

Abstract
Four new X-ray structures of tryptophan synthase (TS) crystallized with varying numbers of the amphipathic N-(4′-trifluoromethoxybenzoyl)-2-aminoethyl phosphate (F6) molecule are presented. These structures show one of the F6 ligands threaded into the tunnel from the β-site and reveal a distinct hydrophobic region. Over this expanse, the interactions between F6 and the tunnel are primarily nonpolar, while the F6 phosphoryl group fits into a polar pocket of the β-subunit active site. Further examination of TS structures reveals that one portion of the tunnel (T1) binds clusters of water molecules, whereas waters are not observed in the nonpolar F6 binding region of the tunnel (T2). MD simulation of another TS structure with an unobstructed tunnel also indicates the T2 region of the tunnel excludes water, consistent with a dewetted state that presents a significant barrier to the transfer of water into the closed β-site. We conclude that hydrophobic molecules can freely diffuse between the α- and β-sites via the tunnel, while water does not. We propose that exclusion of water serves to inhibit reaction of water with the α-aminoacrylate intermediate to form ammonium ion and pyruvate, a deleterious side reaction in the αβ-catalytic cycle. Finally, while most TS structures show βPhe280 partially blocking the tunnel between the α- and β-sites, new structures show an open tunnel, suggesting the flexibility of the βPhe280 side chain. Flexible docking studies and MD simulations confirm the dynamic behavior of βPhe280 allows unhindered transfer of indole through the tunnel, therefore excluding a gating role for this residue.
Keywords: Substrate channeling, Hydrophobic nanopore, Substrate diffusion, Hydrophobic barrier, Dewetted channel, Tryptophan synthase
Graphical abstract
Introduction
Knowledge of the mechanisms by which small molecules are transported has been dependent on the determination of structures for the macromolecular protein assemblages involved in modulating transport. For example, the substantial body of structural information now available for membrane proteins shows that transport mechanisms are fundamentally dependent on the presence of macromolecular structures which form pores with dimensions and polarity designed to provide selective transfer of the desired small molecule or ion.[1–6] These pores or “channels” are capable of considerable selectivity, viz. the potassium channel.[1, 2] It is now apparent that similar transport processes take place within assemblages of enzymes in certain metabolic pathways. The functions of these transport processes are postulated to protect labile small molecule intermediates from deleterious side reactions, to enhance catalytic efficiency, and to prevent loss of hydrophobic molecules by adsorption into hydrophobic environments such as the membranes of the organelles comprising the cell.
Substrate channeling plays an important role in many different enzymatic reactions wherein a common metabolite must be transferred from one enzyme active site to another within a multi-enzyme complex.[7–9] Channeling typically involves diffusion through a largely hydrophobic tunnel, and the phenomenon has been implicated in many multi-enzyme complexes. For example, the structurally well-described amidotransferase family of enzymes employs tunnels to channel ammonia and other substrates between the active sites of the protein complexes.[9–12] The most remarkable member of this family, the carbamoyl phosphate synthase complex, transfers NH3, carboxyphosphate, and carbamate among three active sites through a 100 Å-long network of tunnels.[9, 11, 12] Gating mechanisms have been postulated to play a role in the regulation of substrate channeling in some enzymes, for example imidazole glycerol phosphate synthase. Yet, it has been demonstrated by Amaro et al.[13, 14] that gating is not significant, and, interestingly, molecular dynamics studies indicate that the channel in this complex disfavors the conduct of water while optimizing ammonia transport.[15]
In the pyridoxal-5′-phosphate-requiring family of enzymes, the 143 kDa tryptophan synthase complex (TS, EC 4.2.1.20) has been shown to transfer the common metabolite indole from one site to another via a 30 Å tunnel.[16] Bacterial TS[16] catalyzes the biosynthesis of L-tryptophan from substrates indole-3-glycerol phosphate (IGP) and L-serine (reactions 1–3).[17] The enzyme consists of two α-subunits (29 kDa each) and two β-subunits (43 kDa each), arranged in a nearly linear assembly of αβ dimeric units to give an αββα structure. TS was the first enzyme system demonstrated to use substrate channeling to connect two active sites for sequential reactions.[16, 18, 19] The TS catalytic mechanism depicted in Scheme 1ab shows indole is produced in the α-site reaction and consumed in the β-reaction.[17, 20, 21]
Scheme 1.


Chemical processes catalyzed by the α- and β-sites of the tryptophan synthase bienzyme complex. (a) α-Reaction. (b) β-Reaction. (c) Structure of F6. (d) Structure of F9.
Early studies of TS showed that free indole is not found in solution during the overall αβ reaction, leading to the hypothesis that the active sites of both subunits are buried in the interior of the complex adjacent to each other.[17, 19, 22, 23] This hypothesis provided a structural basis for the lack of any build-up of indole during the αβ reaction. In the late 1980s and 1990s, investigations of the tunnel hypothesis involving both solution and structural studies were undertaken in parallel. The 2.5 Å resolution crystal structures of the TS internal aldimine, E(Ain), and the complex of E(Ain) with indole propanol phosphate (IPP) were published in 1988.[16] These structures indicated that rather than aligned adjacent to each other, the active sites within each αβ dimeric unit are actually separated by a distance of approximately 30 A and are connected by a 25 Å long hydrophobic tunnel. The dimensions of the tunnel appeared sufficient to accommodate the passage of indole from the α-site to the β-site. Kinetic analyses revealed that formation of the α-aminoacrylate intermediate at the β-site increased IGP cleavage at the α-site nearly 30-fold and that indole channeling is rapid relative to the turnover of substrates in the αβ reaction. A series of kinetic studies[24–29] demonstrated that the tunnel is functional in the transfer of indole from the α-site to the β-site and that, in the αβ-reaction, the transfer occurs within αβ dimeric units wherein the α- and β-subunits have closed conformations, postulated to prevent the escape of indole.[25, 27, 28, 30] Thus, allosteric communication between the α- and β-sites within αβ-dimeric units of the tetrameric assembly switches the subunits between open (inactive) and closed (active) conformations that synchronize the catalytic cycles of the α- and β-sites and prevent the build-up and escape of indole during the αβ-catalytic cycle.[12, 20, 25, 30–32]
Computational simulations have provided important contributions to understanding both the flexibility of the tunnel and the conformational allostery that play a large role in regulation of channeling in TS. Bahar and Jernigan[33] explored allosteric communication between subunits in both wild type and mutant forms of the enzyme and identified specific flexible residues integral to structural changes necessary for catalysis. Spyrakis et al.[34] undertook MD studies to focus on the conformational transition between the open and closed conformations of the α-subunit, the changes this conformational transition brings about in the αβ-subunit interface, and the pathway for the entry of substrate into the α-site from solution. Fatmi and Chang[35] employed MD and Brownian dynamics simulations to probe the effect of conformational changes in the protein on the channeling of indole. These simulations suggested that indole can freely pass through the tunnel with little steric clashing.
Efforts have been made to prepare TS variants with site-directed mutations designed to block the tunnel.[36–39] The results of these efforts were ambiguous in that the βA169L/βC170W mutant exhibited altered substrate reactivities and specificities as well as a perturbed three dimensional structure. This mutant may block the transfer of indole, but the crystal structure shows a ~2 Å shift in the position of the COMM domain relative to wild-type enzyme that likely destroys the allosteric interactions between the sites[38] and perturbs the subunit interface in the vicinity of the tunnel. Further experiments with Nile Red led to the proposal that it was sequestered within the nonpolar region of the tunnel between βC170 and βF280, yet no crystal structure was reported.[39]
The evidence for the switching of αβ dimeric units between open and closed states during the αβ catalytic cycle[24–29] was strongly reinforced by the determination of the crystal structure of the complex of G3P with the E(A-A) complex.[40] This structure showed that both the α- and β-subunits have conformations wherein the α- and β-active sites are closed; closing of the α-subunit is associated with motions in α-loop L6 (αL6, residues α176–196) that switch the subunit from a disordered (open) to an ordered (closed) state, while closing of the β-subunit involves motion of the communication (COMM) domain (residues β102–189) and concomitant formation of a salt-bridge between βArg141 and βAsp305. Evidence that small molecules with dimensions similar to indole could be sequestered within the confines of the tunnel and the α- and β-sites when the αβ-dimeric unit is in the closed state was provided by the demonstration that the indole analogue, indoline, could be trapped within the complex by stabilizing the closed conformations of the αβ-dimeric unit via binding of high affinity ligands to the α-site of the α-aminoacrylate form.[41, 42] The studies of Brzovic et al.[27, 28] provided strong evidence that the switching from the open to the closed conformation occurs when L-Ser reacts with the PLP cofactor to form E(A-A), and Leja et al.[43] demonstrated that conversion of the L-Trp quinonoid, E(Q3), to the L-Trp external aldimine, E(Aex2), triggers both the conformational switch back to the open form and deactivation of the α-site. These conclusions have been further extended using 19F NMR to investigate the binding of the ligand N-(4′-trifluoromethoxybenzoyl)-2-aminoethyl phosphate (F6, an inhibitive analogue of IGP) to probe conformational switching and catalysis in the structure and function of tryptophan synthase.[44] This switching between open and closed states likely brings about synchronization of the α- and β-reactions and prevents the loss of indole during the αβ catalytic cycle.[12, 20, 25, 30–32, 43, 44]
Although the existence of an interconnecting tunnel between the α- and β-sites has been proposed based on the X-ray structures of TS, only one structure has been published showing a ligand bound within this tunnel. In 2007,[45] Ngo et al. reported that at relatively low concentrations one F6 molecule binds to the α-site of TS (PDB ID: 2CLE). When present in high concentrations, F6 gave an internal aldimine crystal structure wherein F6 was bound both to the α-site and to the tunnel within the β-subunit (PDB ID: 2CLF). The structural studies presented herein show that depending on the concentrations of F6 present, crystals containing one, two or three molecules of F6 per αβ-dimeric unit can be formed wherein one F6 molecule is threaded into the tunnel from the β-site. Thus, F6 can bind to three well-defined sites: the α-subunit catalytic site,[45] a site located along the αβ-subunit interface adjacent to the α-site near the tunnel (this work), and a site within the β-subunit portion of the tunnel[45] (this work). We present here a detailed analysis of the binding of F6 to these three sites via single crystal X-ray crystallography, the transfer of indole via a flexible docking study, and the dynamic modeling of water along the tunnel via molecular dynamics (MD) simulations using another crystal structure with a clear tunnel (PDBID: 4HN4). We also discuss the implications of these binding interactions to the structure and function of TS.
Experimental Methods
Crystallization, Data Collection, and Structural Determination
TS was purified as previously described.[46] About 10 mg of pure TS was applied to a HiPrep 16/60 Sephacryl S-200 HR column (GE Healthcare) equilibrated in 50 mM bicine-NaOH, pH 7.80, 20 μM PLP, and 100 mM NaCl to ensure sodium ion in the metal coordination site of the β-chain. Fractions with λmax 280 nm were analyzed in 15% SDS-polyacrylamide gel electrophoresis[47] stained with Coomassie Brilliant Blue R250. Fractions of interest containing high amounts of pure TS were pooled and concentrated using a 30 kDa Amicon tube at 293 K. Protein concentration was determined,[48] and the sample concentration was adjusted to 10 mg/mL in the column buffer. All crystallization experiments were carried out as sitting-drop vapor-diffusion experiments with equal volumes (5+5 μL) of protein solution and reservoir solution droplets using 24-well, 500 μL reservoir Cryschem plates (Hampton Research). The plates were sealed with sealing film and incubated at 293 K in the dark for up to 2 weeks. Optimized large, plate-like single crystals were grown in a solution containing 50 mM Na-bicine adjusted to pH 7.8, containing 100 mM NaCl, 8–10% PEG-8000, and 2% spermine. To determine the TS crystal structures in complex with the F6 inhibitor, a single plate-like crystal was harvested and soaked in reservoir buffer containing 1 mM F6 inhibitor and PEG-400 as cryoprotectant agent (F6-PEG buffer). In order to prevent cracks or fissures in the diffraction-quality crystals promoted by sudden exposure to high cryoprotectant concentration, the loop-mounted crystal was consecutively equilibrated for 3 minutes each in F6-PEG buffer enriched with increasing concentrations (5, 10, 15, and 20%) of PEG-400 (PDB ID: 4WX2). A second crystal (PDB ID: 5BW6) was soaked in the same buffers but equilibrated for 1 minute each in F6-PEG buffer, as described above. A third crystal was soaked in 10 mM F6 in F6-PEG buffer, and the loop-mounted crystal was consecutively equilibrated for 1 minute in F6-PEG buffers containing higher PEG-400 concentration as described above (PDB ID: 4Y6G). A fourth crystal was consecutively equilibrated for 2 minutes in reservoir buffer containing 10 mM F6 inhibitor containing DMSO as cryoprotectant agent (F6-DMSO buffer) and the loop-mounted crystal was soaked for 2 minutes in F6-DMSO buffers containing 10, 20, and 30% of DMSO (PDB ID: 4ZQC). The crystals were then flash-cooled at 100 K in a stream of gaseous nitrogen, and a single-wavelength near-complete X-ray data set was collected using a Rigaku MicroMax-007HF rotating-anode X-ray generator and the Rigaku RAXIS IV++ image plate detector (UC-Riverside, Biochemistry Department) or using synchrotron radiation at SIBYLS (Advance Light Source beamline 12.3.1) and the ADSC QUANTUM 315 CCD detector (Berkeley-CA, USA). All further data and model manipulation was carried out using the CCP4 suite of programs.[49] All X-ray diffraction data were indexed and merged with iMOSFLM[50] and then scaled with SCALA.[51] Phases for the native crystal structure were determined using the molecular-replacement method as implemented in MOLREP[52] using the TS crystal structure from S. typhimurium with the F6 inhibitor in the α-site as a search model (PDB ID: 4KKX).[44] The resulting model was further refined with REFMAC5[53] to generate the initial electron-density maps. Models were improved after extensive cycles of manual modeling in Coot[54] and restrained refinements in REFMAC. Water molecules were added in REFMAC5, and their occupancies were manually checked in Coot. The TLS protocol was implemented in the final stage of refinement using TLS groups generated by the TLSMD web server.[55] After final refinement, the stereochemical quality of the protein structure data was checked using PROCHECK.[56]
Since the orientation of F6-1 in both PDB IDs: 4WX2, 4ZQC, and 4Y6G is flipped relative to that of F6-1 in PDB ID: 2CLF, we were interested in re-refining the model of PDB ID: 2CLF with rotation of the F6-1 molecule to the orientation we observed in our models PDB ID: 4WX2 and PDB ID: 4Y6G (see Results). Re-refinement of model PDB ID: 2CLF (rr2CLF) was accomplished using the coordinate file and the structure factor (SF) file of model PDB ID: 2CLF downloaded from the PDB web-site and then uploaded in the Structure Factor Conversion and Validation (sf-valid) version 2.03 (http://sf-tool.wwpdb.org/) to convert the SF file from the mmCIF format to MTZ format, using 5.0% of the reflection data (free R) for cross-validation (See Supplemental Material).
Publication quality images were drawn using the PyMOL molecular-graphics program.[57] The coordinates of the refined structures were deposited in the Protein Data Bank. The crystallographic data-collection and structure refinement statistics are summarized in Table 1.
Table 1.
Crystallographic data and model refinement. Values in parentheses are for the highest resolution shell.
| PDB ID: | 5BW6 | 4Y6G | 4WX2 | 4ZQC |
|---|---|---|---|---|
| X-ray source | MicroMax-007HF | MicroMax-007HF | MicroMax-007HF | ALS-SIBYLS |
| Wavelength (Å) | 1.5418 | 1.5418 | 1.5418 | 0.9793 |
| C 1 2 1 (No. 5) | C 1 2 1 (No. 5) | C 1 2 1 (No. 5) | C 1 2 1 (No. 5) | C 1 2 1 (No. 5) |
| Unit-cell parameters | ||||
| a, b, c (Å) | 183.03, 58.90, 67.23 | 184.17, 59.84, 67.30 | 183.81, 58.89, 67.27 | 183.53, 58.63, 67.22 |
| α, β, γ (°) | 90.00, 94.91, 90.00 | 90.00, 94.91, 90.00 | 90.00, 95.21, 90.00 | 90.00, 94.92, 90.00 |
| Rotation (Å) | 122 | 160 | 175 | 200 |
| No. of images | 244 | 320 | 350 | 200 |
| Resolution range (Å) | 20.00–1.82 (1.92–1.82) | 21.69–1.65 (1.74–1.65) | 20.95–1.75 (1.84–1.75) | 30.00–1.54 (1.62–1.54) |
| No. of reflections | 100061 (13824) | 276270 (37626) | 250959 (35470) | 376716 (53926) |
| No. of unique reflections | 46573 (6445) | 87302 (12471) | 72402 (10537) | 99870 (14139) |
| Rmerge † (%) | 4.3 (45.4) | 4.6 (45.8) | 6.9 (46.1) | 10.5 (55.6) |
| Completeness (%) | 72.8 (69.7) | 99.4 (98.0) | 99.9 (100.0) | 95.0 (92.4) |
| Redundancy | 2.1 (2.1) | 3.2 (3.0) | 3.5 (3.4) | 3.8 (3.8) |
| Mean I/σ(I) | 13.1 (2.00) | 11.9 (2.3) | 9.9 (2.10) | 7.2 (2.1) |
| CC1/2 ╪ (%) | 99.9 (75.6) | 99.9 (85.0) | 99.8 (89.9) | 98.7 (81.3) |
| Mosaicity (°) | 1.68 | 0.63 | 0.81 | 0.64 |
| Refinement statistics | ||||
| Resolution (Å) | 19.83–1.82 | 21.69–1.65 | 20.95–1.75 | 29.31–1.54 |
| Total of reflections | 44334 | 82910 | 72402 | 94912 |
| Rwork/Rfree ¥ (%) | 16.82/21.43 | 15.82/17.91 | 15.84/18.85 | 13.57/18.66 |
| No. of non-H atoms | ||||
| Protein | 4924 | 4943 | 4905 | 5018 |
| PLP | 15 | 15 | 15 | 15 |
| F6F | 21 | 42 | 63 | 63 |
| EDO | - | - | 4 | - |
| PEG | - | - | 7 | - |
| Na | 1 | 1 | 1 | 1 |
| DMSO | - | - | - | 48 |
| Water | 650 | 732 | 604 | 831 |
| Average B-factors (Å2) | ||||
| Protein | 24.57 | 25.90 | 25.97 | 17.84 |
| PLP | 14.09 | 16.62 | 18.20 | 11.29 |
| F6F | 39.18 | 33.77 | 34.35 | 21.16 |
| EDO | - | - | 53.59 | - |
| PEG | - | - | 40.82 | - |
| Na | 12.55 | 16.85 | 14.62 | 12.33 |
| DMSO | - | - | - | 30.15 |
| Water | 35.23 | 38.36 | 35.62 | 34.89 |
| R.m.s. deviations from ideality | ||||
| Bond lengths (Å) | 0.014 | 0.020 | 0.016 | 0.016 |
| Bond angles (°) | 1.659 | 1.951 | 1.720 | 1.736 |
| Ramachandran plot ‡ | ||||
| Most favored regions | 516 (93.8%) | 521 (94.2%) | 514 (93.8%) | 529 (94.1%) |
| Allowed regions | 33 (6.0%) | 31 (5.6%) | 33 (6.0%) | 32 (5.7%) |
| Generously allowed regions | 1 (0.20%) | 1 (0.20%) | 1 (0.20%) | 0 (0%) |
| Disallowed | 0 (0%) | 0 (0%) | 0 (0%) | 1 (0.20%) |
Rmerge = Σhkl Σi |Ii(hkl) − <I(hkl)>| / Σhkl Σi Ii(hkl), where Ii(hkl) and <I(hkl)> are the observed individual and mean intensities of a reflection with the indices hkl, respectively, Σi is the sum over i measurements of a reflection with the indices hkl, and Σhkl is the sum over all reflections.
CC1/2 indicates the percentage of correlation between intensities of random half data sets.[73]
R = Σhkl ||Fobs| − |Fcalc|| / Σhkl|Fobs|. Rfree is the R value calculated for a random 5% of the data set not included in the refinement.
Percentage of residues in Ramachandran plot regions were determined using PROCHECK.[56]
MD Simulations
The MD simulation performed with a closed α-aminoacrylate structure (PDB ID: 4HN4) was used to investigate the dynamic behavior of residues in the tunnel.[58] We performed MD simulations using Amber14,[59] with the Amber 99SB force field for the protein and general amber force field (GAFF) for the ligands. First, we checked the protonation state using the MCCE program.[60, 61] We then conducted energy minimization for hydrogen atoms, protein side chains, and the whole system for 500, 5000, and 5000 steps, respectively. The systems were then solvated in a rectangular box of 12 Å explicit TIP3P water model by the tleap program in the Amber14 package.[62] Placement of the counter ion of Na+ was based on the Coulombic potential to keep the whole system neutral, and particle mesh Ewald was used to consider long-range electrostatic interactions.[63] Before equilibration, we ran energy minimization of 10000 and 20000 steps for the waters and the system, respectively, and then gradually heated the systems from 250 K for 20 ps, 275 K for 20 ps, and 300 K for 160 ps. We collected the resulting trajectories every 1 ps with a time step of 2 fs in the isothermic–isobaric (NPT) ensemble. The Langevin thermostat with a collision frequency of 2 ps−1 was employed to maintain a temperature of 300 K. We also used the SHAKE procedure to constrain hydrogen atoms during MD simulations.[64] Finally, the production runs were performed for 50 ns at 300 K.
We manually replaced the ligand N-(4′-trifluoromethoxybenzenesulfonyl)-2-aminoethyl phosphate (F9, Scheme 1) in the α-site of 4HN4 with G3P and indole molecules and performed MD simulations for 10 ns. We used the structure formed after 10 ns MD simulation to perform a docking simulation. Indole docking was performed by the Autodock[65] program with the Lamarckian genetic algorithm, and the side chains of nearby residues were set flexible. The Vcharge[66] program was used to assign partial charges to the indole molecule. The Autodock scoring function is a subset of the AMBER force field that treats molecules using the United Atom model. We performed docking of indole at five different sites along the tunnel. The final docking result was obtained by 10 runs of simulation with 2.5 million rounds of energy evaluation in each run. Each round of energy evaluation was for one possible orientation of the indole molecule at the docking site.
Accession Numbers
The Worldwide Protein Data Bank accession file names for the new TS X-ray crystal structures presented in this work are as follows: structures with three, three, two, and one molecule(s), respectively, of F6 bound per αβ dimer, PDB ID models 4WX2; 4ZQC; 4Y6G, and 5BW6. The crystal parameters, data collection, and refinement statistics are summarized in Table 1.
Results and Discussion
The Binding of F6 to Tryptophan Synthase
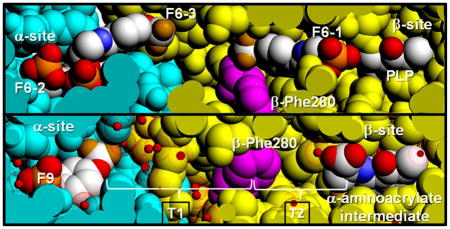
The crystal structure PDB ID: 4WX2 contains one F6 molecule (F6-1) in the β-subunit, and two F6 molecules (F6-2 and F6-3) within the α-subunit near the αβ-subunit interface region (Figure 1). The F6 ligand is an amphipathic compound; the hydrophobic head located at one end of the molecule consists of the trifluoromethoxy and phenyl groups; the polar tail is composed of the carboxamido and the ethyl phosphoryl groups. Fluorine atoms covalently bound to carbon atoms are very poor hydrogen bonders and rarely act as hydrogen-bond acceptors.[67] Consequently, the hydrophobic trifluoromethoxy group of F6 in these sites mainly interacts through weak contacts with neighboring amino acid residues. In each of the three sites hydrogen bonding interactions occur through the carboxamido and phosphoryl groups (Figure 1b,c) and are listed in Table S1A–C. The conformations of bound F6 determined from different sites and different crystal structures are compared in Figure 1d. Inspection of Figure 1a shows that the aromatic ring and carboxamido nitrogen of F6-3 appears solvent-exposed in the crystal structure (left side of the structure). This surface of F6 is the region that would be expected to be enclosed by the amino acid residues of loop αL6. As discussed earlier, this portion of the loop is disordered.
Figure 1.

(a) Stereo view showing F6 bound to the three sites identified in PDB ID: 4WX2 designated as F6-1, F6-2, and F6-3. The location of βPhe280 is also shown. Color scheme: α-subunit green, β-subunit gold, C yellow, and other atoms shown in CPK colors. (b) Details of the atom contacts in the β-subunit between F6-1, site residues, waters and PLP. (c) Details of the atom contacts in the α-subunit between F6-2 and F6-3 and with site residues and waters. Color schemes in (b) and (c); waters shown as red dot surfaces, α-subunit residues, orange; β-subunit residues, green; F6-1, F6-2 and F6-3, C yellow; all other atoms shown in CPK colors; PLP is shown in standard CPK colors. (d) Overlay aligning aromatic rings of F6 molecules from crystal structures PDB IDs: 2CLE, 2CLF, 4WX2, 4ZQC, and 4Y6G. For better visualization, F6 molecules were colored differently; PDB ID: 2CLE (F6-2, cyan), PDB ID: 2CLF (F6-1, orange; F6-2, yellow), PDB ID: 4WX2 (F6-1, blue; F6-2, marine; F6-3, light blue), PDB ID: 4Y6G (F6-1, green; F6-2, limon),PDB ID: 4ZQC (F6-1, red; F6-2, warm pink; F6-3, salmon).
The F6-1 Binding Site
Prior to this work, out of the more than 70 X-ray structures reported for TS in the PDB, only one structure (PDB ID: 2CLF) includes a ligand bound in the interconnecting tunnel. The binding site for F6-1 in PDB ID: 4WX2 (Figure 1b; Figure 2) resides almost completely within the tunnel connecting the β-site with the α-site. The hydrophobic head makes weak contacts in the β-subunit with residues Cys170, Leu174, Leu188, Tyr186, Pro194, Phe280, Phe306, and water β894. The polar tail makes extensive hydrogen bonding contacts with amino acid residues Lys87, Glu109, Thr110, Gly111, Ala112, and His115, neighboring water molecules β641, β667, and β874, and the PLP C-4′ carbon (Table S1A). These amino acid residues form part of the tunnel wall in the β-subunit and extend from Lys87 and Gln114 in the β-catalytic site to the side chains of Leu174 and Phe280 approximately 18 Å away. Two of the waters are hydrogen-bonded to the F6 phosphoryl group, and one is hydrogen-bonded to the oxygen of the F6 carboxamido group.
Figure 2.

Representations of the electron density maps for the F6-1 binding site in the β-subunit PDB IDs: 4WX2, 4ZQC, 4Y6G, 2CLF, and rr2CLF. (a) PDB ID: 4WX2; (b) PDB ID: 4ZQC; (c) PDB ID: 4Y6G; (d–e), PDB ID: 2CLF; (e), re-refined model rr2CLF. Sections of the electron densities contoured at 1.2 rms (a–d), 0.5 rms (e), and 0.7 rms (f) from the 2Fo-Fc maps. The coordinate and structure factor files were loaded in Coot 0.8.1. The Fo-Fc electron density maps were deactivated and the clipping planes (a.k.a. “slab”) were adjusted to improve the visualization of the electron density of interest surrounding each F6-1 molecule.
When we compared the X-ray structure PDB ID: 2CLF with PDB ID: 4WX2, we concluded that a careful reevaluation of the orientation and weak ligand-protein interactions of F6 was needed. The position of F6-1 in the tunnel of the model PDB ID: 4WX2 was found to overlap with the position assigned to F6 in the structural model PDB ID: 2CLF[45] (Figure 2). However, the orientation of F6-1 is flipped by approximately 180° in structural models PDB IDs: 4WX2, 4Y6G, and 4ZQC in comparison with that assigned to F6[45] in the model PDB ID: 2CLF. We chose the flipped orientation for two reasons: (1) this orientation gives a significantly better fit to the electron density map (Figure 2; Table S2). See the Supplemental Section for a more detailed account of the fitting of the electron densities in PDB IDs: 4WX2, 4ZQC, 4Y6G, and the re-refinement of PDB ID: 2CLF as rr2CLF. (2) The flipped orientation places the hydrophobic head of F6-1 into a highly nonpolar region of the tunnel, and the carboxamido nitrogen within hydrogen-bonding distance of βGlu109, while the polar phosphoryl group occupies a polar subsite of the β-catalytic site designed for the binding of the carboxylate moieties of the substrates L-Ser and L-Trp (Figure 1b, Table S1). We concluded that the alternative fit in PDB ID: 2CLF[45] contradicts intuitive models of electrostatic interactions.
Interactions with F6-2 and F6-3
Molecules F6-2 and F6-3 in PDB ID: 4WX2 are bound to sites within the α-subunit near the αβ-subunit interface and are hydrogen-bonded to each other (Figure 1c; see below). Weak contacts between protein residues and the hydrophobic head of F6-2 involve α-subunit residues Phe22, Ala59, Asp60, Leu100, Leu127, Ala129, Ile153, Tyr175, and Phe212, and β-subunit residue Pro18, and waters α437 and α626. The α-subunit residues Gly213, Gly234, and Ser235, and water molecules 458 and 523 all form hydrogen-bonds to polar atoms on F6-2 (see Table S1B, C). Molecule F6-3 binds within the α-subunit active site at a locus which overlaps with the substrate IGP binding site (Figure 1c) as previously discussed.[45] The orientation and position of the F6-2 ligand in PDB IDs: 4WX2, 4ZQC, 4Y6G, and 5BW6 matches well with the same molecule in PDB IDs: 2CLE and 2CLF (Figure 1d). The F6-3 ligand was not observed in PDB IDs: 2CLE, 2CLF[45] or 5BW6. Under soaking conditions with lower F6 concentrations (1 mM) we also obtained a crystal structure (PDB ID: 4Y6G) with F6 bound to two loci, corresponding to the locations and orientations of F6-1 and F6-2. However, lowering the soaking time at the same concentration we obtained a crystal structure (PDB ID: 5BW6) with F6 molecule (F6-2) bound exclusively in the α-site.
Effects of F6 Binding on Conformation
The α-subunits of PDB IDs: 4WX2, 4ZQC, 4Y6G, and 5BW6 exhibit the conformation common to many of the TS complexes with substrate analogues of IGP bound to the α-subunit. In this conformation (see Figure S1), the ligand occupies the α-catalytic site, an amphipathic cavity characterized by a phosphoryl group binding subsite defined by the Nα atoms of Gly213, and Gly234, and the Nα and Oγ of Ser235, and a hydrophobic region which accommodates the nonpolar surfaces of the glycerol chain and the indolyl ring system. In comparison, the binding of IGP involves a total of six hydrogen bonding interactions with the protein (Asp60, Tyr175, Glu49, Gly213, Ser235): four with the phosphoryl group, one with a hydroxyl group of the glycerol chain, and one at N-1 of the indolyl ring.[16, 37]
α-Loops 2 and 6 (αL2 and αL6, respectively) play important roles in forming a closed or partially closed conformation of the α-subunit.[27, 28] When the cofactor is in the internal aldimine form (with an open β-subunit), the α-subunit typically gives ligand complexes where the portion of αL6 which folds over the opening to the α-site (residues 179–196) is partially disordered. These complexes are considered to be open or partially open conformations where the substrate for the α-site, IGP, has easy passage between solvent and site. In the structure PDB ID: 4WX2, 15 of 18 αL6 residues (α179–193) are disordered and therefore the site is solvent-exposed (viz. Figure 1a). When the α-subunit of PDB ID: 4WX2 is aligned with the α-subunit of PDB ID: 4Y6G, the folding is highly similar and the overlap is typically within experimental error, except where there are differences due to localized perturbations from ligand-protein interactions and differences in the degree of disorder in αL6 (15 disordered residues in PDB ID: 4WX2 and 11 residues (α182–192) in PDB ID: 4Y6G when the electron density map was contoured at 1.0 rms). At the same map contour level in PDB IDs: 4ZQC and 5BW6, respectively, 12 of 18 (α181–192) and 13 of 18 (α180–192) αL6 main chain residues could not be traced. However, in PDB ID: 4ZQC, partial electron densities are observed for the main chain residues αGly181, αThr183, αGly184, αAla185, αAsn187, αGly189, αAla190, and αPro192 (data not shown). All 18 αL6 residues are traceable when the electron density map was contoured at 0.4 rms. Interestingly, R and Rfree values slightly increased (13.72/18.89%) when residues α181–192 were removed from the current PDB ID: 4ZQC (13.57/18.66%) during the restrained refinement cycles in Refmac5. Consequently, there are small differences in folding, which appear to arise from the binding of F6–2 at this locus (Figure S1).
The β-subunit retains an open conformation in all of the F6 complexes presented herein. However, it appears that in the PDB ID: 4WX2 structure, the COMM domain has undergone a small translation/rotation which alters part of the αβ-subunit interface (see Figure S1). The COMM domain motion is similar to the motion seen in structures where the β-subunit is switched to the closed conformation (i.e., conformations seen in structures of the α-aminoacrylate and quinonoid intermediates[40, 44]). Thus, it appears that the binding of the additional F6 molecule along the subunit interface brings about a partial shift in the conformation of the β-subunit toward the closed β-subunit conformation.
Overall the three TS structures are almost the same despite containing either one (PDB ID: 2CLE or 5BW6), two (PDB ID: 4Y6G or 2CLF), or three (PDB ID: 4WX2 or 4ZQC) F6 molecules. All three structures exhibit significant disorder in the αL6 loop (residues α179-α196). The RMSD for the PDB ID: 4WX2 structure superimposed with PDB IDs: 2CLE, 4ZQC, 4Y6G, and 5BW6 is, respectively, 0.338 Å for 568 Cα atoms, 0.177 Å for 567 Cα atoms, 0.224 Å for 567 Cα atoms, and 0.300 Å for 540 Cα atoms, indicating PDB ID: 4WX2 is more closely related to PDB ID: 4ZQC and PDB ID: 4Y6G than to the PDB IDs: 2CLE and 5BW6. However, these three structures show noticeable differences at the αβ-interface. Compared to the PDB ID: 2CLE structure, the COMM domains in the PDB IDs: 4WX2, 4ZQC, 4Y6G clamp down on to the β-subunit, possibly due to the binding of the F6-1 molecule in the tunnel near the β-active site (Figure S1). In contrast to most TS structures displaying an unobstructed tunnel, all the structures with an F6 molecule in the tunnel show βPhe280 folded into the tunnel wall, opening the tunnel and indicating the flexibility of βPhe280.
Properties of the Tunnel
The interactions observed between F6-1 and the β-subunit residues which comprise the tunnel emphasize the highly nonpolar nature of the tunnel within the β-subunit. Therefore, the new structures described in this work provide important insights into the properties of the tunnel. The tunnel can be divided into two sections: T1, which includes F6-2 and F6-3 in the structures discussed above, is more polar and extends from the α-site to βPhe280 in the β-site; and T2, which extends from βPhe280 to PLP in the β-site, is less polar and binds F6-1 (Figure 3). Section T1 is about 18 Å long and T2 is about 12 Å long. Therefore, indole travels approximately 30 A after it is released from the α-active site to reach the β-active site. The tunnel is widest at the αβ-interface, narrows at the position of βPhe280 near the O-7 of the F6-1 molecule to about 6.7 Å, and then widens again.
Figure 3.

The water networks link βTyr16 to βLys167, and βIle20 to βSer178 along the tunnel (structure PDB ID: 4HN4). All water molecules within 8 Å of F9, βPhe280 and A-A are shown in the Figure. The brackets indicate regions T1 and T2 of the tunnel.
The tunnel section designated T1 is filled with water molecules arranged along the αβ-interface and extends down to the CF3 group of F6-1. In all TS crystal structures with resolutions sufficient to allow identification of water molecules bound to specific sites on the protein, the T1 cavity is occupied by two lattices of water molecules: one with four waters H-bonded to each other and to the protein via the carbonyl oxygens of αLeu58, αGly181, and αArg179, the other with three waters H-bonded to each other and to the carbonyl oxygens of βTyr16 and αAsp56 and to the amide nitrogens of βGly281 and βPhe280. It is important to notice that in the subset of these structures which do not have F6 bound in the T2 section of the tunnel, there is no electron density assignable to water molecules. Consequently, it appears either that T2 contains no water, or that waters in the T2 cavity are highly disordered.
To further investigate the properties of the tunnel, we undertook MD studies on 4HN4 to model the motions of water molecules in the tunnel. During 50 ns MD simulations, a bridge of five water molecules occasionally forms, linking βTyr16 and βLys167 in region T1. More stably, another four water molecules bridge the interaction between βIle20 and βSer178 at the αβ-subunit interface, while most of the time there is either one or no water molecule in region T2 of the tunnel between βPhe280 and the β-catalytic site (Figure 3). Similar MD studies have been reported as a means for investigating the transport of ammonia and water in the imidazole glycerol phosphate synthase complex.[13–15]
Functional Significance of Tunnel Water and Tunnel Vacuum
There is a growing body of literature concerning the properties and function of hydrophobic tunnels in the regulation of water and ion transport across biological membranes. Water in hydrophobic tunnels, both in artificial carbon nanotubes and in biological membranes, can exhibit properties that are quite different from the behavior of bulk water.[3] As to be expected, the transport of water molecules in hydrophobic tunnels becomes increasingly unfavorable as the tunnel narrows. At diameters less than about 14 Å, the unfavorable interaction leads to stochastic liquid–vapor transitions between pore interiors that fluctuate between hydrated or “wetted” and dry or “dewetted” states. [13–15] This effect becomes most pronounced at diameters of 9 to 12 Å. Below 9 Å hydrophobic pores are essentially dry. While the hydrated state is capable of transferring water molecules along the hydrophobic pore, the dry state has a significant energy barrier to the transfer of water molecules (and ions). Thus narrow hydrophobic nanopores generate significant energy barriers to the transfer of water, polar, and ionic small molecules.[3, 13–15]
According to the behavior expected of narrow hydrophobic nanotubes, the tunnel between βPhe280 and the β-site is dewetted or dry, and in agreement with the MD simulations, region T2 provides a high energy barrier for transferring water or ions between the α- and β-sites. Hence, the dry, nonpolar nature of T2 would also preclude transfer of, for example, G3P from the α-site to the β-site. As indicated by the MD docking simulations (see Figure 4), due to its highly nonpolar character, indole can pass through both the wetted and the dewetted regions of the tunnel. We speculate that the barrier to the transfer of polar molecules and ions functions to prevent the transfer of water into the β-site where attack of water on the Cβ of the α-aminoacrylate intermediate results in the formation of pyruvate and ammonium ions.[68]
Figure 4.

Indole docked along the tunnel (PDB ID: 4HN4). (a) Results of docking showing the path of indole movement in the tunnel. (b) αPhe212, βLeu21, βPhe280 side chain rotations giving way for indole. The indole subsite in the β-catalytic site is modeled by the position of benzimidazole bound to the α-aminoacrylate intermediate (PDB ID: 4HPX).
Modeling the Channeling Hypothesis
According to the channeling hypothesis for TS, indole produced by cleavage of IGP in the α-site is channeled to the β-site to react with the α-aminoacrylate intermediate via the interconnecting tunnel. To better understand the mechanism of indole channeling we performed flexible docking studies with indole placed at different positions in the tunnel to model its passage (Figure 4a). This modeling clearly shows that indole can pass through the tunnel with little steric clashing, in agreement with previous computational studies performed with coarse-grained Brownian dynamics simulations.[35] Because both indole and the tunnel are mainly nonpolar, there are no major attractive forces that steer the diffusion of indole from the α-site to the β-site. However, the indole subsite of the β-subunit shows considerable specificity for the binding of indole and structural analogues of indole,[44, 69, 70] and the free energy change associated with indole binding to this site provides a driving force for the transfer of indole. The docking study shows that (a) steric clashing is easily alleviated by the expected thermal motions of protein side chains along the length of the tunnel; (b) indole can be docked in the large cavity between the α-site and the hydrophobic entrance at the αβ-interface; (c) at the interface, the phenyl ring of αPhe212 slightly rotates to give way, allowing indole to move across the αβ-interface; βLeu21 rotates, allowing indole to enter the β-subunit portion of the tunnel; (d) when the side chain of the βPhe280 phenyl ring rotates away due to fluctuations, the tunnel is cleared for the passage of indole; and (e) finally, indole enters the β-catalytic site cavity and binds to the indole ring subsite, positioning indole for reaction with the α-aminoacrylate intermediate to give E(Q2/3) in stage II of the β-reaction. The indole binding cavity is adjacent to the β-carbon of the α-aminoacrylate complex and is bounded by the βGlu109 carboxyl, which hydrogen bonds to N-1 of the indole ring, and by the side chains of βLeu166, βCys170, βThr190, βHis115, and βPhe306, which form weak interactions with the indole rings. After reaching this cavity, adjustments involving diffusion and rotation allow indole to eventually bind and react to form a C-C single bond between C-3 of indole and Cβ of the α-aminoacrylate intermediate. Here we aligned and manually placed benzimidazole (BZI) from crystal structure PDB ID: 4HPX,[44] which shows BZI and the α-aminoacrylate complex in the β-site, to indicate the position of indole just prior to nucleophilic attack of the indole C-3 at Cβ of the α-aminoacrylate intermediate (Figure 4b). Investigation of the flexibility of the tunnel was also undertaken by comparing the B factors of Cα of residues along the tunnel from two different TS structures (Figure S3).
One of the controversial features of the mechanism proposed for TS channeling has been the postulated gating of the tunnel by the side chain of βPhe280. Crystal structures show that this side chain takes up two conformations: one where the phenyl ring partially blocks the tunnel (most structures) and one where it is folded into the tunnel wall giving an open conformation. We performed 50 ns molecular dynamics (MD) simulations using the PDB ID: 4HN4 crystal structure of the α-aminoacrylate intermediate.[44] Its α-site ligand F9, an inhibitive analogue of IGP, is a close match to F6-2 both structurally and conformationally. The simulation trajectory indicates that the βPhe280 side chain will occasionally swing back and forth between blocked and open conformations within the 50 ns period (Figure 5). The dihedral angle χ(N-Cα-Cβ-Cγ) has two main rotamers at 101° (tunnel open) and 190° (tunnel blocked), respectively. To confirm the rotamer switch of this dihedral angle, we performed another MD simulation with a different random number seed and came to similar results (Figure S4). Following cleavage from IGP, the docking study shows that indole enters into the tunnel from the α-site into the space surrounded by β-subunit residues Leu21, Leu174, and Phe280, waiting for the phenyl ring to move aside, and then quickly goes through into the β-site when the open conformation forms. The MD simulation shows the flipping of the Phe280 side chain occurs on a nanosecond timescale, while the experimentally measured rates of indole transfer from the α-site to β-site, covalent reaction, and turnover occurs on timescales of microseconds, milliseconds, and seconds, respectively,[25, 27, 30, 41, 42, 71] indicating that steric hindrance by the Phe280 side chain cannot play a gating role in the regulation of channeling in tryptophan synthase. Consequently, both dynamic modeling simulations and experimental measurements suggest that the gating effect from residues positioned along the length of the tunnel is insignificant to tunnel function.[72] This finding is similar to the conclusions drawn by Amaro et al.[13–15] concerning the ammonia-channeling imidazole glycerol phosphate synthase complex.
Figure 5.

Side chain rotation of βPhe280 during the passage of indole through the tunnel (structure PDB ID: 4HN4). (a) Representation of two averaged positions (yellow blocked position and red open position) of βPhe280 in the tunnel. (b) Histogram of the main rotamers of the βPhe280 side chain. (c) βPhe280 side chain dihedral angle χ(N-Cα-Cβ-Cγ) changes during a 50 ns MD simulation.
In summary, the results of these structural studies unambiguously establish that the interconnecting tunnel between the α- and β-sites of tryptophan synthase can accommodate small hydrophobic molecules, such as the amphipathic F6, with cross-sections and polarities similar to indole; the flexible docking and molecular dynamics studies confirm that transfer of indole is not significantly hindered by steric clashing with the phenyl ring of βPhe280. Therefore, we conclude βPhe280 does not play a gating role in the regulation of channeling. Inspection of high resolution structures taken from the tryptophan synthase structural data base and molecular dynamics analyses indicate that a portion of the tunnel is too hydrophobic to allow passage of water molecules between the α- and β-catalytic sites but will accommodate the non-polar head of F6. We propose that this hydrophobic section of the tunnel restricts access of water into the β-site and thereby inhibits the deleterious side reaction of water with the α-aminoacrylate intermediate to form pyruvate and ammonium ions. Thus the tunnel is a hydrophobic, dewetted nanopore, which selectively transfers indole but discriminates against the passage of water. Work is now underway to further investigate the proposed function of the dewetted portion of the tunnel as a selectivity filter which rejects water but transfers nonpolar small molecules.
Supplementary Material
Highlights.
X-ray structures of a ligand threaded into the tunnel connecting the α- and β-sites
βPhe280 appears not to play a gating role in transfer of indole through the tunnel
X-ray structures and MD simulations indicate a dewetted channel
Hydrophobic indole freely diffuses between the α- and β-sites via the tunnel
Exclusion of water inhibits a deleterious side reaction in the catalytic cycle
Acknowledgments
We thank and acknowledge the staff at the SIBYLS. Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health (NHI) under award number R01GM097569. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number CHE-130009.
Abbreviations
- TS
tryptophan synthase from S. typhimurium
- F6
N-(4′-trifluoromethoxybenzoyl)-2-aminoethyl phosphate
- F9
N-(4′-trifluoromethoxybenzenesulfonyl)-2-aminoethyl phosphate
- PLP
pyridoxal-5′-phosphate
- IGP
3-indole-D-glycerol 3′-phosphate
- G3P
D-glyceraldehyde 3-phosphate
- E(Ain)
the internal aldimine (Schiff base) intermediate
- E(GD1)
the L-Ser gem diamine species
- E(Aex1)
the external aldimine intermediate formed between the PLP cofactor and L-Ser
- E(Q1)
the L-Ser quinonoid intermediate
- E(A-A)
the α-aminoacrylate Schiff base intermediate
- E(Q3)
the quinonoid intermediate that accumulates in the reaction between E(A-A) and indole
- E(Aex2)
the L-Trp external aldimine intermediate
- E(GD2)
the L-Trp gem diamine species
- F6-1, F6-2, F6-3
the binding sites for F6 in the β- and α-subunits
- MD
molecular dynamics
- loop αL2
residues α53-α60
- loop αL6
residues α179-α193
- COMM domain
residues β102-β189
- section T1
the tunnel extending from the α-site to βPhe280 in the β-subunit
- section T2
the tunnel extending from βPhe280 to PLP in the β-site
- B factor
indicator of the relative vibrational motion of atoms in crystal structures
- MR
molecular replacement. The Worldwide Protein Data Bank accession numbers for all published structures are provided in the text
Footnotes
Supporting Information Available
The Supplemental Information provides more technically detailed descriptions of the crystallographic work regarding the following: (1) refinement of electron density for the molecule bound to site F6-1 in the β-subunit of TS for the model, PDB ID: 2CLF; (2) comparison of the conformations of TS containing one, two, or three molecules of F6 bound to the αβ-dimeric units of models, PDB IDs: 5BW6, 4Y6G, 4WX2, and 4ZQC; (3) protein-water interactions in the tunnel; and (4) additional MD simulations. This material is available free of charge via the Internet
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baker LA, Baldus M. Characterization of membrane protein function by solid-state NMR spectroscopy. Curr Opin Struc Biol. 2014;27:48–55. doi: 10.1016/j.sbi.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 2.Horn R, Roux B, Aqvist J. Permeation Redux: Thermodynamics and Kinetics of Ion Movement through Potassium Channels. Biophys J. 2014;106:1859–1863. doi: 10.1016/j.bpj.2014.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aryal P, Sansom MSP, Tucker SJ. Hydrophobic Gating in Ion Channels. J Mol Biol. 2015;427:121–130. doi: 10.1016/j.jmb.2014.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ziervogel BK, Roux B. The Binding of Antibiotics in OmpF Porin. Structure. 2013;21:76–87. doi: 10.1016/j.str.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galdiero S, Falanga A, Cantisani M, Tarallo R, Della Pepa ME, D’Oriano V, Galdiero M. Microbe-Host Interactions: Structure and Role of Gram-Negative Bacterial Porins. Curr Protein Pept Sc. 2012;13:843–854. doi: 10.2174/138920312804871120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaffer PL, Goehring A, Shankaranarayanan A, Gouaux E. Structure and Mechanism of a Na+-Independent Amino Acid Transporter. Science. 2009;325:1010–1014. doi: 10.1126/science.1176088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis RH. Channeling in Neurospora Metabolism. In: Vogel HJ, Lampen JO, Bryson V, editors. Organizational Biosynthesis. Academic Press; New York, New York: 1967. pp. 303–325. [Google Scholar]
- 8.Friedrich P. Supramolecular Enzyme Organization: Quaternary Structure and Beyond. Pergamon Press; New York: 1984. [Google Scholar]
- 9.Holden HM, Thoden JB, Raushel FM. Carbamoyl phosphate synthetase: a tunnel runs through it. Curr Opin Struc Biol. 1998;8:679–685. doi: 10.1016/s0959-440x(98)80086-9. [DOI] [PubMed] [Google Scholar]
- 10.Raushel FM, Thoden JB, Holden HM. Enzymes with molecular tunnels. Accounts Chem Res. 2003;36:539–548. doi: 10.1021/ar020047k. [DOI] [PubMed] [Google Scholar]
- 11.Raushel FM, Thoden JB, Holden HM. The amidotransferase family of enzymes: Molecular machines for the production and delivery of ammonia. Biochemistry-Us. 1999;38:7891–7899. doi: 10.1021/bi990871p. [DOI] [PubMed] [Google Scholar]
- 12.Miles EW, Rhee S, Davies DR. The molecular basis of substrate channeling. J Biol Chem. 1999;274:12193–12196. doi: 10.1074/jbc.274.18.12193. [DOI] [PubMed] [Google Scholar]
- 13.Amaro R, Tajkhorshid E, Luthey-Schulten Z. Developing an energy landscape for the novel function of a (beta/alpha)8 barrel: ammonia conduction through HisF. Proc Natl Acad Sci U S A. 2003;100:7599–7604. doi: 10.1073/pnas.1331150100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amaro RE, Luthey-Schulten Z. Molecular dynamics simulations of substrate channeling through an alpha-beta barrel protein. Chem Phys. 2004;307:147–155. [Google Scholar]
- 15.Amaro RE, Myers RS, Davisson VJ, Luthey-Schulten ZA. Structural elements in IGP synthase exclude water to optimize ammonia transfer. Biophys J. 2005;89:475–487. doi: 10.1529/biophysj.104.058651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hyde CC, Ahmed SA, Padlan EA, Miles EW, Davies DR. 3-Dimensional Structure of the Tryptophan Synthase Alpha-2-Beta-2 Multienzyme Complex from Salmonella-Typhimurium. J Biol Chem. 1988;263:17857–17871. [PubMed] [Google Scholar]
- 17.Yanofsky C, Rachmeler M. The Exclusion of Free Indole as an Intermediate in the Biosynthesis of Tryptophan in Neurospora-Crassa. Biochim Biophys Acta. 1958;28:640–641. doi: 10.1016/0006-3002(58)90533-x. [DOI] [PubMed] [Google Scholar]
- 18.Tatum EL, Bonner D. Indole and serine in the biosynthesis and breakdown of tryptophane. P Natl Acad Sci USA. 1944;30:30–37. doi: 10.1073/pnas.30.2.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crawford IP, Yanofsky C. On the Separation of the Tryptophan Synthetase of Escherichia-Coli into 2 Protein Components. P Natl Acad Sci USA. 1958;44:1161–1170. doi: 10.1073/pnas.44.12.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dunn MF, Niks D, Ngo H, Barends TRM, Schlichting I. Tryptophan synthase: the workings of a channeling nanomachine. Trends Biochem Sci. 2008;33:254–264. doi: 10.1016/j.tibs.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 21.Miles EW. Tryptophan Synthase: Structure, Function, and Subunit Interaction. In: Meister A, editor. Advances in Enzymology and Related Areas of Molecular Biology. Vol. 49. John Wiley & Sons, Inc; USA: 1979. [DOI] [PubMed] [Google Scholar]
- 22.Demoss JA. Studies on Mechanism of Tryptophan Synthetase Reaction. Biochim Biophys Acta. 1962;62:279. doi: 10.1016/0006-3002(62)90041-0. [DOI] [PubMed] [Google Scholar]
- 23.Creighton TE. A Steady-State Kinetic Investigation of the Reaction Mechanism of the Tryptophan Synthase of Escherichia coli. Eur J Biochem. 1970;13:1–10. doi: 10.1111/j.1432-1033.1970.tb00892.x. [DOI] [PubMed] [Google Scholar]
- 24.Dunn MF, Roy M, Robustell B, Aguilar V. Biochemistry of Vitamin B6. In: Korpela TK, Christen P, editors. 7th International Congress on Chemical and Biological Aspects of Vitamin B6 Catalysis; Proceedings of the 7th International Congress on Chemical and Biological Aspects of Vitamin B6 Catalysis; Turku, Finland: Birkhauser; 1987. [Google Scholar]
- 25.Dunn MF, Aguilar V, Brzovic P, Drewe WF, Houben KF, Leja CA, Roy M. The Tryptophan Synthase Bienzyme Complex Transfers Indole between the Alpha-Sites and Beta-Sites Via a 25–30 a Long Tunnel. Biochemistry-Us. 1990;29:8598–8607. doi: 10.1021/bi00489a015. [DOI] [PubMed] [Google Scholar]
- 26.Lane AN, Kirschner K. Mechanism of the Physiological Reaction Catalyzed by Tryptophan Synthase from Escherichia-Coli. Biochemistry-Us. 1991;30:479–484. doi: 10.1021/bi00216a025. [DOI] [PubMed] [Google Scholar]
- 27.Brzovic PS, Sawa Y, Hyde CC, Miles EW, Dunn MF. Evidence That Mutations in a Loop Region of the Alpha-Subunit Inhibit the Transition from an Open to a Closed Conformation in the Tryptophan Synthase Bienzyme Complex. J Biol Chem. 1992;267:13028–13038. [PubMed] [Google Scholar]
- 28.Brzovic PS, Hyde CC, Miles EW, Dunn MF. Characterization of the Functional-Role of a Flexible Loop in the Alpha-Subunit of Tryptophan Synthase from Salmonella-Typhimurium by Rapid-Scanning, Stopped-Flow Spectroscopy and Site-Directed Mutagenesis. Biochemistry-Us. 1993;32:10404–10413. doi: 10.1021/bi00090a016. [DOI] [PubMed] [Google Scholar]
- 29.Anderson KS, Miles EW, Johnson KA. Serine Modulates Substrate Channeling in Tryptophan Synthase - a Novel Intersubunit Triggering Mechanism. J Biol Chem. 1991;266:8020–8033. [PubMed] [Google Scholar]
- 30.Brzovic PS, Ngo K, Dunn MF. Allosteric Interactions Coordinate Catalytic Activity between Successive Metabolic Enzymes in the Tryptophan Synthase Bienzyme Complex. Biochemistry-Us. 1992;31:3831–3839. doi: 10.1021/bi00130a014. [DOI] [PubMed] [Google Scholar]
- 31.Pan P, Woehl E, Dunn MF. Protein architecture, dynamics and allostery in tryptophan synthase channeling. Trends Biochem Sci. 1997;22:22–27. doi: 10.1016/s0968-0004(96)10066-9. [DOI] [PubMed] [Google Scholar]
- 32.Dunn MF. Allosteric regulation of substrate channeling and catalysis in the tryptophan synthase bienzyme complex. Arch Biochem Biophys. 2012;519:154–166. doi: 10.1016/j.abb.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bahar I, Jernigan RL. Cooperative fluctuations and subunit communication in tryptophan synthase. Biochemistry-Us. 1999;38:3478–3490. doi: 10.1021/bi982697v. [DOI] [PubMed] [Google Scholar]
- 34.Spyrakis F, Raboni S, Cozzini P, Bettati S, Mozzarelli A. Allosteric communication between alpha and beta subunits of tryptophan synthase: Modelling the open-closed transition of the alpha subunit. Bba-Proteins Proteom. 2006;1764:1102–1109. doi: 10.1016/j.bbapap.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 35.Fatmi MQ, Chang CEA. The Role of Oligomerization and Cooperative Regulation in Protein Function: The Case of Tryptophan Synthase. Plos Comput Biol. 2010;6 doi: 10.1371/journal.pcbi.1000994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ahmed SA, Ruvinov SB, Kayastha AM, Miles EW. Mechanism of Mutual Activation of the Tryptophan Synthase-Alpha and Beta-Subunits - Analysis of the Reaction Specificity and Substrate- Induced Inactivation of Active-Site and Tunnel Mutants of the Beta-Subunit. J Biol Chem. 1991;266:21548–21557. [PubMed] [Google Scholar]
- 37.Schlichting I, Yang XJ, Miles EW, Kim AY, Anderson KS. Structural and Kinetic-Analysis of a Channel-Impaired Mutant of Tryptophan Synthase. J Biol Chem. 1994;269:26591–26593. [PubMed] [Google Scholar]
- 38.Weyand M, Schlichting I. Structural basis for the impaired channeling and allosteric inter-subunit communication in the beta A169L/beta C170W mutant of tryptophan synthase. J Biol Chem. 2000;275:41058–41063. doi: 10.1074/jbc.C000479200. [DOI] [PubMed] [Google Scholar]
- 39.Ruvinov SB, Yang XJ, Parris KD, Banik U, Ahmed SA, Miles EW, Sackett DL. Ligand-mediated changes in the tryptophan synthase indole tunnel probed by nile red fluorescence with wild type, mutant, and chemically modified enzymes. J Biol Chem. 1995;270:6357–6369. doi: 10.1074/jbc.270.11.6357. [DOI] [PubMed] [Google Scholar]
- 40.Ngo H, Kimmich N, Harris R, Niks D, Blumenstein L, Kulik V, Barends TR, Schlichting I, Dunn MF. Allosteric regulation of substrate channeling in tryptophan synthase: Modulation of the L-Serine reaction in stage I of the ss-reaction by alpha-site ligands. Biochemistry-Us. 2007;46:7740–7753. doi: 10.1021/bi7003872. [DOI] [PubMed] [Google Scholar]
- 41.Harris RM, Dunn MF. Intermediate trapping via a conformational switch in the Na+-activated tryptophan synthase bienzyme complex. Biochemistry-Us. 2002;41:9982–9990. doi: 10.1021/bi0255672. [DOI] [PubMed] [Google Scholar]
- 42.Harris RM, Ngo H, Dunn MF. Synergistic effects on escape of a ligand from the closed tryptophan synthase bienzyme complex. Biochemistry-Us. 2005;44:16886–16895. doi: 10.1021/bi0516881. [DOI] [PubMed] [Google Scholar]
- 43.Leja CA, Woehl EU, Dunn MF. Allosteric Linkages between Beta-Site Covalent Transformations and Alpha-Site Activation and Deactivation in the Tryptophan Synthase Bienzyme Complex. Biochemistry-Us. 1995;34:6552–6561. doi: 10.1021/bi00019a037. [DOI] [PubMed] [Google Scholar]
- 44.Niks D, Hilario E, Dierkers A, Ngo H, Borchardt D, Neubauer TJ, Fan L, Mueller LJ, Dunn MF. Allostery and Substrate Channeling in the Tryptophan Synthase Bienzyme Complex: Evidence for Two Subunit Conformations and Four Quaternary States. Biochemistry-Us. 2013;52:6396–6411. doi: 10.1021/bi400795e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ngo H, Harris R, Kimmich N, Casino P, Niks D, Blumenstein L, Barends TR, Kulik V, Weyand M, Schlichting I, Dunn MF. Synthesis and characterization of allosteric probes of substrate channeling in the tryptophan synthase bienzyme complex. Biochemistry-Us. 2007;46:7713–7727. doi: 10.1021/bi700385f. [DOI] [PubMed] [Google Scholar]
- 46.Yang XJ, Ruvinov SB, Miles EW. Overexpression and Purification of the Separate Tryptophan Synthase Alpha and Beta Subunits from Salmonella-Typhimurium. Protein Expres Purif. 1992;3:347–354. doi: 10.1016/1046-5928(92)90011-k. [DOI] [PubMed] [Google Scholar]
- 47.Laemmli UK. Cleavage of Structural Proteins during Assembly of Head of Bacteriophage-T4. Nature. 1970;227:680. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 48.Hartree EF. Determination of Protein - Modification of Lowry Method That Gives a Linear Photometric Response. Anal Biochem. 1972;48:422. doi: 10.1016/0003-2697(72)90094-2. [DOI] [PubMed] [Google Scholar]
- 49.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 Suite and Current Developments. Acta Crystallogr D. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Battye TGG, Kontogiannis L, Johnson O, Powell HR, Leslie AGW. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D. 2011;67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Evans P. Scaling and assessment of data quality. Acta Crystallogr D. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 52.Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr D. 2010;66:22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 53.Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 55.Painter J, Merritt EA. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 56.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. Procheck - a Program to Check the Stereochemical Quality of Protein Structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 57.DeLano WL. Use of PYMOL as a communications tool for molecular science. Abstr Pap Am Chem S. 2004;228:U313–U314. [Google Scholar]
- 58.Huang YM, You W, Caulkins BG, Dunn MF, Mueller LJ, Chang CA. Protein science: a publication of the Protein Society. 2015. Protonation states and catalysis: Molecular dynamics studies of intermediates in tryptophan synthase. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Case DA, Babin V, Berryman JT, Betz RM, Cai Q, Cerutti DS, Cheatham ITS, Darden TA, Duke RE, Gohlke H, Goetz AW, Gusarov S, Homeyer N, Janowski P, Kaus J, Kolossváry I, Kovalenko A, Lee TS, LeGrand S, Luchko T, Luo R, Madej B, Merz KM, Paesani F, Roe DR, Roitberg A, Sagui C, Salomon-Ferrer R, Seabra G, Simmerling CL, Smith W, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Kollman PA. AMBER 14. University of California; San Francisco: 2014. [Google Scholar]
- 60.Song YF, Gunner MR. Using Multiconformation Continuum Electrostatics to Compare Chloride Binding Motifs in alpha-Amylase, Human Serum Albumin, and Omp32. J Mol Biol. 2009;387:840–856. doi: 10.1016/j.jmb.2009.01.038. [DOI] [PubMed] [Google Scholar]
- 61.Song YF, Mao JJ, Gunner MR. MCCE2: Improving Protein pK(a) Calculations with Extensive Side Chain Rotamer Sampling. J Comput Chem. 2009;30:2231–2247. doi: 10.1002/jcc.21222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 63.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A Smooth Particle Mesh Ewald Method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 64.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical-Integration of Cartesian Equations of Motion of a System with Constraints - Molecular-Dynamics of N-Alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 65.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 66.Gilson MK, Gilson HSR, Potter MJ. Fast assignment of accurate partial atomic charges: An electronegativity equalization method that accounts for alternate resonance forms. J Chem Inf Comp Sci. 2003;43:1982–1997. doi: 10.1021/ci034148o. [DOI] [PubMed] [Google Scholar]
- 67.Dunitz JD, Taylor R. Organic Fluorine Hardly Ever Accepts Hydrogen Bonds. Chem Eur J. 1997;3:89–98. [Google Scholar]
- 68.Xu Y, Abeles RH. Inhibition of tryptophan synthase by (1-fluorovinyl)glycine. Biochemistry-Us. 1993;32:806–811. doi: 10.1021/bi00054a010. [DOI] [PubMed] [Google Scholar]
- 69.Rhee S, Parris KD, Hyde CC, Ahmed SA, Miles EW, Davies DR. Crystal structures of a mutant (betaK87T) tryptophan synthase alpha2beta2 complex with ligands bound to the active sites of the alpha- and beta-subunits reveal ligand-induced conformational changes. Biochemistry-Us. 1997;36:7664–7680. doi: 10.1021/bi9700429. [DOI] [PubMed] [Google Scholar]
- 70.Dierkers AT, Niks D, Schlichting I, Dunn MF. Trptophan synthase: structure and function of the monovalent cation site. Biochemistry-US. 2009;48:10997–11010. doi: 10.1021/bi9008374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Drewe WF, Jr, Dunn MF. Characterization of the reaction of L-serine and indole with Escherichia coli tryptophan synthase via rapid-scanning ultraviolet-visible spectroscopy. Biochemistry-Us. 1986;25:2494–2501. doi: 10.1021/bi00357a032. [DOI] [PubMed] [Google Scholar]
- 72.McDowell LM, Lee MS, McKay RA, Anderson KS, Schaefer J. Intersubunit communication in tryptophan synthase by carbon-13 and fluorine-19 REDOR NMR. Biochemistry-Us. 1996;35:3328–3334. doi: 10.1021/bi9518297. [DOI] [PubMed] [Google Scholar]
- 73.Karplus PA, Diederichs K. Linking Crystallographic Model and Data Quality. Science. 2012;336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.