

Abstract
Acute myocardial ischemia/reperfusion (MIR) injury leads to severe arrhythmias and a high lethality. We aim to determine the effect of heat shock protein A12B (HSPA12B), a newly discovered member of the Hsp70 family, on heart injury parameters following MIR surgery. We used HSPA12B transgenic mice to determine its effects on heart function parameters, infarct size and cellular apoptosis following MIR surgery. Proinflammatory cytokines, oxidative products and anti-oxidative enzymes in the myocardium were measured to evaluate the anti-inflammatory and anti-oxidative effects of HSPA12B over-expression. The role of PPARs/eNOS/PI3k/Akt pathway was investigated using their inhibitors. The alteration of hemodynamic parameters, histopathological, apoptotic and infarct size caused by MIR was greatly attenuated in HSPA12B over-expressed mice. HSPA12B also greatly mitigated the inflammatory response, demonstrated by the decrease in the levels of IL-1β, IL-6, TNF-a and MPO. Anti-oxidative enzymes (SOD, Catalase and GPx) were restored by HSPA12B; oxidative products (8-OHdG, MDA and protein carbonyl) were decreased. HSPA12B activated the PPARγ-dependent eNOS/PI3k/Akt pathway, and the influence of HSPA12B on cardiac function was reversed by the inhibitors of eNOS, PPARγ, Akt and PI3K. Our results present a novel signaling mechanism that HSPA12B protects MIR injury through a PPARγ-dependent PI3K/Akt/eNOS pathway.
Keywords: HSPA12B, myocardium ischemic/reperfusion injury, PPARγ, PI3K, Akt, eNOS
Introduction
Acute myocardial ischemia/reperfusion (MIR) injury leads to severe arrhythmias; endothelial dysfunction; myocardial stunning; cell death, either by necrosis or apoptosis; and a high lethality rate [1,2]. MIR may occur when removing the aortic cross-clamp during cardiac surgery or when the balloon is deflated after inflation in the primary PCI. It can induce acute consequences, such as low cardiac output and death, or chronic results, including heart failure [3]. MIR injury is a complex pathophysiological process that involves various factors and pathways. Impaired cardiovascular function and endothelial cell injury are the main courses of MIR injury. Among the key contributors to MIR injury are neutrophil infiltration, cytokine release and oxidative stress. There is substantial evidence that reperfusion injury in the myocardium is an acute inflammatory reaction, which involves multiple cytokines. It has been reported that the production of tumor necrosis factor (TNF)-α, interleukin (IL)-6, IL-1β, and myeloperoxidase (MPO) are increased during MIR [4,5]. The oxygen free-radical system has been implicated in the pathogenesis of MIR injury [6]. Such free radicals are generated by injured myocytes, endothelial cells and neutrophils in the ischemic zone, and they become activated by reperfusion. The over-production of reactive oxygen species (ROS) exacerbates membrane damage, which leads to calcium loading and causes tissue damage through cell membrane lipid peroxidation, protein denaturation and DNA damage [7]. Some anti-oxidative enzymes, such as superoxide dismutase (SOD), catalase and glutathione peroxidase (GPx), can be consumed by these excess ROS.
Heat shock protein A12B (HSPA12B), a member of the Hsp70 family, was newly discovered in human atherosclerotic lesions by Han et al. in 2003 [8]. The HSPA12 family, which consists of HSPA12A and HSPA12B, is the least-conserved subfamily in the HSP70 superfamily [8,9]. The HSPA12B mRNA transcript was detected at the highest levels in the heart, and HSPA12B is predominantly expressed in endothelial cells as an angiogenesis regulator [10]. Some studies have found that HSPA12B is induced in LPS-induced inflammation in the central nervous system, which provides important clues to the biological functions of HSPA12B in inflammation [11]. Others have shown that the over-expression of HSPA12B attenuates LPS-induced cardiac dysfunction by limiting leukocyte infiltration into the myocardium [9]. Recently, several studies have confirmed an attenuation of heart and brain injury after over-expression of HSPA12B in experimental models [12-14]. Over-expression of HSPA12B attenuated cardiac dysfunction during endotoxemia. Tg mice given HSPA12B exhibited improvements in cardiac dysfunction and remodeling after myocardial infarction, accompanied by a significant decrease in cardiomyocyte apoptosis and increase in their capillary and arteriolar densities [9].
Endothelial NOS (eNOS), also known as nitric oxide synthase 3 or constitutive NOS, generates NO in blood vessels and regulates vascular tone by inhibiting smooth muscle contraction and platelet aggregation [15]. It has been reported that eNOS is protective against MIR injury and regulates myocardium blood flow, mediating the vascular response to oxidative stress and inhibiting neutrophil adhesion to the vascular endothelium [16-18]. Peroxisome proliferator-activated receptors (PPARs), a group of nuclear receptor proteins that function as transcription factors regulating the expression of genes, are essential for the regulation of cellular differentiation, development, and metabolism (carbohydrate, lipid, protein) [19]. Three types of PPARs (PPAR-α, PPAR-γ, and PPAR-β) have been identified. PPAR-α mainly exists in the liver, kidney, heart, muscle, and adipose tissue; PPAR-γ is mostly expressed in the heart, muscles, colon, kidney, pancreas and spleen.PPAR-β is expressed in many tissues, but is found markedly in the brain, adipose tissue, and skin [20]. PPAR-γ is closely related to eNOS in endothelial cells and acts in the regulation of vessel endothelium [21,22]. Bi et al. suggested that PPAR-γ could promote endothelial cell proliferation and migration by inducing eNOS, which produces the vasodilator NO from amino acid l-arginine in endothelial cells [23].
Some investigators have studied the protective effects of HSPA12B on various types of injuries, such as cerebral ischemia/reperfusion [12], neuronal ischemic injury [24], and myocardial infarction [14]. However, the effects of HSPA12B on MIR injury have not yet been explored. Hence, in the present study, we aim to study the protective effects of HSPA12B against MIR injury and their underlying mechanisms. We observed the effects of HSPA12B over-expression on heart function parameters, infarct size and cellular apoptosis following MIR surgery. Proinflammatory cytokines, oxidative products and anti-oxidative enzymes in the myocardium were measured to evaluate the anti-inflammatory and anti-oxidative effects of HSPA12B over-expression. Next, we investigated theexpression of the PPARs, phosphorylated-eNOS and PI3k/Akt pathways in the myocardium and the relationshipsamong these pathways using their inhibitors.
Materials and methods
Animals and reagents
The generation of HSPA12B transgenic mice (Tg) and wild type mice (WT) has been previously described by Zhou et al. [9]. The human hspa12b transgene consists of 20.4 kb of coding sequence (gene ID: ENSG number 00000132622), 7.3 kb of 5’ flanking sequence and promoter, and 3.6 kb of 3’ flanking sequence. The animals were all housed in Shanghai Jiaotong University School of Medicine at a constant 22°C temperature, 41% relative humidity, and 12-/12-hour light/dark cycle in the animal care wing, and they were allowed access to water and food ad libitum. The experiments were performed in accordance with the guidelines in the “Principles of Laboratory Animal Care” and the “Guide for the Care and Use of Laboratory Animals” (NIH Publication No. 85-23, revised 1996). The eNOS inhibitor L-NIO, PPARγ inhibitor GW9662, PI3K pathway inhibitor LY294002, and Akt pathway inhibitor triciribine were purchased from Sigma-Aldrich (St. Louis, MO, USA). L-NIO (20 mg/kg of body weight); GW9662 (2 mg/kg of body weight), LY294002 (100 mg/kg of body weight) or triciribine (2 mg/kg of body weight) was administered intraperitoneally 30 min prior to the MIR surgery.
Model of MIR and following procedures
The myocardial I/R injuries were generated as previously described by Yang et al. [25]. Briefly, mice were first intraperitoneally anesthetized with pentobarbital sodium (100 mg/kg, Abbott Laboratories). A heating pad was used to keep the temperature at 37°C. Then, the left coronary artery was located, exposed and ligated for 45 min. Reperfusion was then initiated by releasing the ligature. Sham-operated mice underwent a similar procedure, except that the left coronary artery was not ligated. When the reperfusion was finished, for hemodynamic measurements, the muscle layer and the skin were closed and the animals were allowed to recover for three weeks. Buprenorphine hydrochloride was injected (0.65 mg/kg, intramuscularly) to reduce postoperative pain. Analyses of oxidative products, anti-oxidative enzymes and inflammatory factors, using TdT-mediated dUTP nick end labeling (TUNEL) staining and Hematoxylin-eosin (H&E), were performed on the same day of hemodynamic measurements. Hearts were harvested and cut into slices for TUNEL and H&E staining. Sirius Red staining was performed after the reperfusion was completed.
Cardiac function measurements
Cardiac function measurements were similar to Gao’s study [26]. Mice were intraperitoneally anesthetized with chloral hydrate (300 mg/kg) three weeks after the ischemia/reperfusion treatment. The external right carotid artery was exposed, and a micro-tipped transducer catheter (1.4F, Millar Instrument Inc.) was placed into the artery and then advanced into the LV. The other end of the catheter was connected to an electrostatic chart recorder (model ES 2000, Gould, Cleveland, USA). The mean arterial pressure (MAP), LV end-diastolic pressure (LVEDP), LV systolic pressure (LVSP) and maximal rates of rise and fall in LV pressure (dP/dtmax, dP/dtmin) were measured and averaged from consecutive 10 beats.
H&E staining
The hearts were harvested and embedded in paraffin. The paraffin was then cut into 4-μm thickness slices and stained with H&E staining. Briefly, hearts were deparaffinized with xylene, re-hydrated in alcohol, stained in a hematoxylin solution, differentiated in 1% acid alcohol and counterstained in an eosin-phloxine solution. Finally, they were mounted with xylene-based mounting medium and then observed under a microscope for histological changes, including ardiomyocyte hydropic changes, neutrophilic infiltrate, hemorrhage, lymphohistiocytic infiltrate, and acute myocardial necrosis.
TUNEL assay
For the TUNEL assay, the paraffin-treated hearts were fixed in 4% methanol-free formaldehyde solution in PBS and then cut into 4-μm thick slices. The slices were incubated with terminal deoxynucleotidyl transferase and fluorescein-labeled dUTP (Sigma-Aldrich, St. Louis, MO). They were then analyzed under a fluorescence microscope. The numbers of TUNEL-positive and normal cardiomyocytes were counted across 10 fields per section by an expert in a double-blind manner.
Infarct size determination by Sirius Red staining
After the 2-hour reperfusion, the infarct size was determined with Sirius Red staining. Briefly, the mice were sacrificed, and their hearts were fixed overnight in para-formaldehyde and cut into five 1-mm thick slices. These slices were flat embedded in paraffin and cut into 4-μm thick sections, which were then stained with Sirius Red to determine the infarct volume. Subsequently, the slices were placed on a light table and photographed on both sides. The different areas were then delineated. The infarct size was calculated as a percentage of the volume of the infarct area versus the LV wall volume.
Measurement of the IL-1β, IL-6, TNF-a and MPO levels in the myocardium tissues
Transmural tissues from AAR were harvested and washed in normal saline and then homogenized in saline (25 mg/ml) on ice. The homogenates were centrifuged at 3000 g at 4°C for 15 min. The IL-1β, IL-6, TNF-a and MPO levels were then measured with an enzyme-linked immunosorbent assay (ELISA) kit, according to the manufacturer’s instructions (Sigma, St. Louis, USA). The levels were calculated with the absorbance read on a microplate reader. The protein concentration was determined using a standard BCA protein assay kit (Beyotime Institute of Biotechnology, Shanghai, China), and the results were expressed per microgram of protein.
Measurement of malondialdehyde (MDA), protein carbonyl and 8-hydroxy-2-deoxyguanosine (8-OHdG) in myocardium tissues
Transmural tissue from AAR (50 mg) was dissected out and carefully trimmed, weighed, and homogenized in a potassium phosphate buffer solution (50 mM, pH 7.5). After centrifugation at 1500 g for 10 min at 4°C, the supernatant was recovered, placed on ice and measured for the levels of MDA, protein carbonyl and 8-OHdG with the previously mentioned methods [27-29]. The protein concentration was determined using a standard BCA protein assay kit (Beyotime Institute of Biotechnology, Shanghai, China).
Measurement of superoxide dismutase (SOD), catalase and glutathione peroxidase (Gpx) activities in myocardium tissues
After the termination of the experiment, the mice were sacrificed and the myocardium tissues kidney tissues were immediately collected for experiments. They were homogenized immediately on ice in 1 mL of saline. The homogenates were centrifuged at 1500 g at 4°C for 10 min. Then, the supernatant was collected for SOD, catalase and Gpx assays using the corresponding assay kits from the Beyotime Institute of Biotechnology (Shanghai, China). All of these enzymes were expressed as units per milligram tissue (U/mg).
Western blotting
Cardiac tissues were harvested, washed in ice-cold saline, and then homogenized in lysis buffer (50 mmol/L Tris, 15 mmol/L EDTA, 150 mmol/L NaCl, 0.1% Triton X-100, pH 8.0) with 1 mM PMSF on ice. After centrifugation at 12,000 g at 4°C for 20 min, the supernatant was collected. A 50-mg protein sample was loaded per lane, separated by 12% dodecyl sulfatepolyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to nitrocellulose membranes electrophoretically. Membranes were blocked in blocking buffer containing 5% non-fat dry milk for 2 h at room temperature, and they were then blocked with primary antibodies in 5 ml of 5% BSA wash buffer at 4°C overnight. The membrane was washed with wash buffer and incubated with horseradish peroxidase-conjugated secondary antibody anti-rabbit IgG (1:2000) in TBST solution for 1 h. Finally, the membranes were washed three times in TBST. They were exposed to ECL chemiluminescence reagents for 2 min, and the blots were exposed to X-ray film for radiographic detection. The protein bands were then quantified by densitometry using Quantity One (Bio-Rad Laboratories, CA, USA).
Statistical analysis
All statistical analyses were performed using the SPSS software, version 17.0, with one-way analysis of variance followed by the Student-Newman-Keuls post-hoc test. Differences were considered significant at p<0.05.
Results
Cardiac function changes with HSPA12B
As shown in Table 1, there were significant changes after MIR plus over-expression of HSPA12B on all of the hemodynamic parameters. Over-expression of HSPA12B alone (without MIR treatment) had no significant impact on the LVEDP, LVSP, (dP/dt) max, -(dP/dt) minor MAP. MIR treatment alone significantly increased LVEDP and decreased the LVSP, (dP/dt) max, -(dP/dt) min and MAP (p<0.05 compared to the Sham+WT group). However, in HSPA12B over-expressed mice, alterations in the hemodynamic parameters induced by MIR were greatly attenuated (p<0.05 compared to the MIR+WT group).
Table 1.
Cardiac function changes by HSPA12B
| Sham | Myocardial ischemia-reperfusion | |||
|---|---|---|---|---|
|
|
||||
| WT | Tg | WT | Tg | |
| LVEDP (mmHg) | 3.8±0.4 | 3.9±0.4 | 7.9±0.4& | 4.6±0.3# |
| LVSP (mmHg) | 112.2±9.6 | 109.3±6.2 | 57.5±3.5& | 96.6±6.5# |
| (dP/dt) max (mmHg/s) | 3695.3±321.2 | 3654.2±304.2 | 2134.5±314.2& | 3257.3±295.4# |
| -(dP/dt) min (mmHg/s) | 3864.3±260.3 | 4021.2±311.8 | 2231.1±263.2& | 3869.4±257.4# |
| MAP (mmHg) | 86.5±10.2 | 80.2±10.3 | 44.2±8.5& | 63.5±9.3# |
Note: LVEDP, LV end-diastolic pressure; LVSP, LV systolic pressure; dP/dtmax, dP/dtmin, maximal rates of rise and fall in LV pressure; MAP, mean arterial pressure. Values are expressed as the Mean ± SD.
p<0.05 compared to the Sham+WT;
p<0.05 compared to the MIR+WT.
N = 12 per group.
Histopathological, apoptotic and infarct size changes with HSPA12B
Figure 1 shows the histopathological (A), apoptotic (B, C) and infarct size (D) changes in each group. age, neutrophilic infiltrate and spindle-shaped interstitial cells compared to As shown in Figure 1A, H&E staining showed that the MIR+WT group exhibited significant necrosis, hemorrh the Sham+WT and Sham+Tg groups, but the injury degree was greatly lower in the MIR+Tg mice. Figure 1B and 1C show apoptotic mouse heart cells stained with TUNEL. The TUNEL-positive cell proportions in the Sham+WT and Sham+Tg groups were below 2.3% and 2.7%, respectively; in the MIR+WT group, the proportion was 36.2%, which was significantly higher than either the Sham+WT or Sham+Tg group (p<0.05). However, the TUNEL-positive cell proportion dropped to 18.2% in the MIR+Tg group (p<0.05 compared to the MIR+WT group). Figure 1D shows the MIR-induced myocardium infarction, but the infarction percentage in the MIR+Tg group (16.4%) was only about half of that in the MIR+WT group (33.1%) (p<0.05).
Figure 1.
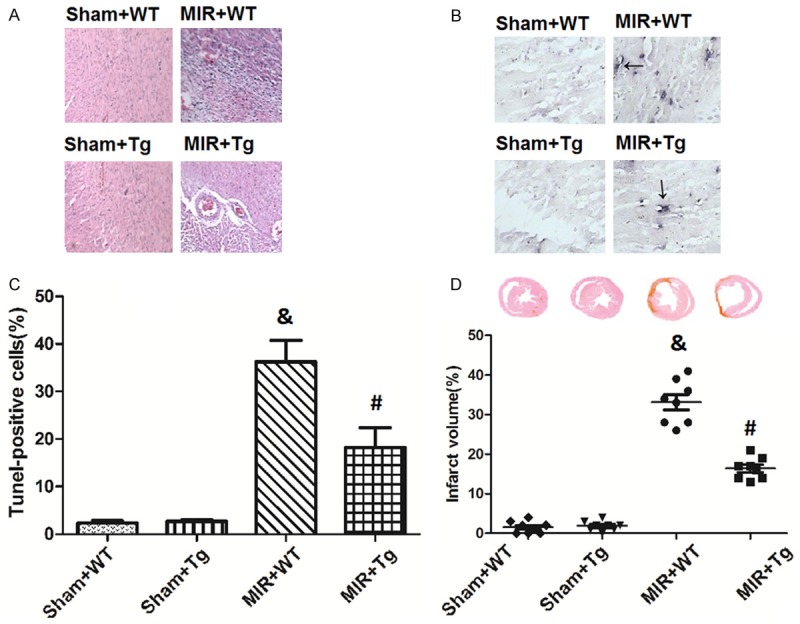
Histopathological, apoptotic and infarct size changes by HSPA12B. Microscopic examinations of the myocardium tissues were stained with hematoxylin and eosin (A). Cellular apoptosis were evaluated via TUNEL staining (B and C). Myocardium infarction was stained with Sirius Red (D). Values are expressed as the Mean ± SD. &p<0.05 compared to the Sham+WT; #p<0.05 compared to the MIR+WT. N = 8 per group.
Changes in the myocardial inflammation markers (IL-1β, IL-6, TNF-a and MPO)
As shown in Figure 2, MIR caused significant increases in the myocardial inflammation markers (IL-1β, IL-6, TNF-a and MPO) compared to the Sham+WT groups (p<0.05). The IL-1β, IL-6, TNF-a and MPO levels in the Sham+WT group are 19.34 pg/mg protein, 5.31 pg/g protein, 4.67 pg/mg protein and 52.36 ng/mg protein, respectively, while in the MIR+WT group they were increased to 116.35 pg/mg protein, 15.67 pg/g protein, 34.67 pg/mg protein and 163.55 ng/mg protein. These changes were reversed by HSPA12B over-expression, as shown by dramatic decreases in the IL-1β, IL-6, TNF-a and MPO levels (53.67 pg/mg protein, 10.36 pg/g protein, 16.34 pg/mg protein and 98.68 ng/mg protein; P<0.05 compared to the MIR+WT group).
Figure 2.
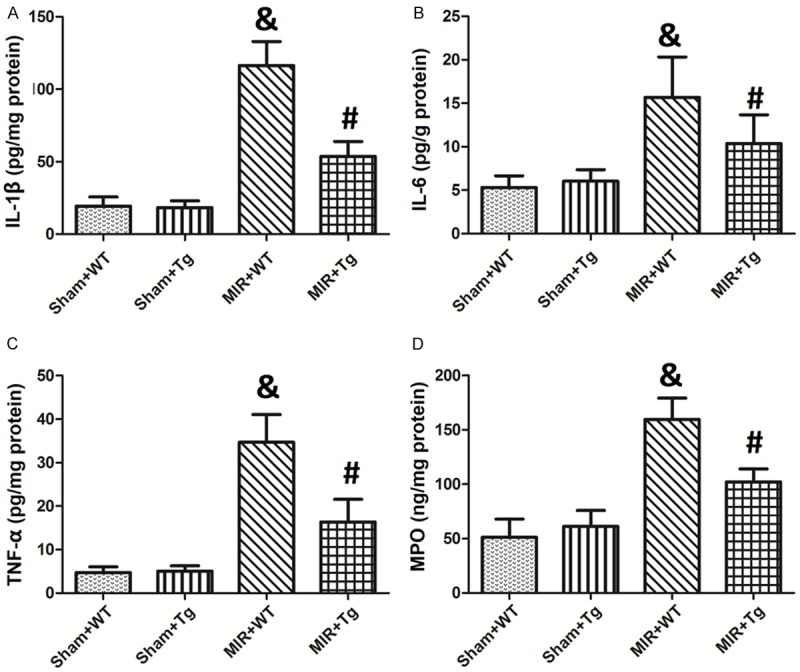
Changes in the myocardial inflammation markers (IL-1β, IL-6, TNF-a and MPO). The levels of IL-1β, IL-6, TNF-a and MPO levels in the myocardium tissues are shown in A-D, respectively. Values are expressed as the Mean ± SD. &p<0.05 compared to the Sham+WT; #p<0.05 compared to the MIR+WT. N = 12 per group.
Changes in anti-oxidative enzymes (SOD, Catalase and GPx) and oxidative products (8-OHdG, MDA and protein carbonyl) in heart tissue
Figure 3 shows changes in the myocardial anti-oxidative enzymes (SOD, Catalase and GPx) and oxidative products (8-OHdG, MDA and protein carbonyl). As shown in Figure 2A-C, in mice in the MIR+WT group the SOD, Catalase and GPx levels were all decreased compared to the Sham+WT groups. SOD level was decreased from 1.26 U/mg protein to 0.44 U/mg protein; Catalase level was decreased from 10.13 U/mg protein to 3.67 U/mg protein; GPx level was decreased from 37.26 U/mg protein to 13.29 U/mg protein. The over-expression of HSPA12B, however, significantly inhibited the consumption of these anti-oxidative enzymes. The SOD, Catalase and GPxlevel were restored to 1.34 U/mg protein, 10.32 U/mg proteinand 30.22 U/mg protein, respectively. Figure 3D-F demonstrated that MIR caused significant oxidative stress (the 8-OHdG, MDA and protein carbonyl levels were increasedfrom 2.16 pg/g to 7.91 pg/g, 1.92 μmol/mg to 5.76 μmol/mg, 3.81 nmol/mg to 11.02 nmol/mg). HSPA12B over-expression significantly mitigated this oxidative stress, as shown by dramatic decreases in these oxidative products compared to the MIR+WT group (p<0.05). The 8-OHdG was decreased to 2.46 pg/g, MDA was decreased to 2.87 μmol/mg, and the protein carbonyl wasdecreased to 4.01 nmol/mg.
Figure 3.
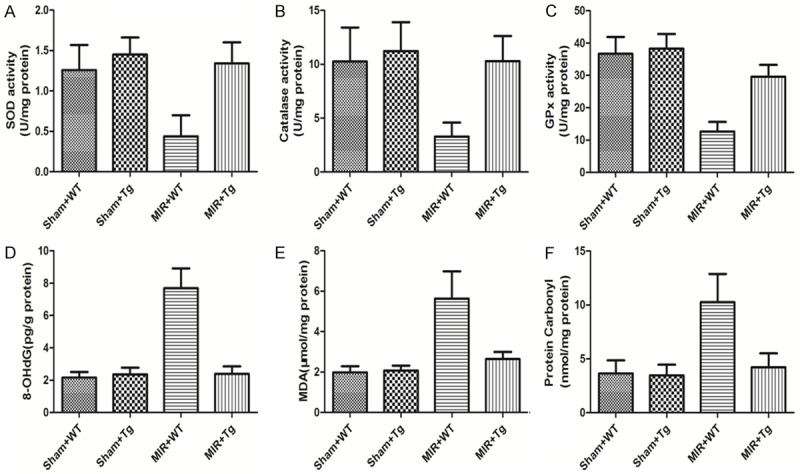
Changes in anti-oxidative enzymes (SOD, Catalase and GPx) and oxidative products (8-OHdG, MDA and protein carbonyl) in the myocardium tissues. The levels of SOD, Catalase, GPx, 8-OHdG, MDA and protein carbonyl in the myocardium tissues are shown in A-F, respectively. Values are expressed as Mean ± SD. &p<0.05 compared to the Sham+WT; #p<0.05 compared to the MIR+WT. N = 12 per group.
Changes in PPARα, PPARγ and PPARβ expression, eNOS and Akt phosphorylation regulation by MIR and HSPA12B
Figure 4 shows western blots of changes in PPARα, PPARγ and PPARβ, eNOS and Akt phosphorylation regulation by MIR and HSPA12B. As shown in Figure 4A and 4D-F, PPARα and PPARβ expression was not altered by MIR or HSPA12B over-expression, but PPARγ expression was significantly activated by MIR and HSPA12B over-expression. MIR caused 2.25 fold increase in the PPARγ expression, while MIR+Tg caused 4.31 fold increase in the PPARγ expression. Figure 4B and 4G show that eNOS phosphorylation was slightly increased in the MIR+WT group (1.62 fold) and significantly up-regulated by HSPA12B over-expression (2.81 fold). Figure 4C and 4F show that Akt phosphorylation was greatly increased in the MIR+WT and MIR+Tg groups (3.46 fold and 6.53 fold, respectively).
Figure 4.
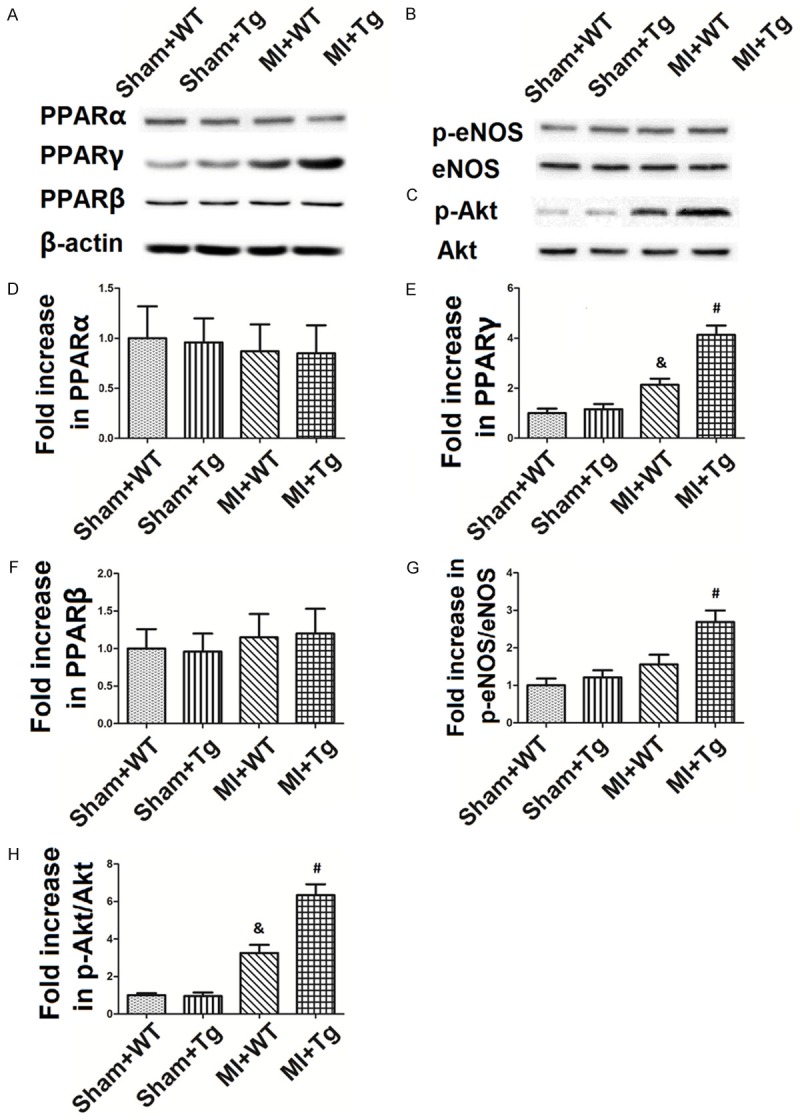
Changes in PPARα, PPARγ and PPARβ expression, eNOS and Akt phosphorylation. The blots of PPARα, PPARγ and PPARβ expression, eNOS and Akt phosphorylation are shown in (A-C), while the value changes are shown in (D-H). The values are expressed as Mean ± SD. &p<0.05 compared to the Sham+WT; #p<0.05 compared to the MIR+WT. N = 8 per group.
Changes in eNOS and Akt phosphorylation and PPARγ expression regulation by eNOS, PPARγ, Akt and PI3K inhibitors
As shown in Figure 5A, eNOS phosphorylation was greatly enhanced by the over-expression of HSPA12B, but all eNOS, PPARγ, Akt and PI3K inhibitors were able to inhibit it. L-NIO, GW9662, LY294002 and triciribine decreased the ratio of p-eNOS/eNOS from 1.69 fold to 0.68 fold, 0.82 fold, 0.71 fold and 0.80 fold, respectively. Figure 5B shows that Akt phosphorylation was not inhibited by eNOS inhibitor L-NIO but was greatly inhibited by PPARγ, Akt or PI3K inhibitors (from 2.82 fold to 1.13 fold, 0.77 fold and 1.23 fold, respectively). Figure 5C demonstrates that PPARγ expression was only inhibited by the PPARγ inhibitor GW9662 (from 2.05 fold to 0.66 fold), but was not significantly impacted by eNOS, Akt or PI3K inhibitors.
Figure 5.
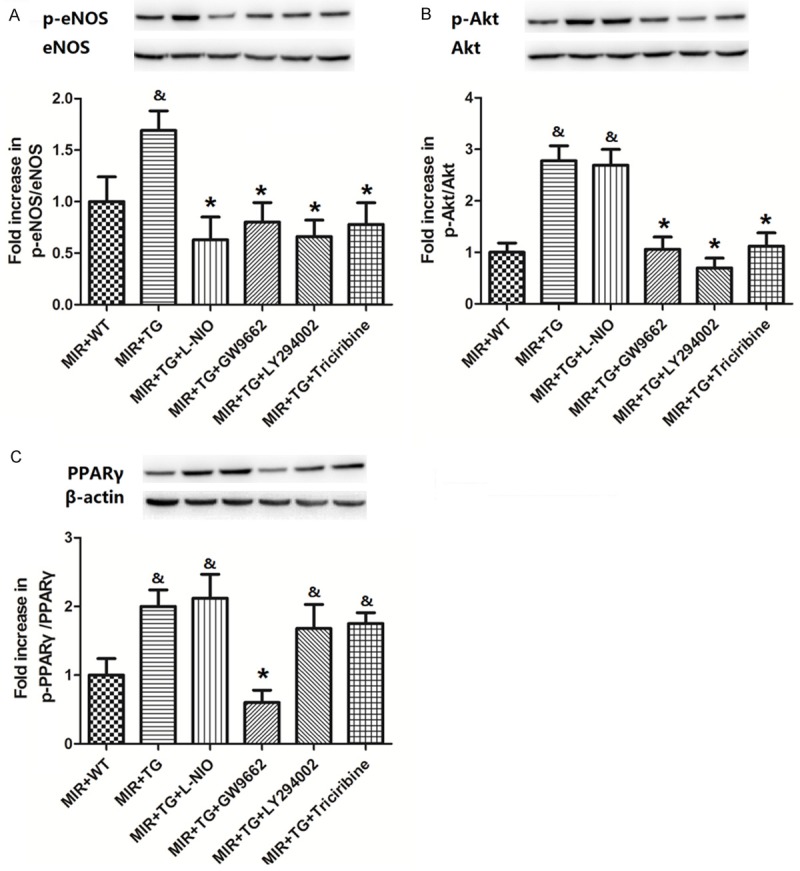
Changes in eNOS and Akt phosphorylation and PPARγ expression by eNOS, PPARγ, Akt and PI3K inhibitors. A-C. Show the blots and value changes in eNOS and Akt phosphorylation and PPARγ expression by eNOS, PPARγ, Akt and PI3K inhibitors. The values are expressed as Mean ± SD. &p<0.05 compared to the Sham+WT; *p<0.05 compared to the MIR+Tg. N = 8 per group.
Cardiac function changes by eNOS, PPARγ, Akt and PI3K inhibitors
Table 2 shows cardiac functional changes after eNOS, PPARγ, Akt and PI3K inhibition (L-NIO, GW9662, LY294002 and triciribine, respectively). Significant changes were caused by eNOS, PPARγ, Akt and PI3K inhibitors on all of the hemodynamic parameters. These inhibitors significantly increased LVEDP and decreased the LVSP, (dP/dt) max, -(dP/dt) min and MAP (p<0.05 compared to the MIR+Tg group).
Table 2.
Cardiac function changes by eNOS, PPARγ, Akt and PI3K inhibitors
| MIR+Tg | MIR+Tg+L-NIO | MIR+Tg+GW9662 | MIR+Tg+LY294002 | MIR+Tg+Triciribine | |
|---|---|---|---|---|---|
| LVEDP (mmHg) | 4.7±0.5 | 7.6±0.7* | 7.2±0.4* | 6.8±0.5* | 6.9±0.4* |
| LVSP (mmHg) | 97.2±8.2 | 64.5±8.1* | 65.6±6.8* | 67.5±5.9* | 61.2±8.2* |
| (dP/dt) max (mmHg/s) | 3326.4±256.8 | 2259.4±301.2* | 2236.4±311.4* | 2351.6±259.3* | 2567.8±266.5* |
| -(dP/dt) min (mmHg/s) | 3459.4±278.6 | 2351.2±266.4* | 2410.2±268.5* | 2311.5±284.5* | 2634.5±261.4* |
| MAP (mmHg) | 68.5±5.9 | 41.6±4.9* | 39.8±6.6* | 41.5±7.2* | 40.2±5.9* |
Note: The influences of eNOS, PPARγ, Akt and PI3K inhibitors (L-NIO, GW9662, LY294002 and triciribine, respectively) on cardiac function are shown in the table. Values are expressed as the Mean ± SD.
p<0.05 compared to the MIR+WT.
N = 12 per group.
Discussion
In recent years, myocardial infarction has become the major cause of death worldwide. The current standard treatment is to undergoreperfusion as soon as possible [30], but reperfusion itself can paradoxically causeserious injury to the myocardium, including neutrophil infiltration, cytokine release, ROS overproductionand calcium overload. After numerous laboratory studies and clinical trials, there are still no effective treatments to protect the myocardium from reperfusion injury. Thus, the present study aims to identify a novel therapy that may at least minimize the extent of MIRinjury. Here, we demonstrated that MIR treatment significantly increased LVEDP and decreased LVSP, (dP/dt) max, -(dP/dt) min and MAP, but that these alterations in hemodynamic parameters were greatly attenuated in HSPA12B over-expressed mice. The histopathological, apoptotic and infarct size changes induced by MIR were suppressed by HSPA12B. Over-expression of HSPA12B also greatly mitigated the inflammatory response, as demonstrated by a decrease in the myocardial inflammation markers (IL-1β, IL-6, TNF-a and MPO). Moreover, the levels of anti-oxidative enzymes (SOD, catalase and GPx) were significantly restored in HSPA12B over-expressed mice, and these increases in oxidative products (8-OHdG, MDA and protein carbonyl) were all attenuated by HSPA12B. The western blottingresults showed that PPARγ expression, phosphorylated-eNOSand phosphorylated-Akt expression were activated by both MIR and HSPA12B. All of the measured eNOS, PPARγ, Akt and PI3K inhibitors (L-NIO, GW9662, LY294002 and triciribine, respectively) successfully inhibitedeNOS phosphorylation. Akt phosphorylation was inhibited by PPARγ, Akt or PI3K inhibitors, but PPARγ expression was not impacted by eNOS, Akt or PI3K inhibitors. Finally, our cardiac function measurements demonstratedthat all of the eNOS, PPARγ, Akt and PI3K inhibitors significantly reversed the influence of HSPA12B on the LVEDP, LVSP, (dP/dt) max, -(dP/dt) min, MAP and EF.
HSPA12B, a new member of the HSP70 protein family, which plays a key role in protecting cells, tissues, organs and animals from various noxious conditions, was first discovered in human atherosclerotic lesions by Han et al. in 2002 [8]. It contains an atypical HSP70 ATPase domain and exerts effects during cellular adhesion and cell migration [10]. HSPA12A and HSPA12B form the HSPA12 family, which is the least conserved subfamily that belongs to the HSP70 superfamily [8,9]. HSPA12B was first found in endothelial cells, but it is also found in the heart, brain, kidney and lungs [8,10]. It has beenshown by multiple studies that HSPA12B plays an important role in the proliferation and migration of human umbilical vein endothelial cells and in the vascular development of zebra fish [31]. For the first time, our results demonstrate that HSPA12B over-expressed mice exhibit significantly improved hemodynamic parameters, such as LVEDP, LVSP, (dP/dt) max, -(dP/dt) min and MAP. Our H&E results additionally showed that the myocardium of MIR+Tgmice showed less necrosis, hemorrhage, neutrophilic infiltrate and spindle-shaped interstitial cells, primarily indicating the protective effects of HSPA12B against MIR injury.
Many studies have suggestedthat HSPA12B might regulate cellular apoptosis and inflammation in the process of ischemia and/or reperfusion, which is important in the pathogenesis of MIR. Cui et al. found that HSPA12B was expressed after LPS-induced inflammation in the brain, indicating that HSPA12B may play an important role in inflammation [11]. Others have found that HSPA12B attenuates LPS-induced cardiac dysfunction and limits leucocytes infiltration into the myocardium [9]. Moreover, Ma et al. confirmed that HSPA12B improved neurological deficits, decreased infarct volumes, and inhibited the expression of active caspase-3 and neural apoptosis aftercerebral ischemia/reperfusion injury [12,13]. The results from Li et al. demonstrate that HSPA12B attenuates cardiac dysfunction and remodeling after myocardial infarction by the prevention of cardiomyocyte apoptosis and the promotion of myocardial angiogenesis [14]. Our results in the present study suggestthat apoptotic cells in HSPA12B over-expressed mice were greatly reduced and that infarct size changes induced by MIR were also suppressed by HSPA12B. Over-expression of HSPA12B also greatly mitigated the inflammatory response, as demonstrated by the decrease in myocardial inflammation markers (IL-1β, IL-6, TNF-a and MPO), which are all activated by MIR. Oxidative stress is another crucial factor initiating reperfusion injury.The endogenous anti-oxidative capacity consists of SOD, catalase and GPx enzyme systems, which defend against the deleterious effects of ROS and protect against various oxidative-related cardiovascular injuries [8]. Abnormal accumulations of intracellular ROS overwhelm the natural anti-oxidative capacities of the body, causing detrimental modifications of important cellular macromolecules (such as lipids, proteins and DNA) and induce apoptosis. The present study confirmed that MIR significantly consumed the anti-oxidative enzymes (SOD, Catalase and GPx), indicating that thisanti-oxidative capacity in mice was overwhelmed. The disturbance of the oxidative/anti-oxidative balance caused macromolecule modification, demonstrated by increased oxidative products (8-OHdG, MDA and protein carbonyl). Interestingly, over-expression of HSPA12B significantly inhibited the consumption of these anti-oxidative enzymes and mitigated the oxidative stress, as shown by changes in anti-oxidative enzymesand oxidative products.
Nitric oxide synthases (NOS) form a family of enzymes catalyzing the production of nitric oxide (NO) from L-arginine. The family includes three subtypes, including neuronal NOS (nNOS), eNOS and inducible NOS (iNOS). NO may have both toxic and protective effects after MIR injury. High levels of NO produced by iNOS and nNOS induce cell injury or death, but NO induced by eNOS may protect cardiomyocytes. Many studies have revealed that eNOS is protective against MIR injury by regulating myocardium blood flow, mediating the vascular response to oxidative stress and inhibiting neutrophil adhesion to the vascular endothelium [16-18]. In our study, eNOS phosphorylation was slightly increased by MIR and significantly up-regulated by HSPA12B over-expression, suggesting the possible involvement of eNOS in the actions of HSPA12B. The interactions of HSP70 and eNOS have been demonstrated in the prevention of various diseases [32-35]. Karpe et al. determined that HSP72 prevents insulin resistance-induced vascular complications by augmenting angiotensin-(1-7) signaling via the Mas/eNOS/SIRT1 pathway [32]. Uryash et al. revealed that periodic acceleration preconditioning provides cardioprotection, with increased survival, reduced infarct size, and improved contractility, by regulation of HSP70 and the PI3/Akt/eNOS pathway [34]. Moreover, Li et al. discovered that over-expression of HSPA12B attenuates cardiac dysfunction by preventing cardiomyocyte apoptosis and promoting myocardial angiogenesis via an eNOS-dependent mechanism [14]. In addition, Hu et al. reportedthat this mechanism might involve the activation of Akt during the HSPA12B-induced migration of endothelial cells [31]. Their findings suggested that the phosphorylation of Akt was consistently reduced by siRNAs against HspA12B and that the over-expression of active Akt rescued the inhibitory effects of the knockdown of HspA12B on migration of human umbilical vein endothelial cells. Hence, to further explore the possible role of the PI3K/Akt pathway in the regulation of HSPA12B viaeNOS, we also examined the levels of activation of Akt. The results showed that Akt phosphorylation was greatly increased in both the MIR+WT group and the MIR+Tg group. These findings revealed that the PI3K/Akt pathway could be activated under the stress of MIR and could be regulated by HSPA12B.
Recent studies have shown that PPARγ, a member of the nuclear hormone receptor superfamily of ligand-activated transcription factors, is associated with the release of vasoactive substances from vascular endothelial cells. The administration of PPARγ activators could reversevascular remodeling, reduce vascular inflammation, and improve endothelial function [36]. Treating human endothelial cellswith PPARγ ligands increased the HSP-eNOS interaction and eNOS phosphorylation, as well as NO release [37]. Moreover, activation of PPARγ has been shown to increase eNOS activity and to improve left ventricular remodeling of infracted hearts [38,39]. In the present study, we found that of three types of PPARs (PPAR-α, PPAR-γ, and PPAR-β), only PPAR-γ expression was significantly activated by MIR and HSPA12B over-expression, which is consistent with previous studies.
To further investigate the relationships among HSPA12B, eNOS, PPAR-γand the PI3K/Akt pathway, we gave the animalseNOS, PPARγ, Aktor PI3K inhibitors (L-NIO, GW9662, LY294002 and triciribine, respectively); then,we examined their protein expressions or phosphorylation. All of the eNOS, PPARγ, Akt and PI3K inhibitors inhibited eNOS phosphorylation. Akt phosphorylation was inhibited by PPARγ, Akt or PI3K inhibitors, but not by the eNOS inhibitor L-NIO. PPARγ expression, however, was not significantly impacted by eNOS, Akt or PI3K inhibitors. These results indicate the following: (1) PPARγand the PI3K/Aktpathway are required for eNOS activation; (2) PPARγ and PI3K, but not eNOS, are required for Akt activation; and (3) PPARγis activated by HSPA12B, but not eNOS or the PI3K/Akt pathway. These findings suggest that PPARγ is an upstream modulator of PI3K/Akt and the eNOSpathway. Finally, our cardiac function measurements showed that all of the eNOS, PPARγ, Akt and PI3K inhibitors significantly reversed the effects of HSPA12B on hemodynamic parameters, indicating their roles in the actions of HSPA12B. Taken together, these data present a novel signaling mechanismthrough which HSPA12B protects against MIR injury in a PPARγ-dependent fashion via the PI3K/Akt/eNOS pathway.
In conclusion, we demonstrated that over-expression of HSPA12B protected cardiac functional andhistological changes, reduced the apoptosis induced by MIR, and decreased oxidative injury and inflammation. PPARγ, eNOS and the PI3K/Akt pathway were activated by both MIR and HSPA12B. Treatment with eNOS, PPARγ, Akt and PI3K inhibitors showed that HSPA12B might induce PPARγ-dependent PI3K/Akt/eNOS activation. Taken together, our results present a novel signaling mechanismthrough which HSPA12B protects against MIR injury in a PPARγ-dependent fashion via the PI3K/Akt/eNOS pathway.
Acknowledgements
This study was funded byNational Basic Research Program of China (No.2013CB945304).
Disclosure of conflict of interest
None.
References
- 1.Hausenloy DJ, Yellon DM. New directions for protecting the heart against ischaemia-reperfusion injury: targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway. Cardiovasc Res. 2004;61:448–460. doi: 10.1016/j.cardiores.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 2.Monassier JP. Reperfusion injury in acute myocardial infarction. From bench to cath lab. Part I: Basic considerations. Arch Cardiovasc Dis. 2008;101:491–500. doi: 10.1016/j.acvd.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 3.Bolli R, Becker L, Gross G, Mentzer R Jr, Balshaw D, Lathrop DA NHLBI Working Group on the Translation of Therapies for Protecting the Heart from Ischemia. Myocardial protection at a crossroads: the need for translation into clinical therapy. Circ Res. 2004;95:125–134. doi: 10.1161/01.RES.0000137171.97172.d7. [DOI] [PubMed] [Google Scholar]
- 4.Wei G, Guan Y, Yin Y, Duan J, Zhou D, Zhu Y, Quan W, Xi M, Wen A. Anti-inflammatory effect of protocatechuic aldehyde on myocardial ischemia/reperfusion injury in vivo and in vitro. Inflammation. 2013;36:592–602. doi: 10.1007/s10753-012-9581-z. [DOI] [PubMed] [Google Scholar]
- 5.Yang J, Jiang H, Yang J, Ding JW, Chen LH, Li S, Zhang XD. Valsartan preconditioning protects against myocardial ischemia-reperfusion injury through TLR4/NF-kappaB signaling pathway. Mol Cell Biochem. 2009;330:39–46. doi: 10.1007/s11010-009-0098-1. [DOI] [PubMed] [Google Scholar]
- 6.Park JL, Lucchesi BR. Mechanisms of myocardial reperfusion injury. Ann Thorac Surg. 1999;68:1905–1912. doi: 10.1016/s0003-4975(99)01073-5. [DOI] [PubMed] [Google Scholar]
- 7.Bozlu M, Eskandari G, Cayan S, Canpolat B, Akbay E, Atik U. The effect of poly (adenosine diphosphate-ribose) polymerase inhibitors on biochemical changes in testicular ischemiareperfusion injury. J Urol. 2003;169:1870–1873. doi: 10.1097/01.ju.0000049228.37887.4d. [DOI] [PubMed] [Google Scholar]
- 8.Han Z, Truong QA, Park S, Breslow JL. Two Hsp70 family members expressed in atherosclerotic lesions. Proc Natl Acad Sci U S A. 2003;100:1256–1261. doi: 10.1073/pnas.252764399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou H, Qian J, Li C, Li J, Zhang X, Ding Z, Gao X, Han Z, Cheng Y, Liu L. Attenuation of cardiac dysfunction by HSPA12B in endotoxin-induced sepsis in mice through a PI3K-dependent mechanism. Cardiovasc Res. 2011;89:109–118. doi: 10.1093/cvr/cvq268. [DOI] [PubMed] [Google Scholar]
- 10.Steagall RJ, Rusinol AE, Truong QA, Han Z. HSPA12B is predominantly expressed in endothelial cells and required for angiogenesis. Arterioscler Thromb Vasc Biol. 2006;26:2012–2018. doi: 10.1161/01.ATV.0000235720.61091.c7. [DOI] [PubMed] [Google Scholar]
- 11.Cui Z, Wang P, Sun L, Liu H, Yang J, Li X, Kang L, Huang Y, Shen A, Cheng C. Lipopolysaccharide-evoked HSPA12B expression by activation of MAPK cascade in microglial cells of the spinal cord. J Neurol Sci. 2010;294:29–37. doi: 10.1016/j.jns.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 12.Ma Y, Lu C, Li C, Li R, Zhang Y, Ma H, Zhang X, Ding Z, Liu L. Overexpression of HSPA12B protects against cerebral ischemia/reperfusion injury via a PI3K/Akt-dependent mechanism. Biochim Biophys Acta. 2013;1832:57–66. doi: 10.1016/j.bbadis.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 13.Kang L, Zhang G, Yan Y, Ke K, Wu X, Gao Y, Li J, Zhu L, Wu Q, Zhou Z. The role of HSPA12B in regulating neuronal apoptosis. Neurochem Res. 2013;38:311–320. doi: 10.1007/s11064-012-0922-y. [DOI] [PubMed] [Google Scholar]
- 14.Li J, Zhang Y, Li C, Xie J, Liu Y, Zhu W, Zhang X, Jiang S, Liu L, Ding Z. HSPA12B attenuates cardiac dysfunction and remodelling after myocardial infarction through an eNOS-dependent mechanism. Cardiovasc Res. 2013;99:674–684. doi: 10.1093/cvr/cvt139. [DOI] [PubMed] [Google Scholar]
- 15.Marsden PA, Schappert KT, Chen HS, Flowers M, Sundell CL, Wilcox JN, Lamas S, Michel T. Molecular cloning and characterization of human endothelial nitric oxide synthase. FEBS Lett. 1992;307:287–293. doi: 10.1016/0014-5793(92)80697-f. [DOI] [PubMed] [Google Scholar]
- 16.Li Q, Cai H. Induction of cardioprotection by small netrin-1-derived peptides. Am J Physiol Cell Physiol. 2015;309:C100–106. doi: 10.1152/ajpcell.00332.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qian GQ, Ding J, Zhang X, Yin X, Gao Y, Zhao GP. Preconditioning with glycyrrhizic, ferulic, paeoniflorin, cinnamic prevents rat hearts from ischemia/reperfusion injury via endothelial nitric oxide pathway. Pharmacogn Mag. 2015;11:292–296. doi: 10.4103/0973-1296.153081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shinmura K, Tamaki K, Ito K, Yan X, Yamamoto T, Katsumata Y, Matsuhashi T, Sano M, Fukuda K, Suematsu M, Ishii I. Indispensable role of endothelial nitric oxide synthase in caloric restriction-induced cardioprotection against ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2015;308:H894–903. doi: 10.1152/ajpheart.00333.2014. [DOI] [PubMed] [Google Scholar]
- 19.Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, Grimaldi PA, Kadowaki T, Lazar MA, O’Rahilly S, Palmer CN, Plutzky J, Reddy JK, Spiegelman BM, Staels B, Wahli W. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev. 2006;58:726–741. doi: 10.1124/pr.58.4.5. [DOI] [PubMed] [Google Scholar]
- 20.Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- 21.Kielstein JT, Bode-Boger SM, Hesse G, Martens-Lobenhoffer J, Takacs A, Fliser D, Hoeper MM. Asymmetrical dimethylarginine in idiopathic pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol. 2005;25:1414–1418. doi: 10.1161/01.ATV.0000168414.06853.f0. [DOI] [PubMed] [Google Scholar]
- 22.Martin-Nizard F, Furman C, Delerive P, Kandoussi A, Fruchart JC, Staels B, Duriez P. Peroxisome proliferator-activated receptor activators inhibit oxidized low-density lipoproteininduced endothelin-1 secretion in endothelial cells. J Cardiovasc Pharmacol. 2002;40:822–831. doi: 10.1097/00005344-200212000-00003. [DOI] [PubMed] [Google Scholar]
- 23.Bi R, Bao C, Jiang L, Liu H, Yang Y, Mei J, Ding F. MicroRNA-27b plays a role in pulmonary arterial hypertension by modulating peroxisome proliferator-activated receptor gamma dependent Hsp90-eNOS signaling and nitric oxide production. Biochem Biophys Res Commun. 2015;460:469–475. doi: 10.1016/j.bbrc.2015.03.057. [DOI] [PubMed] [Google Scholar]
- 24.Chi W, Meng F, Li Y, Wang Q, Wang G, Han S, Wang P, Li J. Downregulation of miRNA-134 protects neural cells against ischemic injury in N2A cells and mouse brain with ischemic stroke by targeting HSPA12B. Neuroscience. 2014;277:111–122. doi: 10.1016/j.neuroscience.2014.06.062. [DOI] [PubMed] [Google Scholar]
- 25.Yang Z, Day YJ, Toufektsian MC, Xu Y, Ramos SI, Marshall MA, French BA, Linden J. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation. 2006;114:2056–2064. doi: 10.1161/CIRCULATIONAHA.106.649244. [DOI] [PubMed] [Google Scholar]
- 26.Gao XM, Dart AM, Dewar E, Jennings G, Du XJ. Serial echocardiographic assessment of left ventricular dimensions and function after myocardial infarction in mice. Cardiovasc Res. 2000;45:330–338. doi: 10.1016/s0008-6363(99)00274-6. [DOI] [PubMed] [Google Scholar]
- 27.Weng D, Lu Y, Wei Y, Liu Y, Shen P. The role of ROS in microcystin-LR-induced hepatocyte apoptosis and liver injury in mice. Toxicology. 2007;232:15–23. doi: 10.1016/j.tox.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 28.Levine RL, Wehr N, Williams JA, Stadtman ER, Shacter E. Determination of carbonyl groups in oxidized proteins. Methods Mol Biol. 2000;99:15–24. doi: 10.1385/1-59259-054-3:15. [DOI] [PubMed] [Google Scholar]
- 29.Umemura T, Tasaki M, Kijima A, Okamura T, Inoue T, Ishii Y, Suzuki Y, Masui N, Nohmi T, Nishikawa A. Possible participation of oxidative stress in causation of cell proliferation and in vivo mutagenicity in kidneys of gpt delta rats treated with potassium bromate. Toxicology. 2009;257:46–52. doi: 10.1016/j.tox.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 30.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 31.Hu G, Tang J, Zhang B, Lin Y, Hanai J, Galloway J, Bedell V, Bahary N, Han Z, Ramchandran R, Thisse B, Thisse C, Zon LI, Sukhatme VP. A novel endothelial-specific heat shock protein HspA12B is required in both zebrafish development and endothelial functions in vitro. J Cell Sci. 2006;119:4117–4126. doi: 10.1242/jcs.03179. [DOI] [PubMed] [Google Scholar]
- 32.Karpe PA, Tikoo K. Heat shock prevents insulin resistance-induced vascular complications by augmenting angiotensin-(1-7) signaling. Diabetes. 2014;63:1124–1139. doi: 10.2337/db13-1267. [DOI] [PubMed] [Google Scholar]
- 33.Sharma S, Aramburo A, Rafikov R, Sun X, Kumar S, Oishi PE, Datar SA, Raff G, Xoinis K, Kalkan G, Fratz S, Fineman JR, Black SM. Lcarnitine preserves endothelial function in a lamb model of increased pulmonary blood flow. Pediatr Res. 2013;74:39–47. doi: 10.1038/pr.2013.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uryash A, Wu H, Bassuk J, Kurlansky P, Adams JA. Preconditioning with periodic acceleration (pGz) provides second window of cardioprotection. Life Sci. 2012;91:178–185. doi: 10.1016/j.lfs.2012.06.031. [DOI] [PubMed] [Google Scholar]
- 35.Garcia IM, Mazzei L, Benardon ME, Oliveros L, Cuello-Carrion FD, Gil Lorenzo A, Manucha W, Valles PG. Caveolin-1-eNOS/Hsp70 interactions mediate rosuvastatin antifibrotic effects in neonatal obstructive nephropathy. Nitric Oxide. 2012;27:95–105. doi: 10.1016/j.niox.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 36.Li H, Lu W, Cai WW, Wang PJ, Zhang N, Yu CP, Wang DL, Liu BC, Sun W. Telmisartan attenuates monocrotaline-induced pulmonary artery endothelial dysfunction through a PPAR gamma-dependent PI3K/Akt/eNOS pathway. Pulm Pharmacol Ther. 2014;28:17–24. doi: 10.1016/j.pupt.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 37.Polikandriotis JA, Mazzella LJ, Rupnow HL, Hart CM. Peroxisome proliferator-activated receptor gamma ligands stimulate endothelial nitric oxide production through distinct peroxisome proliferator-activated receptor gammadependent mechanisms. Arterioscler Thromb Vasc Biol. 2005;25:1810–1816. doi: 10.1161/01.ATV.0000177805.65864.d4. [DOI] [PubMed] [Google Scholar]
- 38.Yuen CY, Wong WT, Tian XY, Wong SL, Lau CW, Yu J, Tomlinson B, Yao X, Huang Y. Telmisartan inhibits vasoconstriction via PPARgamma-dependent expression and activation of endothelial nitric oxide synthase. Cardiovasc Res. 2011;90:122–129. doi: 10.1093/cvr/cvq392. [DOI] [PubMed] [Google Scholar]
- 39.Maejima Y, Okada H, Haraguchi G, Onai Y, Kosuge H, Suzuki J, Isobe M. Telmisartan, a unique ARB, improves left ventricular remodeling of infarcted heart by activating PPAR gamma. Lab Invest. 2011;91:932–944. doi: 10.1038/labinvest.2011.45. [DOI] [PubMed] [Google Scholar]