these new loci were similar in a logistic regression model that analyzed only adenocarcinoma cases with controls (Supplementary Material, Table S5). In addition, we found no evidence of association (P > 0.05) for these three loci in a lung cancer GWAS study (5713 cases and 5736 controls) comprised mostly of smokers of European decent (37) (results not shown), although there was a limited number of non-smokers (355 cases). Further work is needed to fine-map each region to identify the optimal alleles for laboratory studies that could further our understanding of the biological mechanism underlying these susceptibility alleles and their interactions with environmental factors such as coal, which is widely used in this region.
Materials and Methods
Study population
The discovery stage included lung cancer studies in Asian never-smoking women with subjects drawn from four independent GWAS, namely NCI FLCCA (5), two other GWAS studies from Japan (6,8) and one from China (7). Details about each GWAS can be found in previous publications. For FLCCA, we excluded 53 GELAC cases and 51 GELAC controls that were genotyped on the Illumina 370 K SNP microarray, resulting in a slightly smaller total number of individuals (5457 cases and 4493 controls) as compared with the original paper (5) but these remaining samples were all genotyped on comparable SNP microarrays (Illumina 660 W or Illumina 610 K). For the other three GWAS studies, the never-smoking women component was extracted for this analysis. The number of cases and controls is listed in Supplementary Material, Table S1. All lung cancer cases were histologically confirmed. Each study was approved by their local institutional review board and all study participants provided informed consent prior to participation. We cannot make the full meta-analysis results publicly available mainly because we included one GWAS study from China and two GWAS studies from Japan in addition to the NCI GWAS, which has already been deposited into dbGaP (Accession: phs000716.v1.p1).
Genotype imputation
Genotype imputation was conducted by each center but followed a similar protocol as detailed below.
For both NCI and Nanjing studies, SNPs with a call rate < 95% or Hardy–Weinberg proportion test P-value < 0.000001 or minor allele frequency (MAF) < 1% were further removed prior to imputation for the current analysis. Imputation was conducted by using IMPUTE2 software version 2.2.2 (see URLs) and version 3 of the 1000 Genomes Project Phase 1 data as the reference set. First, the genomic coordinates were lifted over from NCBI human genome build 36 to build 37 using the UCSC lift over tool (see URLs). Second, the strand of the inference data was aligned with the 1000 Genomes data by simple allele state comparison or allele frequency matching for A/T and G/C SNPs. A pre-phasing strategy with SHAPEIT software version 1 (see URLs) was adopted to improve the imputation performance. The phased haplotypes from SHAPEIT were input directly into the IMPUTE2 program. Two Japanese studies were imputed slightly differently. For quality control, we removed SNPs with call rates <99% or Hardy–Weinberg proportion test P-values < 0.000001 or that were monomorphic (i.e. MAF = 0). SNPs with large allele frequency difference between reference and inference sets were also excluded (threshold was set to 0.16). Imputation used MaCH (38) and minimac2 (39), and the same version of 1000 Genomes as reference set, but only included Asian individuals (n = 286; including JPT, CHB and CHS). For all four imputed sets, imputed loci with INFO score (r2 for MaCH) < 0.3 or MAF < 0.01 were excluded from further association analysis.
For the 6p21.1 locus harboring rs7741164, we also attempted imputation of the NCI data set using minimac2 (39) for a 4 Mb window ranging from 39 493 412 to 43 493 412 (hg19) using only the ASN subset (n = 286) from the same version of 1000 Genomes data as the reference with the phased inference haplotypes either from SHAPEIT (40) or MaCH (38) program. In either approach, we obtained very similar imputed genotypes. When the imputed genotypes were compared with the TaqMan data generated for technical validation, the squared correlation of allelic dosage improved to 0.33 from 0.21 for data generated from the IMPUTE2 approach detailed in the paragraph above. We found the LD between all the genotyped SNPs and the imputed SNP rs7741164 is moderately low. The best genotyped SNP rs2477842 has a pair-wise r2 of only 0.3. The low LD makes the imputation of the SNP rs7741164 intrinsically difficult.
Replication genotyping
The TaqMan custom genotyping assay (Applied Biosystems, CA, USA) was used to genotype all the samples except for BBJ_NCCH, where Invader assays were used, for the set of 13 significant SNPs from the discovery meta-analysis on an additional 5878 cases and 7046 controls. The replication samples consisted of subjects from China (seven centers), Japan (four centers) and Taiwan (one center) that were not previously included in the FLCCA GWAS and meta-analysis. More information on each replication data set is found in Supplementary Material, Table S1.
Statistical analysis
For the discovery stage data, the association testing for each SNP (trend effect) was performed using SNPTEST software version 2.2 (see URLs) and based on a multivariate logistic regression model adjusting for age, study group and significant eigenvectors, which controls for population stratification. For the replication stage data, the association testing for each SNP (trend effect) was performed using GLU software (see URLs) and based on a multivariate logistic regression model adjusting for age only. Fixed-effects meta-analysis was used to combine individual association estimates from four imputed GWAS scans as well as each replication data set. Test for genetic effect differences across studies/data sets was assessed by using I2 and P value calculated from the Cochran's Q statistic, which is distributed as a χ2 statistic with (n − 1) degrees of freedom where n is the number of sets included in the meta-analysis.
Technical validation of imputed SNPs
To technically validate our imputation findings, we optimized three TaqMan assays (Applied Biosystems) for rs72658409, rs11610143 and rs7741164, respectively. Because the MAF for rs72658409 is only 7%, we first selected 67 samples with genotypes having one or two rare alleles for rs72658409, and then randomly selected a number of samples that were previously scanned in FLCCA for TaqMan genotyping. For rs7741164, we tried to genotype as many samples as possible because of its relative low imputation quality. The squared correlation (r2) for the allelic dosage between the imputed genotypes and the genotypes measured by TaqMan was calculated.
Recombination hotspot inference
Likelihood ratio statistics for recombination hotspots were estimated by SequenceLDhot (41) software based on background recombination rates inferred by PHASE v2.1 (42,43) using the 1000 Genomes CHB, CHS and JPT data.
In silico bioinformatics analysis
We searched the GTEx database (see URLs) to look for potential eQTLs for the associated SNPs. We used HaploReg v3 (11) and RegulomeDB v1.1 (15) to explore potential functional annotations within the ENCODE database in the genomic region surrounding our index SNPs and all neighboring SNPs having a pair-wise r2 > 0.8 with the index SNP in each of the new regions that we identified (Supplementary Material, Table S3).
URLs
IMPUTE2, http://mathgen.stats.ox.ac.uk/impute/impute_v2.html (20 December 2015, date last accessed)
UCSC lift over tool, http://hgdownload.cse.ucsc.edu/downloads.html (20 December 2015, date last accessed)
glu module, http://code.google.com/p/glu-genetics/ (20 December 2015, date last accessed)
SHAPEIT, http://www.shapeit.fr/ (20 December 2015, date last accessed)
GTEx, http://www.gtexportal.org/ (20 December 2015, date last accessed)
SNPTEST, https://mathgen.stats.ox.ac.uk/genetics_software/snptest/snptest.html (20 December 2015, date last accessed)
Supplementary Material
Funding
The Japan Lung Cancer Study (JLCS) was supported by a Grant-in-Aid from the Japan Agency for Medical Research and Development (AMED) for the Health and Labor Sciences Research Expenses for Commission (the Practical Research for Innovative Cancer Control: H26-practical-general-094) and the Management Expenses Grants from the Government to the National Cancer Center (26-A-1) for Biobank. BioBank Japan was supported by the Ministry of Education, Culture, Sports, Sciences and Technology of the Japanese government. The Japan Public Health Center-based prospective Study (the JPHC Study) was supported by National Cancer Center Research and Development Fund (23-A-31[toku] and 26-A-2) (since 2011) and a Grant-in-Aid for Cancer Research from the Ministry of Health, Labour and Welfare of Japan (from 1989 to 2010). The Taiwan GELAC Study (Genetic Epidemiological Study for Lung AdenoCarcinoma) was supported by grants from the National Research Program on Genomic Medicine in Taiwan (DOH99-TD-G-111-028), the National Research Program for Biopharmaceuticals in Taiwan (MOHW 103-TDU-PB-211-144003, MOST 103-2325-B-400-023) and Bioinformatics Core Facility for Translational Medicine and Biotechnology Development (MOST 104-2319-B-400-002). This work was also supported by the Jinan Science Research Project Foundation (201102051), Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT) in China (IRT_14R40), the National Key Scientific and Technological Project (2011ZX09307-001-04), the National Natural Science Foundation of China (No. 81272293), the State Key Program of National Natural Science of China (81230067), the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (No. NRF-2014R1A2A2A05003665), Agency for Science, Technology and Research (A*STAR), Singapore and the US National Institute of Health Grant (1U19CA148127-01). The overall GWAS project was supported by the intramural program of the US National Institutes of Health/National Cancer Institute.
Authors’ contributions
Q.L., N.R., S.J.C., Zha.W., W.J.S., K.S., C.A.H., K.M., Jie.L., K.C., M.I., Yan.Y., I.-S.C., C.W., P.-C.Y., T.W., Do.L., B.Z., Ji.Y., H.S. and M.K. organized and designed the study. Zha.W., K.S., C.A.H., K.M., Jie.L., K.C., M.I., Yan.Y., I.-S.C., C.W., L.B., K.W., C.C.C., S.A.L., M.Y., A.H., T.K., Ju.Y., H.K., K.A., Y.M., Y.D., T.M., Yas.Y., T.H., Z.H., J.D., H.M., Gu.J., B.S., Zhe.W., S.C., J.C., W.T., C.-J.C., G.-C.C., Y.-H.T., W.-C.S., K.-Y.C., M.-S.H., Y.-M.C., H.Z., H.L., P.C., H.G., P.X., L.L., T.Y., T.S., S.T., J.Z., Ge.J., J.Y.P., Y.H.K., J.S.S., K.H.P., H.N.K., H.-S.J., J.E.C., Y.Y.C., I.-J.O., Y.-C.K., J.S.K., S.-S.K., M.-H.S., S.-J.A., X.-C.Z., J.S., Y.-L.W., R.K., W.P., C.L., C.-F.H., L.-H.C., Y.-H.C., C.-H.C., W.-C.W., C.-Y.C., C.-L.W., C.-J.Y., H.-L.C., Y.-C.S., F.-Y.T., Y.-S.C., Y.-J.L., T.-Y.Y., C.-C.L., P.-C.Y., T.W., Do.L., Ji.Y., H.S., M.K. and Q.L. conducted and supervised the genotyping of samples. Zha.W., W.J.S., K.S., C.A.H., K.M., Jie.L., I.-S.C., M.S., Ni.C., P.-C.Y., T.W., Do.L., B.Z., Ji.Y., H.S., M.K., S.J.C., N.R. and Q.L. contributed to the design and execution of statistical analysis. W.J.S., K.S., C.A.H., K.M., Jie.L., K.C., M.I., Yan.Y., I.-S.C., C.W., Y.-C.H., W.H., Ne.C., M.T.L., Ni.C., J.F.F.J., T.K., Ju.Y., H.K., K.A., Y.M., Y.D., T.M., Yas.Y., T.H., Z.H., J.D., H.M., Gu.J., B.S., Zhe.W., S.C., Z.Y., X.L., Y.R., P.G., J.C., W.T., C.-J.C., G.-C.C., Y.-H.T., W.-C.S., K.-Y.C., M.-S.H., Y.-M.C., H.Z., H.L., P.C., H.G., P.X., L.L., T.Y., T.S., S.T., J.Z., Ge.J., K.F., J.Y.P., Y.H.K., J.S.S., K.H.P., Y.T.K., Y.J.J., C.H.K., I.K.P., H.N.K., H.-S.J., J.E.C., Y.Y.C., J.H.K., I.-J.O., Y.-C.K., S.W.S., J.S.K., H.-I.Y., S.-S.K., M.-H.S., A.S., Y.C., W.-Y.L., Jia.L., M.P.W., V.H.F.L., B.A.B., M.T., S.I.B., W.-H.C., B.-T.J., Junw.W., J.X., A.D.L.S., J.C.H., J.K.C.C., J.-C.W., Da.L., X.Z., Z.Z., Junj.W., H.C., L.J., F.W., G.W., S.-J.A., X.-C.Z., J.S., Y.-L.W., Y.-T.G., Y.-B.X., X.H., Jih.L., W.Z., X.-O.S., Q.C., R.K., W.P., C.L., H.D.H.III, C.-F.H., L.-H.C., Y.-H.C., C.-H.C., W.-C.W., C.-Y.C., C.-L.W., C.-J.Y., H.-L.C., Y.-C.S., F.-Y.T., Y.-S.C., Y.-J.L., T.-Y.Y., C.-C.L., P.-C.Y., T.W., Do.L., B.Z., Ji.Y., H.S., M.K., S.J.C., N.R. and Q.L. conducted the epidemiological studies and contributed samples to the GWAS and/or follow-up genotyping. Zha.W., W.J.S., S.J.C., N.R. and Q.L. wrote the first draft of the manuscript. All authors contributed to the writing of the manuscript.
Supplementary Material
Acknowledgements
We thank Drs Yoko Shimada, Hiromi Sakamoto, Akira Saito, Hidemi Ito and Kazuo Tajima for supporting this study.
Conflict of Interest statement. None declared.
References
- 1.World Cancer Report 2014 (World Health Organization). (2014) IARC Nonserial Publication, distributed by WHO Press.
- 2.Thun M.J., Hannan L.M., Adams-Campbell L.L., Boffetta P., Buring J.E., Feskanich D., Flanders W.D., Jee S.H., Katanoda K., Kolonel L.N. et al. (2008) Lung cancer occurrence in never-smokers: an analysis of 13 cohorts and 22 cancer registry studies. PLoS Med., 5, 1357–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kreuzer M., Heinrich J., Kreienbrock L., Rosario A.S., Gerken M., Wichmann H.E. (2002) Risk factors for lung cancer among nonsmoking women. Int. J. Cancer, 100, 706–713. [DOI] [PubMed] [Google Scholar]
- 4.Hosgood H.D. 3rd, Boffetta P., Greenland S., Lee Y.C., McLaughlin J., Seow A., Duell E.J., Andrew A.S., Zaridze D., Szeszenia-Dabrowska N. et al. (2010) In-home coal and wood use and lung cancer risk: a pooled analysis of the International Lung Cancer Consortium. Environ. Health Perspect., 118, 1743–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Timofeeva M.N., Hung R.J., Rafnar T., Christiani D.C., Field J.K., Bickeboller H., Risch A., McKay J.D., Wang Y., Dai J. et al. (2012) Influence of common genetic variation on lung cancer risk: meta-analysis of 14 900 cases and 29 485 controls. Hum. Mol. Genet., 21, 4980–4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miki D., Kubo M., Takahashi A., Yoon K.A., Kim J., Lee G.K., Zo J.I., Lee J.S., Hosono N., Morizono T. et al. (2010) Variation in TP63 is associated with lung adenocarcinoma susceptibility in Japanese and Korean populations. Nat. Genet., 42, 893–896. [DOI] [PubMed] [Google Scholar]
- 7.Hu Z., Wu C., Shi Y., Guo H., Zhao X., Yin Z., Yang L., Dai J., Hu L., Tan W. et al. (2011) A genome-wide association study identifies two new lung cancer susceptibility loci at 13q12.12 and 22q12.2 in Han Chinese. Nat. Genet., 43, 792–796. [DOI] [PubMed] [Google Scholar]
- 8.Shiraishi K., Kunitoh H., Daigo Y., Takahashi A., Goto K., Sakamoto H., Ohnami S., Shimada Y., Ashikawa K., Saito A. et al. (2012) A genome-wide association study identifies two new susceptibility loci for lung adenocarcinoma in the Japanese population. Nat. Genet., 44, 900–903. [DOI] [PubMed] [Google Scholar]
- 9.Kottgen A., Albrecht E., Teumer A., Vitart V., Krumsiek J., Hundertmark C., Pistis G., Ruggiero D., O'Seaghdha C.M., Haller T. et al. (2013) Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat. Genet., 45, 145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kedra D., Pan H.Q., Seroussi E., Fransson I., Guilbaud C., Collins J.E., Dunham I., Blennow E., Roe B.A., Piehl F. et al. (1998) Characterization of the human synaptogyrin gene family. Hum. Genet., 103, 131–141. [DOI] [PubMed] [Google Scholar]
- 11.Ward L.D., Kellis M. (2012) HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res., 40, D930–D934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lonsdale J., Thomas J., Salvatore M., Phillips R., Lo E., Shad S., Hasz R., Walters G., Garcia F., Young N. et al. (2013) The Genotype-Tissue Expression (GTEx) project. Nat. Genet., 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davydov E.V., Goode D.L., Sirota M., Cooper G.M., Sidow A., Batzoglou S. (2010) Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol., 6, e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garber M., Guttman M., Clamp M., Zody M.C., Friedman N., Xie X. (2009) Identifying novel constrained elements by exploiting biased substitution patterns. Bioinformatics, 25, i54–i62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boyle A.P., Hong E.L., Hariharan M., Cheng Y., Schaub M.A., Kasowski M., Karczewski K.J., Park J., Hitz B.C., Weng S. et al. (2012) Annotation of functional variation in personal genomes using RegulomeDB. Genome Res., 22, 1790–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin G.F., Ma H.X., Wu C., Dai J.C., Zhang R.Y., Shi Y.Y., Lu J.C., Miao X.P., Wang M.L., Zhou Y.F. et al. (2012) Genetic variants at 6p21.1 and 7p15.3 are associated with risk of multiple cancers in Han Chinese. Am. J. Hum. Genet., 91, 928–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu C., Hu Z., He Z., Jia W., Wang F., Zhou Y., Liu Z., Zhan Q., Liu Y., Yu D. et al. (2011) Genome-wide association study identifies three new susceptibility loci for esophageal squamous-cell carcinoma in Chinese populations. Nat. Genet., 43, 679–684. [DOI] [PubMed] [Google Scholar]
- 18.Takata R., Akamatsu S., Kubo M., Takahashi A., Hosono N., Kawaguchi T., Tsunoda T., Inazawa J., Kamatani N., Ogawa O. et al. (2010) Genome-wide association study identifies five new susceptibility loci for prostate cancer in the Japanese population. Nat. Genet., 42, 751–754. [DOI] [PubMed] [Google Scholar]
- 19.Wilusz J.E., Sunwoo H., Spector D.L. (2009) Long noncoding RNAs: functional surprises from the RNA world. Gene Dev., 23, 1494–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaikkonen M.U., Lam M.T.Y., Glass C.K. (2011) Non-coding RNAs as regulators of gene expression and epigenetics. Cardiovasc. Res., 90, 430–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ji P., Diederichs S., Wang W., Boing S., Metzger R., Schneider P.M., Tidow N., Brandt B., Buerger H., Bulk E. et al. (2003) MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene, 22, 8031–8041. [DOI] [PubMed] [Google Scholar]
- 22.Shete S., Hosking F.J., Robertson L.B., Dobbins S.E., Sanson M., Malmer B., Simon M., Marie Y., Boisselier B., Delattre J.Y. et al. (2009) Genome-wide association study identifies five susceptibility loci for glioma. Nat. Genet., 41, 899–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rajaraman P., Melin B.S., Wang Z., McKean-Cowdin R., Michaud D.S., Wang S.S., Bondy M., Houlston R., Jenkins R.B., Wrensch M. et al. (2012) Genome-wide association study of glioma and meta-analysis. Hum. Genet., 131, 1877–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wrensch M., Jenkins R.B., Chang J.S., Yeh R.F., Xiao Y., Decker P.A., Ballman K.V., Berger M., Buckner J.C., Chang S. et al. (2009) Variants in the CDKN2B and RTEL1 regions are associated with high-grade glioma susceptibility. Nat. Genet., 41, 905–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barrett J.H., Iles M.M., Harland M., Taylor J.C., Aitken J.F., Andresen P.A., Akslen L.A., Armstrong B.K., Avril M.F., Azizi E. et al. (2011) Genome-wide association study identifies three new melanoma susceptibility loci. Nat Genet, 43, 1108–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bishop D.T., Demenais F., Iles M.M., Harland M., Taylor J.C., Corda E., Randerson-Moor J., Aitken J.F., Avril M.F., Azizi E. et al. (2009) Genome-wide association study identifies three loci associated with melanoma risk. Nat. Genet., 41, 920–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turnbull C., Ahmed S., Morrison J., Pernet D., Renwick A., Maranian M., Seal S., Ghoussaini M., Hines S., Healey C.S. et al. (2010) Genome-wide association study identifies five new breast cancer susceptibility loci. Nat. Genet., 42, 504–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michailidou K., Hall P., Gonzalez-Neira A., Ghoussaini M., Dennis J., Milne R.L., Schmidt M.K., Chang-Claude J., Bojesen S.E., Bolla M.K. et al. (2013) Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat. Genet., 45, 353–361, 361e351–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bei J.X., Li Y., Jia W.H., Feng B.J., Zhou G., Chen L.Z., Feng Q.S., Low H.Q., Zhang H., He F. et al. (2010) A genome-wide association study of nasopharyngeal carcinoma identifies three new susceptibility loci. Nat. Genet., 42, 599–603. [DOI] [PubMed] [Google Scholar]
- 30.Migliorini G., Fiege B., Hosking F.J., Ma Y., Kumar R., Sherborne A.L., da Silva Filho M.I., Vijayakrishnan J., Koehler R., Thomsen H. et al. (2013) Variation at 10p12.2 and 10p14 influences risk of childhood B-cell acute lymphoblastic leukemia and phenotype. Blood, 122, 3298–3307. [DOI] [PubMed] [Google Scholar]
- 31.Berndt S.I., Skibola C.F., Joseph V., Camp N.J., Nieters A., Wang Z., Cozen W., Monnereau A., Wang S.S., Kelly R.S. et al. (2013) Genome-wide association study identifies multiple risk loci for chronic lymphocytic leukemia. Nat. Genet., 45, 868–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stacey S.N., Sulem P., Gudbjartsson D.F., Jonasdottir A., Thorleifsson G., Gudjonsson S.A., Masson G., Gudmundsson J., Sigurgeirsson B., Benediktsdottir K.R. et al. (2014) Germline sequence variants in TGM3 and RGS22 confer risk of basal cell carcinoma. Hum. Mol. Genet., 23, 3045–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bishop D.T., Demenais F., Goldstein A.M., Bergman W., Bishop J.N., Bressac-de Paillerets B., Chompret A., Ghiorzo P., Gruis N., Hansson J. et al. (2002) Geographical variation in the penetrance of CDKN2A mutations for melanoma. J. Natl. Cancer. Inst., 94, 894–903. [DOI] [PubMed] [Google Scholar]
- 34.Li W.Q., Pfeiffer R.M., Hyland P.L., Shi J., Gu F., Wang Z., Bhattacharjee S., Luo J., Xiong X., Yeager M. et al. (2014) Genetic polymorphisms in the 9p21 region associated with risk of multiple cancers. Carcinogenesis, 35, 2698–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sasaki S., Kitagawa Y., Sekido Y., Minna J.D., Kuwano H., Yokota J., Kohno T. (2003) Molecular processes of chromosome 9p21 deletions in human cancers. Oncogene, 22, 3792–3798. [DOI] [PubMed] [Google Scholar]
- 36.Spitz M.R., Gorlov I.P., Amos C.I., Dong Q., Chen W., Etzel C.J., Gorlova O.Y., Chang D.W., Pu X., Zhang D. et al. (2011) Variants in inflammation genes are implicated in risk of lung cancer in never smokers exposed to second-hand smoke. Cancer Discov., 1, 420–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Landi M.T., Chatterjee N., Yu K., Goldin L.R., Goldstein A.M., Rotunno M., Mirabello L., Jacobs K., Wheeler W., Yeager M. et al. (2009) A genome-wide association study of lung cancer identifies a region of chromosome 5p15 associated with risk for adenocarcinoma. Am. J. Hum. Genet., 85, 679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Y., Willer C.J., Ding J., Scheet P., Abecasis G.R. (2010) MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol., 34, 816–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fuchsberger C., Abecasis G.R., Hinds D.A. (2015) minimac2: faster genotype imputation. Bioinformatics, 31, 782–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delaneau O., Marchini J., Zagury J.F. (2012) A linear complexity phasing method for thousands of genomes. Nat. Methods, 9, 179–181. [DOI] [PubMed] [Google Scholar]
- 41.Fearnhead P. (2006) SequenceLDhot: detecting recombination hotspots. Bioinformatics, 22, 3061–3066. [DOI] [PubMed] [Google Scholar]
- 42.Li N., Stephens M. (2003) Modeling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics, 165, 2213–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crawford D.C., Bhangale T., Li N., Hellenthal G., Rieder M.J., Nickerson D.A., Stephens M. (2004) Evidence for substantial fine-scale variation in recombination rates across the human genome. Nat. Genet., 36, 700–706. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}