

Abstract
Protease-activated receptor-1 (PAR1) is a G protein-coupled receptor that undergoes proteolytic irreversible activation by coagulant and anti-coagulant proteases. Given the irreversible activation of PAR1, signaling by the receptor is tightly regulated through desensitization and intracellular trafficking. PAR1 displays both constitutive and agonist-induced internalization. Constitutive internalization of PAR1 is important for generating an internal pool of naïve receptors that replenish the cell surface and facilitate resensitization, whereas agonist-induced internalization of PAR1 is critical for terminating G protein signaling. We showed that PAR1 constitutive internalization is mediated by the adaptor protein complex-2 (AP-2), whereas AP-2 and epsin control agonist-induced PAR1 internalization. However, the mechanisms that regulate PAR1 recycling are not known. In the present study we screened a siRNA library of 140 different membrane trafficking proteins to identify key regulators of PAR1 intracellular trafficking. In addition to known mediators of PAR1 endocytosis, we identified Rab11B as a critical regulator of PAR1 trafficking. We found that siRNA-mediated depletion of Rab11B and not Rab11A blocks PAR1 recycling, which enhanced receptor lysosomal degradation. Although Rab11A is not required for PAR1 recycling, depletion of Rab11A resulted in intracellular accumulation of PAR1 through disruption of basal lysosomal degradation of the receptor. Moreover, enhanced degradation of PAR1 observed in Rab11B-deficient cells is blocked by depletion of Rab11A and the autophagy related-5 protein, suggesting that PAR1 is shuttled to an autophagic degradation pathway in the absence of Rab11B recycling. Together these findings suggest that Rab11A and Rab11B differentially regulate intracellular trafficking of PAR1 through distinct endosomal sorting mechanisms.
Keywords: autophagy, endothelial cell, lysosome, thrombin, trafficking, G protein-coupled receptor
Introduction
Protease-activated receptor-1 (PAR1)5 is a G protein-coupled receptor (GPCR) that elicits cellular responses to coagulant and anti-coagulant proteases (1, 2). Thrombin, the key effector protease of the coagulation cascade, mediates hemostasis, thrombosis, and inflammatory responses to vascular injury predominantly through PAR1 (3). The mechanisms by which proteases activate PAR1 and subsequent signal regulatory mechanisms are best understood for thrombin. Thrombin binds to and cleaves the N terminus of PAR1, revealing a new N-terminal domain that acts as a tethered ligand by binding intramolecularly to the receptor to initiate signaling and irreversibly activates the receptor (4, 5). In addition to rapid desensitization, trafficking of PAR1 is critical for regulating thrombin signaling and appropriate cellular responses. Unlike classic GPCRs, which are internalized and recycled back to the cell surface after agonist stimulation, activated PAR1 is internalized and sorted from endosomes to lysosomes and degraded. Internalization and lysosomal sorting is critical for terminating activated PAR1 signaling to heterotrimeric G proteins (6, 7). In addition to agonist-induced internalization, PAR1 exhibits constitutive internalization. Unactivated PAR1 cycles continuously between the cell surface and endosomes and generates an internal pool of naïve receptors that is important for replenishing the cell surface with uncleaved receptor, which facilitates cellular resensitization (8–10). Thus, endocytic trafficking of PAR1 is critical for the fidelity of thrombin signaling and appropriate cellular responses.
Previous studies have revealed novel mechanisms that control PAR1 endocytic trafficking. Although β-arrestins mediate desensitization and clathrin-dependent internalization of many GPCRs (11), β-arrestins are only required for PAR1 desensitization and not internalization. After activation, PAR1 signaling is rapidly desensitized by β-arrestins, whereas internalization of PAR1 occurs through a clathrin- and dynamin-dependent pathway independent of β-arrestins (12). Rather than β-arrestins, the clathrin adaptor protein complex-2 (AP-2) mediates constitutive internalization of unactivated PAR1, where the μ2 adaptin subunit binds directly to a tyrosine-based motif localized within the C-tail region (9). Interestingly, AP-2 and epsin are both required for agonist-promoted internalization of PAR1. AP-2 regulates activated PAR1 internalization via recognition of distal C-tail phosphorylation sites and not the tyrosine-based motif, whereas epsin requires its ubiquitin binding motif and ubiquitination of the receptor to promote internalization (13). However, whether other membrane trafficking proteins also regulate constitutive or agonist-induced internalization of PAR1 is not known.
Although constitutive internalization of PAR1 is important for formation of an internal pool of naïve receptors, recycling of PAR1 is critical for re-establishing appropriate amounts of naïve receptors at the cell surface for rapid resensitization (9). However, the mechanisms that mediate recycling of PAR1 from endosomes to the cell surface have not been defined. Most internalized GPCRs recycle back to the cell surface by either bulk membrane flow or through a regulated process mediated by a sequence-dependent interaction with adaptor proteins and actin (14). Recycling of classic GPCRs serves to return de-activated receptors to the cell surface, a process important for cellular resensitization. However, in contrast to constitutive recycling of unactivated PAR1, most classic GPCRs recycle after agonist-induced internalization. Previous studies indicate that the small G protein Rab11 is a key regulator of GPCR vesicle recycling (15). Rab11 proteins interact with adaptor and distinct motor proteins to guide vesicles containing GPCRs along microtubule tracks or actin-filaments to distinct subcellular compartments (16). Three Rab11 proteins are encoded in the mammalian genome including Rab11A, Rab11B, and Rab11C (also known as Rab25) and share high sequence identity. Rab11A is ubiquitously expressed, whereas Rab11B and Rab25 exhibit tissue-specific expression. Rab11A has been implicated in constitutive recycling of the thromboxane receptor-β (17, 18) as well as recycling of agonist-induced internalized β2-adrenergic receptor (19) and the prostacyclin receptor (20) and other GPCRs (15). These studies relied on ectopic expression of wild type and dominant-negative Rab11A to assess function in GPCR recycling. In contrast to Rab11A, the role of Rab11B in GPCR recycling remains elusive, and it is unclear if Rab11B has a distinct function from Rab11A in GPCR recycling in mammalian cells. Interestingly, Rab25 has been implicated in ovarian, breast, and colon cancer (21) and GPCR overexpression resulting from aberrant trafficking contributes to aspects of cancer pathogenesis (6, 22), but whether Rab25 or Rab11 is linked to GPCR trafficking in normal or cancer cells has not been reported.
In this study we sought to identify key mediators that regulate PAR1 trafficking by screening a membrane trafficking siRNA library targeting 140 different proteins. Here, we report that in addition to known regulators of PAR1 internalization including AP-2, epsin, clathrin, and dynamin, Rab11B emerged as a critical regulator of PAR1 expression. We further established that Rab11B and not Rab11A is required for constitutive recycling of PAR1. Moreover, inhibition of PAR1 constitutive recycling by Rab11B knockdown enhanced basal degradation of the receptor at the lysosome. In contrast to Rab11B, we found that Rab11A controls PAR1 lysosomal transport and degradation under basal conditions. In fact, sequential depletion of Rab11B followed by Rab11A restored PAR1 expression in cells deficient in Rab11B expression. These findings suggest that Rab11B and Rab11A function at distinct sites to regulate PAR1 recycling and endosomal-lysosomal sorting, respectively. We also discovered that enhanced degradation of PAR1 observed in Rab11B-deficient cells requires the autophagy-related-5 (ATG5) protein, which mediates formation of autophagosomes, a double membrane vacuole that fuses with lysosomes generating autolysosomes that degrades proteins. Together, this study demonstrates divergent roles for Rab11A and Rab11B in PAR1 trafficking and is the first to show that PAR1 can transit through an autophagic degradation pathway.
Experimental Procedures
Reagents and Antibodies
The PAR1 agonist peptide SFLLRN was synthesized as the carboxyl amide and purified by reverse-phase high pressure liquid chromatography at Tufts University Core Facility (Boston, MA). α-Thrombin was from Enzyme Research Laboratories (South Bend, IN). Rabbit polyclonal anti-FLAG antibody (#600-401-383) was from Rockland Immunochemicals (Pottstown, PA). Mouse monoclonal anti-PAR1 WEDE antibody (#IM2085) was from Beckman Coulter (Fullerton, CA). Monoclonal M1 anti-FLAG (#F3040) and actin antibodies (#A5316) were from Sigma. Rabbit monoclonal anti-EGF receptor (EGFR) (#CS 2232), lysosomal-associated membrane protein-1 (LAMP1)(# CS 9091P), autophagy light chain isoform B (LC3B) (#CS2775), p38 (#CS 9212), phospho-p38 (#CS 4511), Rab11A (#CS2413), and Rab11B (#CS2414) antibodies were from Cell Signaling Technologies (Beverly, MA). Polyclonal rabbit ATG5 (#GTX62601) and monoclonal glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (#GTX10118)antibodies were from GeneTex (Irvine, CA). Mouse monoclonal ErbB2/HER2 (#MS 1072) antibody was from Neomarkers (Fremont, CA). HRP-conjugated goat-anti rabbit (#170-6516) and -anti mouse (#170-6515) antibodies were from Bio-Rad. Goat anti-mouse IgG (#31160), goat anti rabbit AlexaFluor® 488 (#A11008) and 594 (#A11001) secondary antibodies were from Life Technologies (Thermo Fisher Scientific) (Waltham, MA). Polyethyleneimine was purchased from Polysciences Inc. (Warrington, PA).
Cell Lines and cDNAs
HeLa cells were grown in DMEM containing 10% (v/v) fetal bovine serum with 250 μg/ml hygromycin. Human umbilical vein endothelial cell-derived EA.hy926 cells were grown and cultured as previously described (23). MDA-MB-231 cells were obtained from American Type Culture Collection (Manassas, VA) and maintained according to their instructions. N-terminal FLAG-tagged human PAR1 wild type and 0K mutant, in which all cytoplasmic lysines (Lys) were mutated to arginine (Arg) was cloned into pBJ vector and stably expressed in HeLa cells as described (24–26).
Cell Transfections
Cells were seeded at 2.5 × 104 on fibronectin coated 24-well plates and grown overnight. Cells were then transfected with siRNA using Oligofectamine (Life Technologies) per the manufacturer's instructions. The human membrane trafficking siRNA library composed of siRNA SMARTpools targeting 140 different proteins was purchased from Dharmacon (GE Healthcare). The individual nonspecific (ns) siRNA (5′-CUACGUCCAGGAGCGCACC-3′), μ2 siRNA (5′-GUGGAUGCCUUUCGGGUCA-3′). Rab11B siRNA (5′-AAUCGCCAAGCACCUGACCTA-3′), Rab11A siRNA (5′-AAGAGUAAUCUCCUGUCUCGA-3′) were purchased from Qiagen (Germantown, MD). ON-TARGETplus ATG5 (#7, 5′-GCAUUAUCCAAUUGGUUUGCU-3′; #8, 5′-AUACUAUUUGCUUUUGCCAAG-3′; #9, 5′-UGACAGAUUUGACCAGUUUUG-3′; #10, 5′-CAAAGAUGUGCUUCGAGAUGU-3′) was purchased from Thermo Fisher Scientific.
siRNA Library Screen
HeLa cells stably expressing FLAG-PAR1 wild type were seeded at ∼10,000 cells per well of 48-well plates and grown overnight. Cells were washed and then transfected with each siRNA SMARTpool at 50 nm diluted in Opti-MEM in duplicate. Cells were transfected with 50 nm ns- and μ2-adaptin siRNA in parallel as controls. After 24 h, transfection media was replaced with normal growth media, and cells were grown for an additional 48 h.
Cell Surface ELISA
The expression of FLAG-tagged PAR1 wild type or 0K mutant and endogenous PAR1 was measured by ELISA essentially as described (27). Cells were fixed with 4% paraformaldehyde, washed, and incubated with anti-FLAG antibody or anti-PAR1 antibody and followed by incubation with species-specific secondary antibody conjugated to HRP. To determine the amount of antibody bound to PAR1 at the cell surface, cells were washed and incubated with the HRP substrate one-step 2,2′-azinobis-3-ethylbenzthiazoline-6-sulfonic acid (Thermo Fisher Scientific) for 10–20 min at room temperature. An aliquot was removed, and the absorbance at 405 nm was measured using a Molecular Devices SpectraMax Plus microplate reader.
Immunofluorescence Confocal Microscopy
PAR1 expressing HeLa cells were plated at 4.0 × 104 cells per well of 24-well plates containing fibronectin-coated glass coverslips and grown overnight. Cells were transfected, grown for an additional 48 h, fixed in 4% paraformaldehyde, and permeabilized with methanol. Cells were processed and immunostained with the indicated antibodies as described (28). For PAR1 and Rab11B colocalization, cells were incubated with anti-FLAG antibody for 1 h at 37 °C to allow labeling and constitutive internalization of PAR1, fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 diluted in PBS, and then incubated with anti-Rab11B antibody overnight at 4 °C. Cells were then washed and incubated with species-specific secondary antibodies conjugated to AlexaFluor. In some experiments cells were pretreated with 2 mm leupeptin for 24 h at 37 °C to inhibit lysosomal degradation. After processing, coverslips were mounted, and cells were imaged by confocal microscopy using an Olympus IX81 DSU spinning disk confocal microscope fitted with a PlanApo 60× oil objective and a Hamamatsu ORCA-ER digital camera. Fluorescent images of x-y 0.28-μm sections were collected sequentially using SlideBook 5.0 software. Line-scan analysis was performed to assess colocalization using SlideBook 5.0 software.
Immunoblotting
HeLa cells expressing PAR1 were seeded at 2.5 × 104 cells per well of 24-well plates, transfected, and grown as described above. In some experiments cells were pretreated with 2 mm leupeptin for 24 h or 10 μm cycloheximide for 30 min at 37 °C before lysis. MDA-MB-231 cells were plated at 8.0 × 104 cells per well of 24-well plates, transfected, and grown as described above. In some experiments cells were serum-starved for 1 h. Cells were then lysed in 1× Laemmli sample buffer containing 100 mm DTT and sonicated at 10% amplitude, and equivalent amounts of cell lysates were resolved by SDS-PAGE, transferred to PVDF membranes, and incubated with specific antibodies as indicated. Membranes were developed by chemiluminescence and quantified by densitometry using ImageJ software.
Immunoprecipitation of PAR1
Immunoprecipitation of endogenous PAR1 was performed as recently described (29). Human cultured EA.hy926 cells and MDA-MB-231 cells were grown in 6-cm dishes, serum-starved, and then lysed in Triton lysis buffer containing 50 mm Tris-HCl, pH 7.4, 100 mm NaCl, 1% Triton X-100, 5 mm EDTA, 50 mm NaF, 50 mm β-glycerophosphate, supplemented with 10 μg/ml leupeptin, aprotinin, trypsin protease inhibitor, and pepstatin, 100 μg/ml benzamide, and 20 mm N-ethylmaleimide. Cell lysates were passed through a 21-gauge needle and cleared by centrifugation, and protein concentrations were determined by bicinchoninic acid assay. Equivalent amounts of lysates were used for immunoprecipitations using the anti-PAR1 WEDE antibody, and samples were eluted with 2× Laemmli sample buffer containing 200 mm DTT, resolved by SDS-PAGE, and developed by chemiluminescence. Aliquots of cell lysates were also immunoblotted with specific antibodies as indicated.
Constitutive Internalization
PAR1 constitutive internalization was performed essentially as described (12, 30). HeLa cells expressing FLAG-PAR1 were seeded at 0.7 × 105 cells per well of 24-well plates grown overnight and transfected with siRNA. After 24 h of transfection, cells were pre-labeled with M1 anti-FLAG antibody on ice, washed, and warmed to 37 °C to allow internalization of antibody labeled PAR1 for various times. Antibody remaining on the cell surface was removed by washing with PBS (Ca2+- and Mg2+-free) containing 0.04% EDTA. Cells were then lysed in Triton lysis buffer (described above), and the amount of internalized antibody-PAR1 was measured by a sandwich ELISA. Briefly, 96-well plates were coated with goat anti-mouse antibody, cell lysates were applied and incubated for 2 h at room temperature, and plates were washed and then incubated with HRP-coupled goat anti-mouse antibody. The amount of antibody-bound PAR1 was then detected by incubation with HRP substrate 2,2′-azinobis-3-ethylbenzthiazoline-6-sulfonic acid for 10–20 min at room temperature, and the absorbance at 405 nm was measured using a Molecular Devices SpectraMax Plus microplate reader.
Biotinylation Recycling Assay
Biotinylation and recycling of PAR1 was determined as previously described (31). HeLa cells expressing FLAG-PAR1 were seeded at 1.4 × 105 cells per well of 12-well plates grown overnight and transfected with siRNA. After 24 h of transfection, cells were labeled on ice with 0.3 mg/ml EZ Link Sulfo-NHS-SS-biotin (Thermo Fisher Scientific) diluted in PBS on ice for 30 min. Cells were then either left on ice or incubated in media at 37 °C for 30 min to allow constitutive internalization of receptors. The remaining biotin on the cell surface was removed by incubation with ice-cold PBS, pH 8.7, containing 50 mm glutathione, 75 mm NaCl, 75 mm NaOH, and 1% BSA for 10 min, washed with ice-cold PBS, and then re-incubated in media at 37 °C for 15 min to allow receptor recycling. Biotin was then stripped from recycled receptors as described above leaving a “protected” intracellular pool of biotinylated PAR1. Cells were then lysed in Triton lysis buffer (50 mm Tris HCl, pH 7.4, 0.5% Triton X-100, 250 mm NaCl, 5 mm EDTA, 10 μg/ml leupeptin, aprotinin, trypsin protease inhibitor, pepstatin, and 100 μg/ml benzamide). Equivalent amounts of cell lysates were incubated with streptavidin-conjugated agarose beads (Thermo Fisher Scientific), and biotinylated proteins were eluted in 2× Laemmli sample buffer containing 0.2 m DTT, resolved by SDS-PAGE, transferred, and developed by chemiluminescence.
Data Analysis
Data were analyzed using Prism 4.0 software (GraphPad Software, La Jolla, CA), and statistical significance was determined using two-way analysis of variance (ANOVA).
Results
A Membrane Trafficking siRNA Library Screen Identifies Rab11B as a Regulator of PAR1 Surface Expression
We previously showed that depletion of the μ2 adaptin subunit of AP-2 by siRNA blocks PAR1 constitutive and agonist-induced internalization through clathrin-coated pits (9, 13). To identify additional proteins that regulate PAR1 trafficking, a library containing siRNA SMART pools targeting 140 different membrane trafficking proteins was screened. The effect of μ2 adaptin subunit depletion on PAR1 trafficking was examined in parallel as a control. HeLa cells stably expressing FLAG-tagged PAR1 were transiently transfected with nonspecific (ns), μ2 adaptin, or SMARTpool siRNAs. Cells were then left untreated or treated with the PAR1 peptide agonist SFLLRN for 15 min, and the amount of PAR1 remaining on the cell surface was quantified by ELISA. As expected, depletion of the AP-2 α, β, and μ1 adaptin subunits and clathrin light chain caused a significant increase in PAR1 surface expression similar to depletion of the AP-2 μ2 adaptin subunit in both untreated control and agonist-treated cells (Fig. 1, A and B, Table 1). We also observed a substantial increase in PAR1 surface expression in untreated control cells depleted of disabled homolog-2 (DAB2), whereas loss of ALIX (PDCD6IP) caused a modest change in PAR1 surface expression (Fig. 1A and Table 1). Data from the entire siRNA membrane trafficking screen are shown in supplemental Table S1. These findings are consistent with a critical role for AP-2 and clathrin in constitutive and agonist-induced internalization of PAR1 as previously reported (9, 13). Intriguingly, siRNA-mediated depletion of several notable trafficking proteins in untreated control cells including inositol hexakisphosphate kinase-3 (IHPK3), Rab11B, a RAB11B interacting protein GAF, and calcium and integrin binding protein-2 (CIB2) caused a significant decrease in PAR1 surface expression (Fig. 1, A and B, Table 1). Because Rabs are known to function in receptor trafficking and a role for Rab11B in regulation of GPCR recycling had not been previously described, we focused our studies on understanding the underlying mechanisms by which Rab11B regulates PAR1 intracellular trafficking.
FIGURE 1.
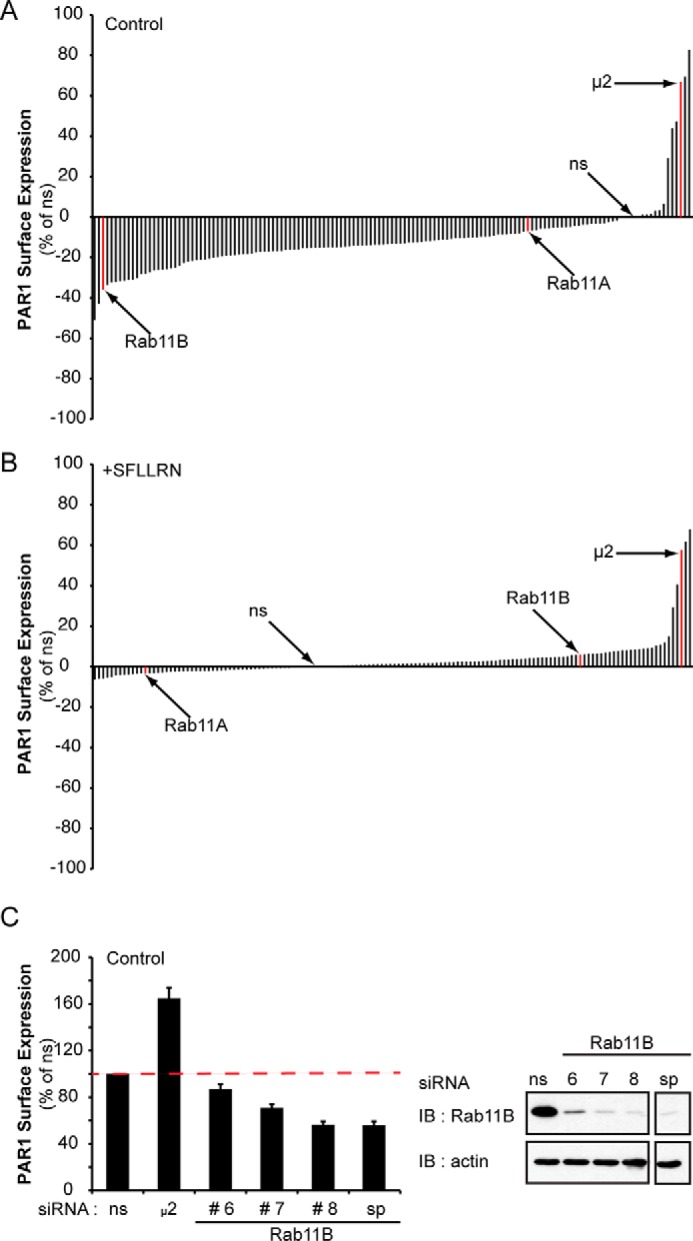
A siRNA library screen reveals an important role for Rab11B in regulation of PAR1 surface expression. A and B, HeLa cells stably expressing FLAG-tagged PAR1 were transfected with ns, μ2 adaptin subunit siRNA, or SMARTpool siRNAs targeting 140 distinct membrane trafficking proteins. After transfection, cells were either left untreated (Control) or treated with 100 μm SFLLRN (PAR1 agonist peptide) for 15 min at 37 °C. Cells were fixed, and the amount of PAR1 remaining on the cell surface was quantified by ELISA. The data (mean, n = 2) are representative of two independent experiments performed in duplicate and are expressed as the percent of ns siRNA control. C, PAR1-expressing HeLa cells were transfected with ns, μ2 adaptin, Rab11B #6, #7, and #8 siRNAs, or the Rab11B SMARTpool (sp) siRNAs, and PAR1 cell surface expression was determined by ELISA. Data (mean ± S.D., n = 3) are expressed as the percent of ns siRNA control. The inset is a representative immunoblot (IB) of cell lysates from similar siRNA transfections probed with anti-Rab11B or anti-actin antibodies.
TABLE 1.
PAR1 surface expression
The data represented in this table lists the targeted proteins that caused the largest increase or decrease in PAR1 surface expression relative to the ns control. AP2A1, adaptor-related protein complex 2, α1 subunit; AP2B1, adaptor-related protein complex 2, β1 subunit; AP1M1, adaptor-related protein complex 1, μ1 subunit; AP2M1, adaptor-related protein complex 2, μ1 subunit, μ2 = control and adaptor-related protein complex 2, μ2 subunit; CIB2, calcium and integrin binding family member 2; CLTB, clathrin light chain B; CLTC, clathrin, heavy chain; CLTCL1, clathrin, heavy chain-like 1; DAB2, disabled homolog 2; DNM2, dynamin-2; EPN, epsin 2; GAF1, Rab11 family-interacting protein 5; IHPK3, inositol hexakisphosphate kinase 3; RAB11B, Ras-related protein Rab-11B; PDCD6IP, programmed cell death 6-interacting protein/ALIX; PICALM, phosphatidylinositol binding clathrin assembly protein; VCP, transitional endoplasmic reticulum ATPase/valosin-containing protein. See also Fig. 1, A and B, and supplemental Table S1.
| Control, PAR1 increase |
Control, PAR1 decrease |
SFLLRN, PAR1 increase |
|||
|---|---|---|---|---|---|
| Gene ID | % of ns | Gene ID | % of ns | Gene ID | % of ns |
| AP2M1 | 82.76% | IHPK3 | −51.08% | CLTC | 61.12% |
| AP2A1 | 69.35% | VCP | −42.95% | AP2M1 | 57.03% |
| μ2 (Ctrl) | 66.88% | RAB11B | −35.90% | μ2 (Ctrl) | 40.55% |
| CLTC | 47.30% | CLTCL1 | −33.57% | AP2A1 | 28.85% |
| AP2B1 | 43.85% | GAF1 | −32.39% | VCP | 14.40% |
| DAB2 | 29.02% | CIB2 | −32.16% | AP2A1 | 11.37% |
| AP1M1 | 6.49% | CLTB | −31.63% | EPN2 | 10.30% |
| PDCD6IP | 3.23% | PICALM | −31.52% | DNM2 | 9.86% |
Rab11A has been reported to regulate mammalian GPCR recycling (17, 19); however, a role for Rab11B in GPCR trafficking has not been described. Moreover, neither Rab11A nor Rab11B has been previously reported to regulate PAR1 trafficking. To confirm that the effect observed with Rab11B siRNA SMARTpools on PAR1 surface expression was specific, the individual Rab11B siRNAs were examined. HeLa cells stably expressing PAR1 were transiently transfected with individual or SMARTpool Rab11B siRNAs and in parallel with siRNA-targeting the μ2-adaptin subunit. A marked reduction in PAR1 surface expression was observed in cells transfected with Rab11B #8 siRNA similar to that observed with SMARTpool siRNAs, whereas depletion of μ2 adaptin subunit resulted in increased PAR1 surface expression as expected (Fig. 1C). To delineate the mechanism by which RAB11B controls PAR1 expression, the Rab11B #8 siRNA was used in all subsequent experiments.
Rab11A and Rab11B Differentially Regulate PAR1 Expression
Next, we examined the effect of Rab11B versus Rab11A individual siRNAs on PAR1 expression. PAR1-expressing HeLa cells were transfected with ns, Rab11A, or Rab11B siRNAs, and the amount of receptor remaining on the cell surface was examined by ELISA. Depletion of Rab11B caused a significant ∼46% reduction in PAR1 surface expression, whereas only a modest ∼14% decrease was observed in Rab11A-transfected cells (Fig. 2A). We also determined whether loss of PAR1 surface expression correlated with changes in expression at the level of receptor protein. Depletion of Rab11B caused a significant ∼39% loss of PAR1 protein (Fig. 2B). However, in contrast to Rab11B, depletion of Rab11A resulted in a significant ∼44% increase in total PAR1 protein expression (Fig. 2B) despite having a minimal effect on receptor surface expression (Fig. 2A). Immunofluorescence confocal microscopy was next used to determine whether the effect of Rab11A versus Rab11B on PAR1 expression was due to accumulation of the receptor in an intracellular compartment. Under basal conditions, constitutive internalization of PAR1 results in the formation of an intracellular pool of receptors (9), which is evident in control ns siRNA-transfected cells (Fig. 2C, top panels). However, cells depleted of Rab11B showed a reduction in PAR1-positive puncta compared with ns siRNA-transfected cells (Fig. 2C, top panels), whereas cells deficient in Rab11A expression displayed a marked accumulation of PAR1 intracellular vesicles (Fig. 2C, top panels).
FIGURE 2.
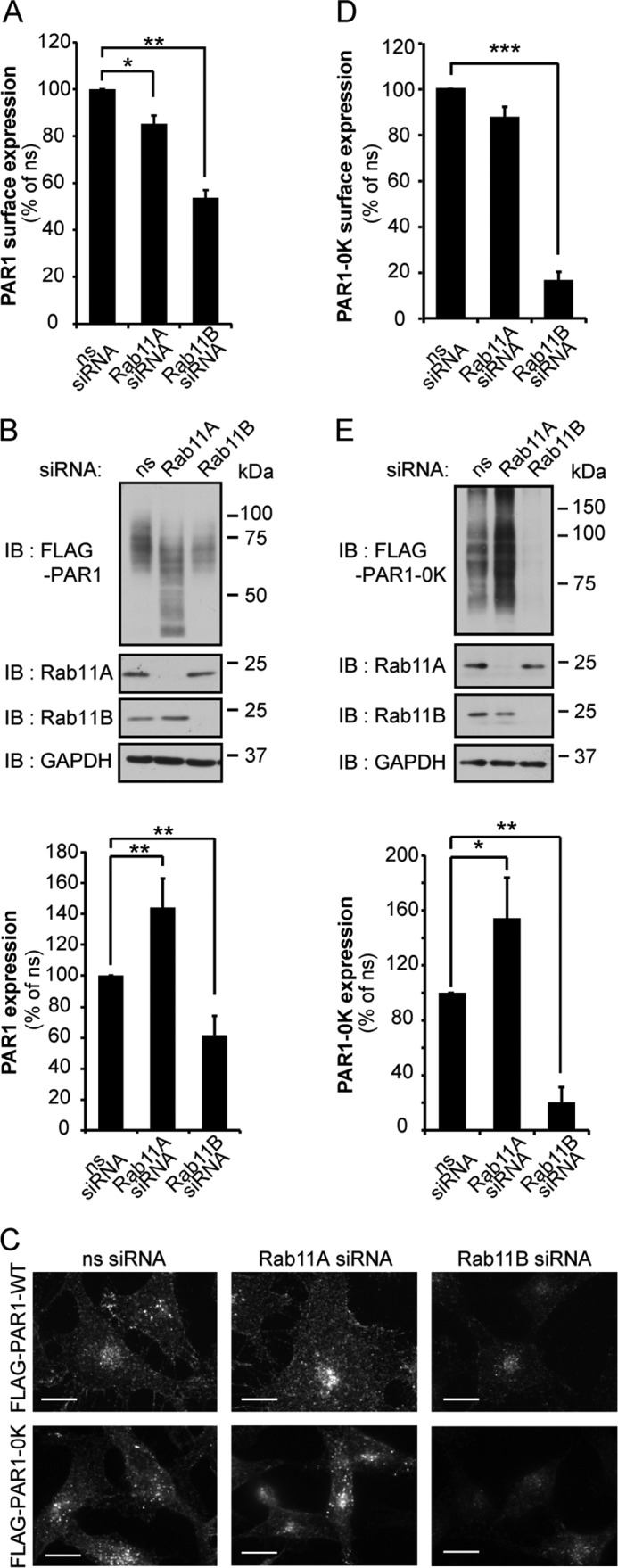
PAR1 expression is differentially regulated by Rab11A and Rab11B. A and B, HeLa cells stably expressing FLAG-PAR1 or FLAG-PAR1 0K mutant were transfected ns, Rab11A, or Rab11B siRNAs for 72 h. A and D, HeLa cells were fixed, and PAR1 wild type and 0K mutant surface expression was quantified by ELISA. Data (mean ± S.D., n = 3) are representative of three independent experiments, expressed as the percent of ns siRNA control, and were analyzed by ANOVA (*, p < 0.05; **, p < 0.01; ***, p < 0.001). B and E, HeLa cell lysates were immunoblotted (IB) for PAR1 wild type and 0K mutant protein expression using anti-FLAG antibodies. Data (mean ± S.D., n = 3) are representative of three separate experiments, expressed as the percent of ns siRNA control, and were analyzed by ANOVA (*, p < 0.05; **, p < 0.01). Cell lysates were also probed with anti-Rab11A, -Rab11B, and -GAPDH antibodies as controls. C, HeLa cells were fixed, permeabilized, and immunostained with anti-FLAG antibody to detect PAR1 wild type and 0K mutant expression. Similar results were observed in three different experiments. Scale bar, 10 μm.
PAR1 is basally ubiquitinated (25), and although PAR1 ubiquitination is enhanced after agonist stimulation (29), the activated receptor undergoes ubiquitin-independent lysosomal sorting (28). To determine if differential regulation of PAR1 expression by Rab11A and Rab11B requires ubiquitination, we used the PAR1 0K mutant, in which all cytoplasmic lysines were converted to arginines, rendering it ubiquitin-deficient (25). Similar to wild type PAR1, cells deficient in Rab11B expression exhibited a marked decrease in PAR1 0K mutant surface expression (Fig. 2D), whereas surface expression was not significantly effected in Rab11A-depleted cells. PAR1 0K mutant protein expression was also significantly decreased in Rab11B-deficient cells and increased in Rab11A-deficient cells (Fig. 2E), like that observed with wild type PAR1. Immunofluorescence microscopy studies were consistent with enhanced degradation of PAR1 0K mutant in Rab11B-depleted cells and receptor accumulation in Rab11A-deficient cells (Fig. 2C, lower panels). These results suggest that differential regulation of PAR1 expression by Rab11A and Rab11B occurs independent of receptor ubiquitination.
To determine whether Rab11A and Rab11B differentially regulate endogenous PAR1, human cultured endothelial cells were examined. HUVEC (human umbilical vein endothelial cell)-derived EA.hy926 endothelial cells were transiently transfected with ns, Rab11A, or Rab11B siRNAs, and the expression of PAR1 was determined. Depletion of Rab11B caused a significant ∼42% decrease in PAR1 surface expression compared with ns siRNA-transfected control cells (Fig. 3A), whereas only a ∼13% reduction in PAR1 surface expression was detected in Rab11A-deficient cells (Fig. 3A). The expression of PAR1 protein was also significantly decreased in Rab11B-depleted cells compared with control siRNA-transfected cells (Fig. 3B, lane 3). However, in cells deficient in Rab11A, PAR1 protein expression was markedly increased (Fig. 3B, lane 1). The differential effects of Rab11A and Rab11B on endogenous PAR1 expression are consistent with those observed in HeLa cells.
FIGURE 3.

Endogenous PAR1 expression is differentially regulated by Rab11A and Rab11B in endothelial and breast cancer cells. A and C, human endothelial EA.hy926 cells or MDA-MB-231 breast carcinoma cells were transfected with ns, Rab11A, or Rab11B siRNAs for 72 h. Cells were fixed, and endogenous PAR1 cell surface expression was quantified by ELISA. Data (mean ± S.D., n = 3) are representative of at least three independent experiments, expressed as the percent of ns siRNA control, and analyzed by ANOVA (*, p < 0.05; **, p < 0.01; ***, p < 0.001). B, D, E, and F, endothelial cells and MDA-MB-231 cells transfected as described above were lysed, equivalent amounts of cell lysates were immunoprecipitated (IP), and PAR1 protein was detected by immunoblotting (IB) with anti-PAR1 antibody. Cell lysates were probed for Rab11A, Rab11B, GAPDH, ErbB2, and EGFR expression using specific antibodies. Data (mean ± S.D., n = 3) are representative of three different experiments, expressed as the percent of ns siRNA control and were analyzed by ANOVA (*, p < 0.05; **, p < 0.01; ***, p < 0.001). G, MDA-MB-231 cells transfected with siRNA were stimulated with α-thrombin (α-Th) for the indicated times and lysed, and equivalent amounts of cell lysates were immunoblotted to detect phosphorylated (phospho) p38, p38, Rab11A, and Rab11B. Data (mean ± S.D., n = 3) are representative of at least three independent experiments, expressed as the percent of ns siRNA control and were analyzed by ANOVA (***, p < 0.001).
Given the importance of Rabs in regulating PAR1 expression, we next examined if Rab11A and Rab11B differentially regulate PAR1 expression in invasive MDA-MB-231 breast carcinoma cells. Dysregulated trafficking results in increased expression of PAR1, persistent transactivation of ErbB family members, breast carcinoma invasion, and tumor growth (6, 32). Overexpression of ErbB2 and EGFR is also correlated with increased metastatic potential (33, 34). We, therefore, examined if Rab11A or Rab11B regulated PAR1 or ErbB family member expression by depleting MDA-MB-231 cells of these proteins using siRNA. The amount of endogenous PAR1 cell surface expression was substantially reduced by ∼50% in Rab11B-deficient cells but only an ∼15% reduction of PAR1 was observed in Rab11A-depleted cells (Fig. 3C). PAR1 protein expression was also markedly reduced by ∼60% in cells lacking Rab11B expression and modestly increased in Rab11A-deficient cells (Fig. 3D). Interestingly, ErbB2 exhibited a similar change in protein expression and was decreased by ∼30% in Rab11B-deficient cells and substantially increased in Rab11A-depleted cells, whereas EGFR expression remained unchanged (Fig. 3, D, E, and F). These data suggest that Rab11A and Rab11B differently regulate PAR1 expression through distinct pathways in multiple cell types. We next examined whether PAR1 signaling was altered in Rab11B-depleted MDA-MB-231 cells. Thrombin activation of PAR1 induced a significant ∼7-fold increase in p38 phosphorylation at 5 min in MDA-MB-231 cells transfected with non- or Rab11A-specific siRNAs (Fig. 3G), whereas thrombin-stimulated p38 activation was reduced significantly in cells lacking Rab11B expression (Fig. 3G). These data suggest that the expression of PAR1 at the cell surface is important for promoting robust p38 signaling in MDA-MB-231breast carcinoma cells.
Rab11B and Not Rab11A Regulates PAR1 Recycling
To understand mechanistically how Rab11B controls PAR1 expression, constitutive internalization and recycling of the receptor was examined. In these experiments, FLAG-PAR1-expressing HeLa cells were transfected with ns, Rab11A, or Rab11B siRNA for 24 h to retain sufficient PAR1 expression at the cell surface. Under these conditions Rab11B expression was ablated, whereas PAR1 surface and protein expression were modestly reduced by 11 and 13%, respectively (Fig. 4, A and B). Rab11A-deficient cells also exhibited no changes in PAR1 surface expression (Fig. 4A); however, receptor protein expression was significantly increased (Fig. 4B). To assess constitutive internalization, the surface cohort of PAR1 was prelabeled with anti-FLAG antibody at 4 °C, and constitutive internalization was determined as described (9). The rate of PAR1 constitutive internalization was comparable in ns and Rab11A- and Rab11B siRNA-transfected cells (Fig. 4C). These results suggest that the observed effects of Rab11A and Rab11B on PAR1 expression are not due to defects in constitutive internalization of the receptor.
FIGURE 4.

Rab11B regulates PAR1 constitutive recycling but not internalization. HeLa cells expressing FLAG-PAR1 were transfected with ns and Rab11A or Rab11B siRNAs for 24 h. A, HeLa cells were fixed, and PAR1 surface expression was determined by ELISA. Data (mean ± S.D., n = 3) are representative of three independent experiments, expressed as the percent of ns siRNA control, and were analyzed by ANOVA (*, p < 0.05). B, PAR1 protein expression was determined from HeLa cell lysates by immunoblotting (IB) with anti-FLAG antibody. Data (mean ± S.D., n = 3) are representative of at least three different experiments, expressed as the percent of ns siRNA control, and were analyzed by ANOVA (*, p < 0.05; ***, p < 0.001). The expression of Rab11A, Rab11B, and GAPDH were determined in cell lysates using the indicated antibodies as controls. C, HeLa cells were prelabeled with anti-FLAG antibody on ice, washed, and warmed to 37 °C for various times to facilitate PAR1 constitutive internalization. Cells were lysed, and internalized PAR1 was detected using a sandwich ELISA. Data (mean ± S.D., n = 3) are representative of three independent experiments, expressed as the percent of ns siRNA control, and were analyzed by ANOVA. D, HeLa cells were labeled with biotin at 4 °C, processed to allow constitutive internalization and recycling of biotinylated PAR1 as described under “Experimental Procedures.” Streptavidin pulldown assays were immunoblotted with anti-PAR1 antibody to detect the internal pool of PAR1. The expression of PAR1, Rab11A, Rab11B, and GAPDH in cell lysates was detected by immunoblotting using the indicated antibodies. Data (mean ± S.D., n = 3) are representative of three independent experiments, expressed as the percent of ns siRNA control, and were analyzed by ANOVA (***, p < 0.001). E, HeLa cells were incubated with anti-FLAG antibody for 1 h at 37 °C to label the internal pool of PAR1 (red). Cells were then fixed and immunolabeled with anti-Rab11B antibody (green). Colocalization is revealed by the yellow color in the merged image and is demarcated by arrowheads. The images are representative of many cells examined in three independent experiments. Insets are magnification of boxed areas. Scale bar = 10 μm. F, line scan analysis of the magnified boxed areas are plotted as pixel intensity in arbitrary units (ADU) versus pixel number (distance) and demonstrates colocalization between PAR1 and Rab11B at puncta in the merged image.
We next examined whether Rab11A or Rab11B regulated PAR1 recycling using a receptor biotinylation assay as we previously described (31). Surface PAR1 was labeled with biotin at 4 °C in cells transfected with ns, Rab11A, or Rab11B siRNAs and then warmed to 37 °C to facilitate constitutive internalization. After PAR1 constitutive internalization, surface biotin was removed, and cells were then either lysed (designated as 0 min) or returned to 37 °C for 15 min to allow receptor recycling. Cells were then chilled to 4 °C to block recycling, surface biotin was removed, and internalized biotinylated PAR1 was captured by streptavidin pulldown and detected by immunoblotting. A substantial amount of biotinylated PAR1 was detected in the internal pool at 0 min in transfected cells under all conditions (Fig. 4D, lanes 1, 3, and 5). These findings are consistent with PAR1 constitutive internalization from the cell surface to an intracellular compartment. However, in control ns siRNA-transfected cells, the amount of biotinylated PAR1 remaining in the internal pool after warming for 15 min to 37 °C was markedly reduced (Fig. 4D, lane 2), suggesting that the majority of internalized PAR1 recycles back to the cell surface. A similar effect was observed in Rab11A-transfected cells (Fig. 4D, lane 4). In contrast, in Rab11B-depleted cells a significant amount of biotinylated PAR1 remained in the intracellular pool after a 15-min incubation at 37 °C (Fig. 4D, lane 6), suggesting that PAR1 is unable to efficiently recycle. In addition, constitutively internalized PAR1 and endogenous Rab11B exhibited substantial colocalization on internal puncta exhibited by the yellow color in the merged image and line-scan analysis (Fig. 4, E and F). These findings suggest that Rab11B rather than Rab11A regulates recycling of PAR1 back to the cell surface. These data also suggest that the accumulation of PAR1 observed in the Rab11A-depleted cells is not due to a defect in receptor recycling.
PAR1 Exhibits Enhanced Lysosomal Degradation in Rab11B-deficient Cells
A defect in PAR1 recycling observed in Rab11B-depleted cells is not sufficient to explain the loss of PAR1 protein expression. To determine whether inhibition of PAR1 recycling promotes receptor trafficking to the lysosome, we used leupeptin, a lysosomal protease inhibitor. HeLa cells were depleted of Rab11B by siRNA for 24 h and incubated with leupeptin for an additional 24 h, and PAR1 expression was determined. In ns siRNA-transfected cells leupeptin treatment resulted in a significant increase in PAR1 surface expression (Fig. 5A), suggesting that PAR1 recycling to the cell surface is enhanced when basal turnover is inhibited. As expected, PAR1 surface expression was significantly reduced in Rab11B siRNA-treated cells (Fig. 5A). However, leupeptin treatment failed to restore PAR1 surface expression to control levels in Rab11B siRNA-treated cells (Fig. 5A), indicating an important role for Rab11B in PAR1 recycling back to the cell surface. In contrast, PAR1 protein expression was significantly increased in both ns and Rab11B siRNA-transfected cells after leupeptin treatment (Fig. 5B). Confocal immunofluorescence microscopy was next used to confirm lysosomal sorting of PAR1 in Rab11B-deficient cells. In ns siRNA control cells treated with or without leupeptin, PAR1 localized to the cell surface and internal puncta that exhibited partial co-localization with LAMP1, a marker of lysosomes, indicated by the yellow color in the merged images (Fig. 5C). Line-scan analysis of PAR1 and LAMP1 indicate a similar extent of co-localization in leupeptin-treated and -untreated cells (Fig. 5C). In contrast, PAR1 expression was virtually abolished in Rab11B-deficient cells not treated with leupeptin (Fig. 5C). However, the addition of leupeptin caused significant accumulation of intracellular PAR1 in Rab11B siRNA-treated cells that exhibited marked co-localization with LAMP1 (Fig. 5C). These results suggest that when recycling is disrupted in Rab11B-deficient cells, PAR1 is re-directed from early endosomes and sorted to lysosomes for degradation.
FIGURE 5.

Depletion of Rab11B results in enhanced basal degradation of PAR1 at the lysosome. A, HeLa cells expressing FLAG-PAR1 were transfected with ns or Rab11B siRNAs for 24 h. Cells were then incubated for an additional 24 h at 37 °C with or without 2 mm leupeptin (Leu), and PAR1 surface expression was determined by ELISA. Data (mean ± S.D., n = 3) are representative of three independent experiments, expressed as the percent of ns siRNA control, and were analyzed by ANOVA (*, p < 0.05; **, p < 0.01; ***, p < 0.001). B, HeLa cells were treated as described above and lysed, and PAR1 protein expression was determined by immunoblotting with anti-FLAG antibody. Data (mean ± S.D., n = 3) are representative of three independent experiments, expressed as the percent of −Leu-treated cells, and analyzed by ANOVA (*, p < 0.01; *, p < 0.05; ***, p < 0.001). The expression of Rab11B and GAPDH were determined by immunoblotting as controls. C, HeLa cells treated as described in panel A were fixed, permeabilized, and incubated with anti-FLAG antibody to detect PAR1 (red) and anti-LAMP1 antibody (green) and imaged by confocal microscopy. The images are representative of many cells examined in three independent experiments. Scale bar, 10 μm. Colocalization is revealed by the yellow color in the merged imaged and by line-scan analysis across the center of cells. Line scan analysis demonstrates colocalization between PAR1 and LAMP1 at puncta in the merged image. ADU, arbitrary units.
Rab11A Regulates PAR1 Basal Degradation and Restores PAR1 Expression in Rab11B-deficient Cells
To understand the mechanism by which Rab11A regulates PAR1 expression, we examined the basal rate of receptor degradation in Rab11A-deficient cells in the presence of cycloheximide, which blocks de novo receptor synthesis. PAR1-expressing HeLa cells transfected with either ns or Rab11A siRNA were incubated with cycloheximide, and the amount of PAR1 protein remaining was then quantified at different time points. The rate of PAR1 degradation was reduced significantly in cells lacking Rab11A expression (Fig. 6A, lanes 5–8) compared with ns siRNA-transfected control cells (Fig. 6A, lanes 1–4). These findings suggest that Rab11A regulates basal degradation of PAR1 in contrast to Rab11B, which promotes receptor recycling and thereby diminishes the rate of basal receptor degradation. To confirm that Rab11A and Rab11B have distinct functions, we examined whether Rab11A depletion could restore PAR1 expression in Rab11B-depleted cells by performing sequential depletion of Rab11B followed by Rab11A. In these studies PAR1 surface expression was significantly decreased in Rab11B siRNA-transfected cells regardless of the order or transfection condition (Fig. 6B), consistent with an important role for Rab11B in regulating PAR1 recycling to the cell surface even in the absence of Rab11A. However, the sequential depletion of Rab11B followed by Rab11A resulted in a marked increase in receptor protein expression and restored PAR1 to levels observed in ns siRNA-treated cells (Fig. 6C, lanes 1 and 6). A similar effect on PAR1 protein expression was observed when the depletion order of Rab11B versus Rab11A was reversed (Fig. 6C, lanes 1 and 3). These data suggest that Rab11A mediates basal degradation of PAR1, which is enhanced when PAR1 recycling is blocked.
FIGURE 6.
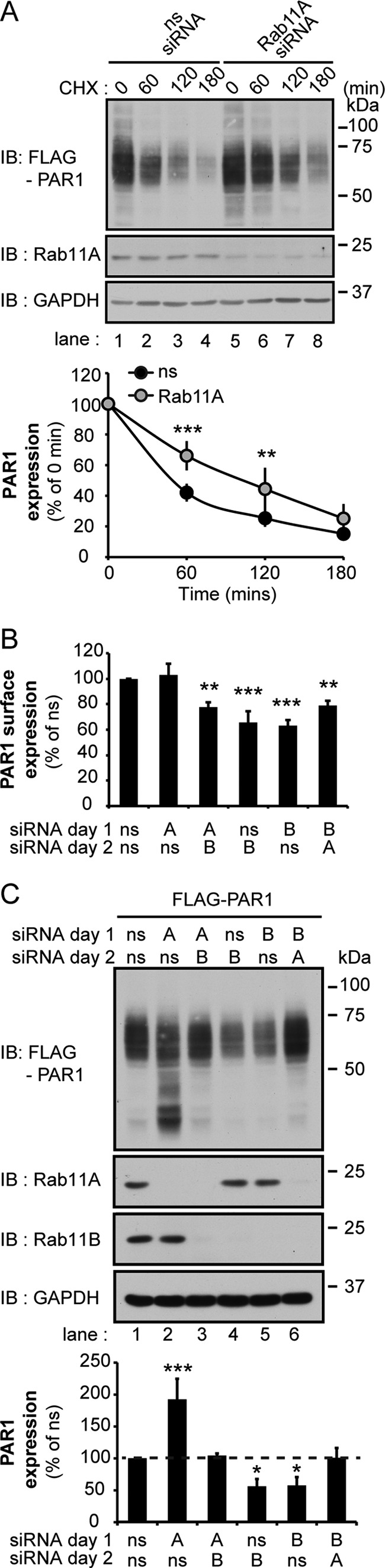
Rab11A mediates PAR1 basal degradation and restores PAR1 expression in Rab11B-depleted cells. A, HeLa cells expressing FLAG-PAR1 were transfected with ns or Rab11A siRNAs. After 48 h cells were incubated with 10 μm cycloheximide (CHX) for the indicated times at 37 °C and lysed, and cell lysates were immunoblotted (IB) with anti-FLAG antibody to detect PAR1. Data (mean ± S.D., n = 3) are representative of three independent experiments, expressed as the percent of ns siRNA control, and were analyzed by ANOVA (**, p < 0.01 and ***, p < 0.001). The expression of Rab11A and GAPDH was detected in cell lysates by immunoblotting as controls. B, HeLa cells expressing FLAG-PAR1 were transfected with ns, Rab11A, or Rab11B siRNA for 24 h (day 1) followed by a second transfection with ns, Rab11A, or Rab11B for 48 h (day 2). Cells were then fixed, and PAR1 surface expression was quantified by ELISA. Data (mean ± S.D., n = 3) are representative of three separate experiments, expressed as the percent of ns siRNA control, and were analyzed by ANOVA (**, p < 0.01 and ***, p < 0.001). C, HeLa cells expressing FLAG-PAR1 were transfected with siRNA as described above and lysed, and cell lysates were immunoblotted using anti-FLAG antibody to detect PAR1. Data (mean ± S.D., n = 3) are representative of three different experiments, expressed as the percent of ns siRNA control, and analyzed by ANOVA (*, p < 0.05 and ***, p < 0.001). Cell lysates were immunoblotted for Rab11A, Rab11B, and GAPDH as controls.
Enhanced PAR1 Degradation after Rab11B Depletion Requires ATG5
Because Rab11 is known to regulate membrane transport from recycling endosomes to light chain-3 (LC3)-positive autophagosomes (35), we investigated whether PAR1 degradation observed in Rab11B-depleted cells occurs through an autophagic pathway by examining the role of ATG5, a protein essential for mammalian autophagy (36). To examine the potential role of ATG5 in Rab11B-deficient cells, Rab11B and ATG5 were depleted from PAR1-expressing HeLa cells individually or simultaneously using siRNAs, and receptor expression was measured. Cells depleted of Rab11B exhibited decreased PAR1 cell surface and protein expression as expected (Fig. 7, A and B). The depletion of ATG5 by siRNA alone had no significant effect on PAR1 cell surface or protein expression (Fig. 7, A and B). The loss of ATG5 expression in cells co-depleted of Rab11B also failed to effect reduced PAR1 surface expression (Fig. 7A). However, ATG5 depletion blocked PAR1 degradation observed in Rab11B-deficient cells and restored receptor expression to levels observed in control ns siRNA-transfected cells (Fig. 7B). These data suggest that the loss of Rab11B expression results in enhanced PAR1 degradation that occurs through an ATG5-dependent autophagic pathway and targeting to the lysosome.
FIGURE 7.
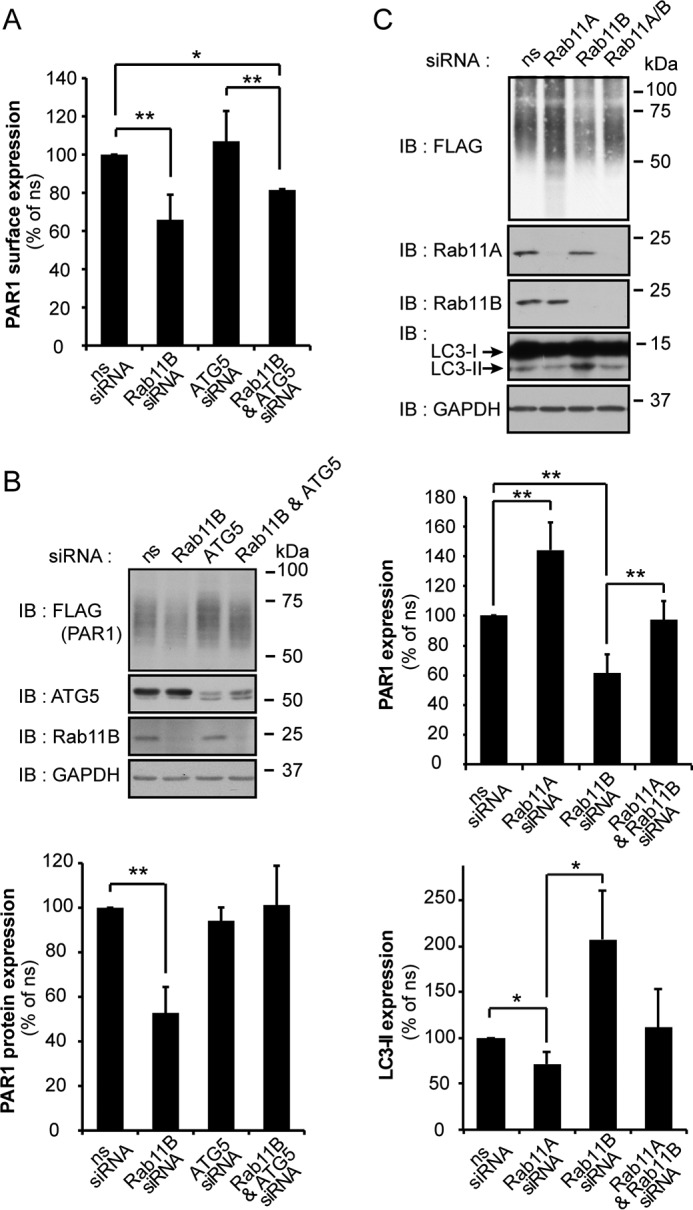
Depletion of Rab11A enhances PAR1 degradation through an ATG5-dependent pathway. A, HeLa cells expressing FLAG-PAR1 were transfected with either ns, Rab11B, or ATG5 siRNA individually or cotransfected with Rab11B and ATG5 siRNAs for 72 h. Cells were then fixed, and PAR1 surface expression was measured by ELISA. Data (mean ± S.D., n = 3) are representative of three independent experiments, expressed as the percent of ns siRNA control, and were analyzed by ANOVA (*, p < 0.05; **, p < 0.01). B, HeLa cells expressing FLAG-PAR1 were transfected as described above and lysed, and cell lysates were immunoblotted (IB) with anti-FLAG antibody to detect PAR1. Data (mean ± S.D., n = 3) are representative of three different experiments, expressed as the percent of ns siRNA control, and analyzed by ANOVA (**, p < 0.01). Cell lysates were immunoblotted for ATG5, Rab11B and GAPDH expression as controls. C, HeLa cells expressing FLAG-PAR1 were transfected with ns, Rab11A, and Rab11B siRNA or cotransfected with Rab11B and Rab11A siRNA. Cell lysates were immunoblotted with anti-FLAG antibody to detect PAR1 and with anti-LC3 antibody to detect LC3-I and -II. Data (mean ± S.D., n = 3) are representative of three different experiments, expressed as the percent of ns siRNA control, and were analyzed by ANOVA (*, p < 0.05; **, p < 0.01). Cell lysates were also immunoblotted for Rab11A, Rab11B, and GAPDH expression as controls.
The microtubule-associated protein LC3 is a known marker of autophagy in mammalian cells (37) The initiation of autophagy results in the conversion of LC3-I to LC3-II via the addition of phosphatidylethanolamine group to the C terminus of LC3-I, which enhances the mobility of LC3-II and distinguishes it from LC3-I. To determine whether Rab11A or Rab11B depletion induced autophagy in PAR1-expressing HeLa cells, we examined LC3 mobility. In Rab11A-deficient cells, PAR1 expression was increased, and LC3-II conversion was modestly reduced (Fig. 7D). In contrast, Rab11B-deficient cells showed substantial loss of PAR1 protein and a significant increase in LC3-II revealed by the detection of a lower molecular weight species of the LC3 doublet (Fig. 7D), suggesting that autophagy is induced in these cells. Interestingly, co-depletion of Rab11A together with Rab11B restored PAR1 expression and reduced the conversion of LC3-I to LC3-II. Together these findings suggest that PAR1 degradation proceeds via an autophagic pathway in cells depleted of Rab11B (Fig. 8).
FIGURE 8.

Differential regulation of PAR1 constitutive recycling and basal degradation by Rab11A and Rab11B. Unactivated PAR1 is constitutively internalized and recycled back to the cell surface through a Rab11B-dependent pathway, whereas Rab11A regulates basal lysosomal degradation of PAR1 (left panel). In the absence of Rab11A expression (right panel), PAR1 constitutively internalizes, and a marked increase in receptor accumulation in early endosomes is observed, as basal degradation of PAR1 is inhibited. Interestingly, in the absence of Rab11B expression (middle panel), PAR1 exhibits enhanced basal degradation that is blocked by Rab11A and ATG5 depletion, suggesting that the receptor traffics through an autophagic pathway before degradation in the autolysosome. These findings suggest that Rab11B and Rab11A serve distinct functions and regulate PAR1 recycling or basal degradation, respectively.
Discussion
Intracellular trafficking of PAR1 is critical for the fidelity of signaling. PAR1 displays both constitutive and agonist-induced internalization. Constitutive internalization of PAR1 is important for formation of an internal pool of naïve receptors that recycle back to the cell surface and mediate resensitization (9), whereas internalization and lysosomal degradation of activated PAR1 is important for termination of G protein signaling (6, 7). Although the mechanisms that control PAR1 constitutive and agonist-induced internalization are well defined, the processes that regulate recycling of constitutively internalized receptor are not known. In this study we define new roles for Rab11A and Rab11B in the regulation of PAR1 intracellular trafficking. We show that Rab11B regulates PAR1 recycling, whereas Rab11A controls basal lysosomal degradation of the receptor. We further discovered that in the absence of Rab11B expression, PAR1 is trafficked to an autophagic pathway mediated by Rab11A and ATG5, suggesting that the receptor is shuttled through autophagosomes before degradation in the autolysosome (Fig. 8). Together these findings identify new and distinct roles for Rab11A and Rab11B in the regulation of GPCR recycling and lysosomal degradation.
This study is the first to demonstrate a specific function for Rab11B in GPCR recycling. After internalization, GPCRs can recycle through either a Rab4-dependent rapid recycling route directly from early endosomes to the plasma membrane or via a Rab11-mediated slow recycling pathway through perinuclear recycling endosomes. Rab4 has been shown to mediate recycling of the angiotensin II type I receptor and the phosphorylated μ-opioid GPCR (38, 39). In contrast to Rab4, a role for Rab11A in recycling of many GPCRs is well documented (15). These previous studies utilized the Rab11A dominant-negative S25N mutant and showed that expression of Rab11A S25N mutant reduced the expression of the β2-adrenergic receptor at the cell surface by blocking recycling, diminished recycling of constitutively internalized thromboxane receptor-β receptor, or blocked agonist-induced internalized M4 muscarinic receptor recycling (17–19, 40). However, an assessment of Rab11A versus Rab11B function on GPCR recycling has not been previously investigated.
Here, we report that rather than Rab4 or Rab11A, Rab11B has emerged as a key regulator of constitutively internalized PAR1 recycling based on a comprehensive siRNA screen of 140 different membrane trafficking proteins. Depletion of Rab11B and not Rab11A caused a marked reduction of PAR1 expression at the cell surface in HeLa cells stably expressing PAR1. A similar effect of Rab11B depletion on endogenous PAR1 surface expression was observed in endothelial cells and MDA-MB-231 breast carcinoma cells. Moreover, Rab11B colocalized with PAR1 on endocytic vesicles and was required for recycling of constitutively internalized receptor back to the cell surface. Interestingly, in cells depleted of Rab11B, basal lysosomal degradation of PAR1 was enhanced, suggesting that Rab11B-dependent recycling plays a critical role in maintaining appropriate amounts of PAR1 at the cell surface. Similar to PAR1, disruption of Rab11B and not Rab11A by siRNA caused a significant reduction in epithelial sodium channel and cystic fibrosis transmembrane conductance regulator expression at the cell surface (41, 42). The expression of epithelial sodium channel and cystic fibrosis transmembrane conductance regulator protein was also significantly reduced in Rab11B-deficient cells, like that observed with PAR1. The mechanisms responsible for enhanced degradation of epithelial sodium channel and cystic fibrosis transmembrane conductance regulator observed in Rab11B-depleted cells is not known. Thus, in addition to ion and anion channels, Rab11B appears to have a selective role in regulating recycling of certain GPCRs.
Rab11 proteins localize to the trans-Golgi network as well as to perinuclear endosomes and have been implicated in a variety of cellular trafficking pathways. Rab11A has been shown to colocalize with PAR2, a GPCR related to PAR1, and appears to regulate PAR2 trafficking from the Golgi to the cell surface (43). In contrast to PAR2, we discovered that Rab11A controls endosomal-lysosomal sorting and basal degradation of PAR1. Although siRNA-mediated depletion of Rab11A failed to effect PAR1 recycling, a marked accumulation of intracellular PAR1 protein was observed in multiple cell types. Moreover, enhanced degradation of PAR1 observed in Rab11B-deficient cells was blocked by co-depletion of Rab11A. These results provide evidence to support a role for Rab11A in PAR1 basal lysosomal sorting in normal cells and in cells depleted of Rab11B expression (Fig. 8). These findings further suggest that Rab11A and Rab11B serve distinct functions in the regulation of PAR1 intracellular trafficking. These distinctions may arise from differential localization of Rab11A versus Rab11B at different vesicular compartments (44) and/or to distinct structural properties observed between Rab11A and Rab11B (45) that may impact their activity and/or interaction with various binding partners. In addition, Rab11A has been shown to interact with the thromboxane A2 receptor, angiotensin II type I receptor, and the prostacyclin receptor (17, 20, 38). However, there is no clearly defined consensus binding motif for Rab11A interaction with GPCRs, suggesting that the interaction may be mediated through adaptor protein binding, but this remains to be determined.
Rab11A and Rab11B have been implicated in autophagosome formation by controlling the transport of membrane from recycling endosomes to forming LC3-positive autophagosomes in mammalian cells (35). We discovered that enhanced basal degradation of PAR1 observed in Rab11B-depleted cells is mediated by ATG5, a protein essential for autophagy (36). In addition, the conversion of LC3-I to LC3-II is significantly increased in Rab11B-deficient cells, strongly suggesting that loss of Rab11B induces autophagy in mammalian cells. These findings suggest that PAR1 is sorted from endosomes to autophagosomes and degraded in autolysosomes when recycling is blocked by depletion of Rab11B. The β2-adrenergic receptor also appears to sort to an autophagic pathway under stress conditions. Stimulation of the β2-adrenergic receptor stably expressed in HEK293 cells with isoproterenol for 4 h in the presence of bafilomycin and chloroquine, which inhibit the vacuolar type H+-ATPase that prevents acidification and inhibit fusion of late endosomes, multivesicular bodies, and autophagosomes, was recently shown to induce β2-adrenergic receptor colocalization and association with LC3-II (46). Colocalization and interaction with LC3-II also requires β2-adrenergic receptor ubiquitination. However, we found that both PAR1 wild type and ubiquitination-deficient 0K mutant exhibit diminished surface expression and enhanced degradation in the absence of Rab11B, whereas depletion of Rab11A caused PAR1 0K mutant accumulation similar to wild type PAR1. These findings suggest that ubiquitin is not required for PAR1 basal turnover by either the Rab11A- or Rab11B-dependent pathways and is consistent with ubiquitin-independent trafficking of activated PAR1 through an ALIX and ESCRT-III mediated lysosomal sorting pathway (28).
In summary, this study clearly demonstrates divergent roles for Rab11A and Rab11B in regulation of PAR1 intracellular trafficking. We further show that in the absence of Rab11B-mediated PAR1 recycling, the receptor is trafficked to an autophagic pathway for degradation. This work also reveals distinct roles for Rab11A and Rab11B in the regulation of PAR1 and ErbB2 but not EGFR expression in invasive breast carcinoma cells. Moreover, loss of PAR1 surface expression in MDA-MB-231 cells deficient in Rab11B expression resulted in reduced signaling. Given the importance of PAR1 trafficking for proper regulation of signaling and appropriate cellular responses, it will be important to determine how modulation of PAR1 and ErbB2 expression by Rab11A and Rab11B effects breast carcinoma invasion and tumor growth in future studies. In addition, the molecular mechanisms responsible for induction of autophagy in Rab11B-depleted cells and the processes that mediate PAR1 sorting to autophagosomes will be important to understand.
Author Contributions
N. J. G. conducted all of the experiments and in some experiments received assistance from L. J. C. and I. C. C. N. J. G. analyzed the results and wrote most of the paper with assistance from J. T.
Supplementary Material
Acknowledgments
We thank members of the Trejo laboratory for comments and advice.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM090689 (to J. T.) The authors declare they have no conflict of interest with the contents of this article.

This article contains supplemental Table S1.
- PAR1
- protease-activated receptor-1
- ATG5
- autophagy related-5 protein
- AP-2
- adaptor protein complex-2
- GPCR
- G protein-coupled receptor
- LAMP1
- lysosomal associated membrane protein-1
- LC3
- microtubule-associated protein light chain-3
- ns
- non-specific
- EGFR
- EGF receptor
- ANOVA
- analysis of variance
- ns
- nonspecific.
References
- 1. Canto I., Soh U. J., and Trejo J. (2012) Allosteric modulation of protease-activated receptor signaling. Mini Rev. Med. Chem. 12, 804–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hollenberg M. D., Mihara K., Polley D., Suen J. Y., Han A., Fairlie D. P., and Ramachandran R. (2014) Biased signalling and proteinase-activated receptors (PARs): targeting inflammatory disease. Br J. Pharmacol. 171, 1180–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Coughlin S. R. (2005) Protease-activated receptors in hemostasis, thrombosis and vascular biology. J. Thromb. Haemost. 3, 1800–1814 [DOI] [PubMed] [Google Scholar]
- 4. Vu T. K., Hung D. T., Wheaton V. I., and Coughlin S. R. (1991) Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 64, 1057–1068 [DOI] [PubMed] [Google Scholar]
- 5. Vu T. K., Wheaton V. I., Hung D. T., Charo I., and Coughlin S. R. (1991) Domains specifying thrombin-receptor interaction. Nature 353, 674–677 [DOI] [PubMed] [Google Scholar]
- 6. Booden M. A., Eckert L. B., Der C. J., and Trejo J. (2004) Persistent signaling by dysregulated thrombin receptor trafficking promotes breast carcinoma cell invasion. Mol. Cell. Biol. 24, 1990–1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Trejo J., Hammes S. R., and Coughlin S. R. (1998) Termination of signaling by protease-activated receptor-1 is linked to lysosomal sorting. Proc. Natl. Acad. Sci. U.S.A. 95, 13698–13702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hein L., Ishii K., Coughlin S. R., and Kobilka B. K. (1994) Intracellular targeting and trafficking of thrombin receptors: a novel mechanism for resensitization of a G protein-coupled receptor. J. Biol. Chem. 269, 27719–27726 [PubMed] [Google Scholar]
- 9. Paing M. M., Johnston C. A., Siderovski D. P., and Trejo J. (2006) Clathrin adaptor AP2 regulates thrombin receptor constitutive internalization and endothelial cell resensitization. Mol. Cell. Biol. 26, 3231–3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ellis C. A., Tiruppathi C., Sandoval R., Niles W. D., and Malik A. B. (1999) Time course of recovery of endothelial cell surface thrombin receptor (PAR-1) expression. Am. J. Physiol. 276, C38–C45 [DOI] [PubMed] [Google Scholar]
- 11. Shukla A. K., Xiao K., and Lefkowitz R. J. (2011) Emerging paradigms of β-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem. Sci. 36, 457–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Paing M. M., Stutts A. B., Kohout T. A., Lefkowitz R. J., and Trejo J. (2002) β-Arrestins regulate protease-activated receptor-1 desensitization but not internalization or down-regulation. J. Biol. Chem. 277, 1292–1300 [DOI] [PubMed] [Google Scholar]
- 13. Chen B., Dores M. R., Grimsey N., Canto I., Barker B. L., and Trejo J. (2011) Adaptor protein complex-2 (AP-2) and epsin-1 mediate protease-activated receptor-1 internalization via phosphorylation- and ubiquitination-dependent sorting signals. J. Biol. Chem. 286, 40760–40770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bowman S. L., and Puthenveedu M. A. (2015) Postendocytic sorting of adrenergic and opioid receptors: new mechanisms and functions. Prog. Mol. Biol. Transl. Sci. 132, 189–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Seachrist J. L., and Ferguson S. S. (2003) Regulation of G protein-coupled receptor endocytosis and trafficking by Rab GTPases. Life Sci. 74, 225–235 [DOI] [PubMed] [Google Scholar]
- 16. Stenmark H. (2009) Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 10, 513–525 [DOI] [PubMed] [Google Scholar]
- 17. Hamelin E., Thériault C., Laroche G., and Parent J. L. (2005) The intracellular trafficking of the G protein-coupled receptor TPβ depends on a direct interaction with Rab11. J. Biol. Chem. 280, 36195–36205 [DOI] [PubMed] [Google Scholar]
- 18. Thériault C., Rochdi M. D., and Parent J. L. (2004) Role of the Rab11-associated intracellular pool of receptors formed by constitutive endocytosis of the β isoform of the thromboxane A2 receptor (TPβ). Biochemistry 43, 5600–5607 [DOI] [PubMed] [Google Scholar]
- 19. Parent A., Hamelin E., Germain P., and Parent J. L. (2009) Rab11 regulates the recycling of the β2-adrenergic receptor through a direct interaction. Biochem. J. 418, 163–172 [DOI] [PubMed] [Google Scholar]
- 20. Reid H. M., Mulvaney E. P., Turner E. C., and Kinsella B. T. (2010) Interaction of the human prostacyclin receptor with Rab11: characterization of a novel Rab11 binding domain within alpha-helix 8 that is regulated by palmitoylation. J. Biol. Chem. 285, 18709–18726 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21. Cheng K. W., Lahad J. P., Kuo W. L., Lapuk A., Yamada K., Auersperg N., Liu J., Smith-McCune K., Lu K. H., Fishman D., Gray J. W., and Mills G. B. (2004) The RAB25 small GTPase determines aggressiveness of ovarian and breast cancers. Nat. Med. 10, 1251–1256 [DOI] [PubMed] [Google Scholar]
- 22. Li Y. M., Pan Y., Wei Y., Cheng X., Zhou B. P., Tan M., Zhou X., Xia W., Hortobagyi G. N., Yu D., and Hung M. C. (2004) Up-regulation of CXCR4 is essential for HER2-mediated tumor metastasis. Cancer Cell 6, 459–469 [DOI] [PubMed] [Google Scholar]
- 23. Russo A., Soh U. J., Paing M. M., Arora P., and Trejo J. (2009) Caveolae are required for protease-selective signaling by protease-activated receptor-1. Proc. Natl. Acad. Sci. U.S.A. 106, 6393–6397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Trejo J., Altschuler Y., Fu H. W., Mostov K. E., and Coughlin S. R. (2000) Protease-activated receptor-1 down-regulation: a mutant HeLa cell line suggests novel requirements for PAR1 phosphorylation and recruitment to clathrin-coated pits. J. Biol. Chem. 275, 31255–31265 [DOI] [PubMed] [Google Scholar]
- 25. Wolfe B. L., Marchese A., and Trejo J. (2007) Ubiquitination differentially regulates clathrin-dependent internalization of protease-activated receptor-1. J. Cell Biol. 177, 905–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lin H., and Trejo J. (2013) Transactivation of the PAR1-PAR2 heterodimer by thrombin elicits β-arrestin-mediated endosomal signaling. J. Biol. Chem. 288, 11203–11215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Soto A. G., and Trejo J. (2010) N-Linked glycosylation of protease-activated receptor-1 second extracellular loop: a critical determinant for ligand-induced receptor activation and internalization. J. Biol. Chem. 285, 18781–18793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dores M. R., Chen B., Lin H., Soh U. J., Paing M. M., Montagne W. A., Meerloo T., and Trejo J. (2012) ALIX binds a YPX(3)L motif of the GPCR PAR1 and mediates ubiquitin-independent ESCRT-III/MVB sorting. J. Cell Biol. 197, 407–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grimsey N. J., Aguilar B., Smith T. H., Le P., Soohoo A. L., Puthenveedu M. A., Nizet V., and Trejo J. (2015) Ubiquitin plays an atypical role in GPCR-induced p38 MAP kinase activation on endosomes. J. Cell Biol. 210, 1117–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shapiro M. J., Trejo J., Zeng D., and Coughlin S. R. (1996) Role of the thrombin receptor's cytoplasmic tail in intracellular trafficking: distinct determinants for agonist-triggered versus tonic internalization and intracellular localization. J. Biol. Chem. 271, 32874–32880 [DOI] [PubMed] [Google Scholar]
- 31. Canto I., and Trejo J. (2013) Palmitoylation of protease-activated receptor-1 regulates adaptor protein complex-2 and -3 interaction with tyrosine-based motifs and endocytic sorting. J. Biol. Chem. 288, 15900–15912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Arora P., Cuevas B. D., Russo A., Johnson G. L., and Trejo J. (2008) Persistent transactivation of EGFR and ErbB2/HER2 by protease-activated receptor-1 promotes breast carcinoma cell invasion. Oncogene 27, 4434–4445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cobleigh M. A., Vogel C. L., Tripathy D., Robert N. J., Scholl S., Fehrenbacher L., Wolter J. M., Paton V., Shak S., Lieberman G., and Slamon D. J. (1999) Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J. Clin. Oncol. 17, 2639–2648 [DOI] [PubMed] [Google Scholar]
- 34. Nicholson R. I., Gee J. M., and Harper M. E. (2001) EGFR and cancer prognosis. Eur. J. Cancer 37, S9–S15 [DOI] [PubMed] [Google Scholar]
- 35. Longatti A., Lamb C. A., Razi M., Yoshimura S., Barr F. A., and Tooze S. A. (2012) TBC1D14 regulates autophagosome formation via Rab11- and ULK1-positive recycling endosomes. J. Cell Biol. 197, 659–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mizushima N., Yamamoto A., Hatano M., Kobayashi Y., Kabeya Y., Suzuki K., Tokuhisa T., Ohsumi Y., and Yoshimori T. (2001) Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 152, 657–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., and Yoshimori T. (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Esseltine J. L., Dale L. B., and Ferguson S. S. (2011) Rab GTPases bind at a common site within the angiotensin II type I receptor carboxyl-terminal tail: evidence that Rab4 regulates receptor phosphorylation, desensitization, and resensitization. Mol. Pharmacol. 79, 175–184 [DOI] [PubMed] [Google Scholar]
- 39. Wang F., Chen X., Zhang X., and Ma L. (2008) Phosphorylation state of mu-opioid receptor determines the alternative recycling of receptor via Rab4 or Rab11 pathway. Mol. Endocrinol. 22, 1881–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Volpicelli L. A., Lah J. J., Fang G., Goldenring J. R., and Levey A. I. (2002) Rab11a and myosin Vb regulate recycling of the M4 muscarinic acetylcholine receptor. J. Neurosci. 22, 9776–9784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Butterworth M. B., Edinger R. S., Silvis M. R., Gallo L. I., Liang X., Apodaca G., Frizzell R. A., Fizzell R. A., and Johnson J. P. (2012) Rab11b regulates the trafficking and recycling of the epithelial sodium channel (ENaC). Am. J. Physiol. Renal Physiol. 302, F581–F590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Silvis M. R., Bertrand C. A., Ameen N., Golin-Bisello F., Butterworth M. B., Frizzell R. A., and Bradbury N. A. (2009) Rab11b regulates the apical recycling of the cystic fibrosis transmembrane conductance regulator in polarized intestinal epithelial cells. Mol. Biol. Cell 20, 2337–2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Roosterman D., Schmidlin F., and Bunnett N. W. (2003) Rab5a and rab11a mediate agonist-induced trafficking of protease-activated receptor 2. Am. J. Physiol. Cell Physiol. 284, C1319–C1329 [DOI] [PubMed] [Google Scholar]
- 44. Lapierre L. A., Dorn M. C., Zimmerman C. F., Navarre J., Burnette J. O., and Goldenring J. R. (2003) Rab11b resides in a vesicular compartment distinct from Rab11a in parietal cells and other epithelial cells. Exp. Cell Res. 290, 322–331 [DOI] [PubMed] [Google Scholar]
- 45. Scapin S. M., Carneiro F. R., Alves A. C., Medrano F. J., Guimarães B. G., and Zanchin N. I. (2006) The crystal structure of the small GTPase Rab11b reveals critical differences relative to the Rab11a isoform. J. Struct. Biol. 154, 260–268 [DOI] [PubMed] [Google Scholar]
- 46. Kommaddi R. P., Jean-Charles P. Y., and Shenoy S. K. (2015) Phosphorylation of the deubiquitinase USP20 by protein kinase A regulates post-endocytic trafficking of β2 adrenergic receptors to autophagosomes during physiological stress. J. Biol. Chem. 290, 8888–8903 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.