
Figure 6.
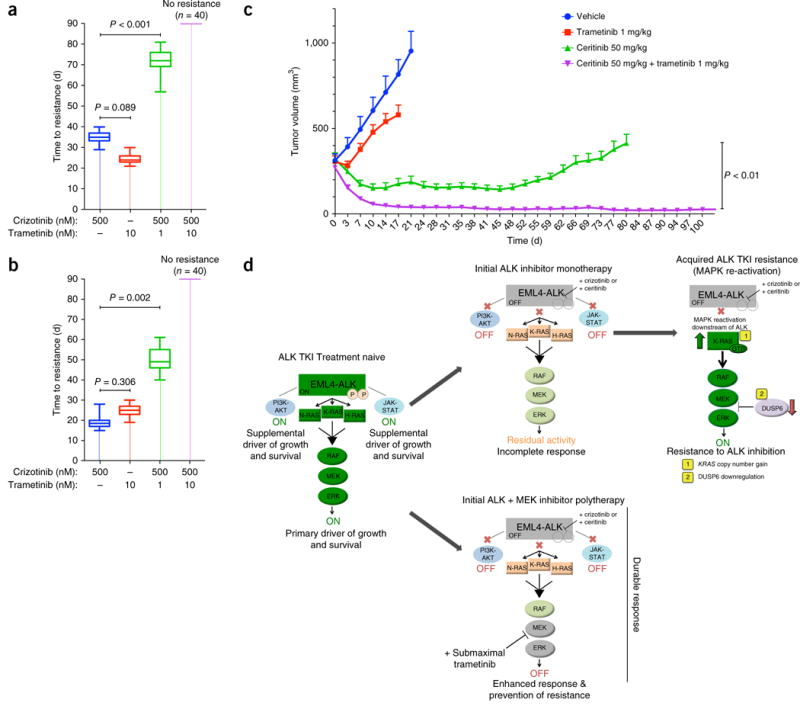
Combined inhibition of ALK and MEK enhances response and eliminates resistance in EML4-ALK lung adenocarcinoma models, in vitro and in vivo. (a,b) Time to acquisition of resistance (defined as days to confluency) in H3122 (a) and STE-1 (b) cells plated in 96-well plates (n = 40) and treated with the indicated drugs. P = 0.002 and P < 0.001, for combination therapy versus control (unpaired t-tests). n = 3, data are presented as box plots, with maximum, minimum and quartile ranges. (c) Tumor volume (mm3) of H3122 xenografts during treatment with ceritinib (50 mg/kg), trametinib (1 mg/kg) or a combination of those, presented as change in tumor volume from baseline (day 0) ±s.e.m., n = 10 tumors per group. P < 0.01, between treatment groups (unpaired t-test). (d) Graphical depiction of the model for EML4-ALK oncogene dependence, in which the tumor cells are dependent primarily on RAS-MAPK signaling. Shown is the mechanism of enhanced efficacy of combined treatment with an ALK inhibitor and a (sub-maximal) MEK inhibitor. EML4-ALK engages RAS-MAPK signaling as the primary downstream effector pathway to drive tumor cell growth and survival (left panel). Upfront ALK monotherapy leads to an incomplete response and tumor-cell survival due to residual MAPK activity (middle top panel). Eventually, these cells acquire resistance to ALK monotherapy by fully rescuing MAPK downstream of EML4-ALK via (1) KRASWT copy number gain or (2) downregulation of the MAPK phosphatase DUSP6 (right top panel). Initial ALK inhibitor–MEK inhibitor polytherapy abrogates this residual MAPK kinase activity to promote greater and more durable upfront responses by minimizing tumor-cell survival and re-activation of MAPK signaling (middle bottom panel).