

Abstract
The mutational profiles of acute myeloid leukemia (AML) with partial tandem duplication of mixed-lineage leukemia gene (MLL-PTD) have not been comprehensively studied. We studied 19 gene mutations for 98 patients with MLL-PTD AML to determine the mutation frequency and clinical correlations. MLL-PTD was screened by reverse-transcriptase PCR and confirmed by real-time quantitative PCR. The mutational analyses were performed with PCR-based assays followed by direct sequencing. Gene mutations of signaling pathways occurred in 63.3% of patients, with FLT3-ITD (44.9%) and FLT3-TKD (13.3%) being the most frequent. 66% of patients had gene mutations involving epigenetic regulation, and DNMT3A (32.7%), IDH2 (18.4%), TET2 (18.4%), and IDH1 (10.2%) mutations were most common. Genes of transcription pathways and tumor suppressors accounted for 23.5% and 10.2% of patients. RUNX1 mutation occurred in 23.5% of patients, while none had NPM1 or double CEBPA mutation. 90.8% of MLL-PTD AML patients had at least one additional gene mutation. Of 55 MLL-PTD AML patients who received standard chemotherapy, age older than 50 years and DNMT3A mutation were associated with inferior outcome. In conclusion, gene mutations involving DNA methylation and activated signaling pathway were common co-existed gene mutations. DNMT3A mutation was a poor prognostic factor in MLL-PTD AML.
Keywords: MLL-PTD, acute myeloid leukemia, gene mutation, DNMT3A mutation
INTRODUCTION
The mixed-lineage leukemia (MLL) gene, now officially named lysine (K)-specific methyltransferase 2A (KMT2A) gene, locates on chromosome 11q23, and encodes a protein of which the SET domain has histone methyltransferase activity that specifically methylates lysine 4 on histone H3, a modification typically associated with transcriptionally active regions of chromatin [1]. The MLL protein acts as a maintenance factor for the homeobox (HOX) group of proteins, which play an important role in specifying cell fate during development and hematopoiesis [2]. MLL gene was initially recognized as a recurrent locus of chromosomal translocation, having more than 85 distinct translocations with different fusion partners [3]. The MLL gene may also be rearranged to generate tandem duplications, the MLL partial tandem duplication (MLL-PTD), which is a cryptic gene rearrangement that most commonly duplicates exons 3 through 9 (e9e3) or exons 3 through 11 (e11e3) giving rise to an in-frame repetition and procured an elongated protein [4]. Like wild-type MLL, the MLL-PTD retained the transcriptional activity of histone H3 lysine 4 methyltransferase within the C-terminal SET domain and impact with the transcriptional regulation of HOX genes and other tumor suppressor genes [5, 6].
MLL-PTD occurred in about 5% to 7% of adult de novo AML, mostly in patients with normal karyotypes [7–10]. In several but not all studies, patients with MLL-PTD had shorter event-free survival (EFS) and overall survivals (OS) [7, 8, 11]. The mechanism of how MLL-PTD contributes to AML and the genetic basis of MLL-PTD associated AML is still unclear. In recent years, novel mutated genes involving epigenetic regulators have been described to occur recurrently in AML [12–14]. Although with its epigenetic regulator function, the cooperation of MLL-PTD with other epigenetic regulator genes such as TET2, IDH1, IDH2, DNMT3A, ASXL1 and EZH2 have not been comprehensively studied. Furthermore, the data on additional molecular alterations in MLL-PTD associated AML is scarce and thus their genetic background is largely unknown. We thus examined a wide spectrum of gene mutations of epigenetic regulators, tumor suppressor genes, signal transactivation (class I) and transcription pathways (class II) on bone marrow cells from patients with de novo MLL-PTD associated AML at the initial diagnosis. The status of gene mutations was correlated with the clinico-hematologic features and outcomes to determine their clinical relevance in patients with MLL-PTD associated AML.
RESULTS
Characterization of MLL-PTD AML patients
Median age of the 98 patients was 55 years (range 6–86). The ratio of male to female was 1:1. The clinical features of 98 MLL-PTD AML patients are listed in Table 1. The estimated median OS was 5.4 months. Fifty-five patients received standard AML protocol (daunomycin 60 mg/m2 for 3 days and cytarabine 100 mg/m2 for 7 days as induction therapy in adults or TPOG-AML97A for children with an age less than 18 years) [18]. Of the 55 patients who received standard AML protocol, 32 achieved complete hematologic remission. Eight patients received allogeneic stem cell transplantation (Supplementary Table S3). The median follow-up time for 55 patients who received standard chemotherapy was 11.0 months (range 0.5–114.2), and the estimated median EFS and OS were 5.2 months (95% CI 0–10.7) and 11.0 months (95% CI 8.6–13.3), respectively.
Table 1. Characteristics of 98 patients with MLL-PTD AML.
| Features | Results |
|---|---|
| Median age (years) | 55.0 (6–86) |
| Age groups | |
| <18 years | 5 (5.1%) |
| 18–59 years | 47 (48.0%) |
| >=60 years | 46 (46.9%) |
| Male/female (ratio) | 49/49 (1.0) |
| Blood counts, median (range) | |
| WBC count (×109/l) | 30.5 (0.8–451.4) |
| Hemoglobin level (g/l) | 74.0 (33.0–137.0) |
| Platelet count (×109/l) | 46.0 (1.0–900.0) |
| Circulating blast (%) | 65.7 (0–98.3) |
| Marrow blast (%) | 79.4 (34.7–99.4) |
| FAB subtype | |
| M0 | 5 (5.1%) |
| M1 | 24 (24.5%) |
| M2 | 43 (43.9%) |
| M4 | 17 (17.3%) |
| M5 | 5 (5.1%) |
| M6 | 3 (3.1%) |
| M7 | 1 (1.0%) |
| ELN cytogenetic risk groups | |
| Favorable | 0 (0%) |
| Intermediate 1 | 56 (73.6%) |
| Intermediate 2 | 19 (25.0%) |
| Adverse | 1 (1.3%) |
| Not evaluated | 22 |
| Number of cytogenetic alterations | |
| 0 (Normal karyotype) | 56 (73.6%) |
| 1 | 13 (17.1%) |
| 2 | 6 (7.9%) |
| 3 or more | 1 (1.3%) |
| Not evaluated | 22 |
FAB: French-American-British, ELN: European LeukemiaNet
Cytogenetic alterations of MLL-PTD AML patients
Seventy-six patients had available cytogenetic data. Twenty of 76 (26.3%) patients had at least one chromosomal abnormality; 13 (17.1%) patients had one chromosome abnormality, and seven (9.2%) had two or more chromosomal abnormalities. The most frequent cytogenetic changes were trisomy 8 (n = 4) and trisomy 11 (n = 3). None of the patients were in the ELN favorable risk group. Fifty-six (73.6%) patients with normal karyotypes were categorized in the European LeukemiaNet (ELN) intermediate-1 group, 19 (25.0%) patients in the ELN intermediate-2 group, and one patient was in the ELN adverse risk group (1.3%; 46, XY, del(3)(p13p22), −4, +8 [12]).
Concomitant gene mutations involving epigenetic regulators in MLL-PTD AML
The gene mutational status of DNMT3A, TET2, IDH1, IDH2, ASXL1, EZH2, and other gene mutations in MLL-PTD associated AML patients is shown in Figure 1.
Figure 1. Diagram of gene mutations in patients with MLL-PTD AML.
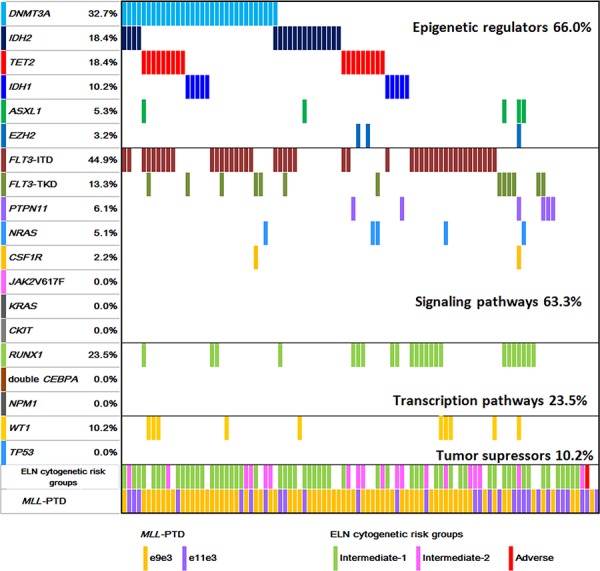
The additional gene mutation status and cytogenetic risk groups in MLL-PTD AML patients at the initial diagnosis are illustrated.
DNMT3A mutations were found in 32 of 98 (32.7%) patients. Most DNMT3A mutations clustered in the methyltransferase domain at amino acid R882. There were 22 mutations (67.7%) at amino acid R882 in exon 23 (R882H, n = 15; R882C, n = 6; R882S, n = 1). Twenty-nine mutations were single nucleotide substitutions leading to missense (n = 28, 87.5%) and nonsense mutations (n = 1, 3.1%). Three patients had frameshift mutations resulted from insertions or deletions (n = 3, 9.4%).
TET2 mutations were identified in 18 of 94 (18.4%) patients. Of the 18 mutated cases, three carried two TET2 mutations. In total, 21 distinct mutations were detected, which were composed of nine point mutations (42.9%), eight deletions (38.1%), and four insertions (19.0%). These alterations led to 12 frame-shift mutations, two nonsense and seven missence mutations. These mutations spread across all the exons of TET2, with the majority of mutations (14/21, 66.7%) located in exons 3 and 8.
IDH2 mutations were present in 18 of 98 (18.4%) patients, with IDH2-R140Q in 14 patients, and IDH2-R172 in four patients (3 R172K, 1 R172S). IDH1 mutations occurred in 10 of 98 (10.2%) patients, with four R132H, three R132C, two R132S, and one R132L.
ASXL1 and EZH2 mutations were detected less frequently. Five of 94 (5.3%) patients had ASXL1 mutations. Three patients carried protein-truncating (two frame-shifts and one nonsense) mutations and two patients had missence mutations. Three out of 94 (3.2%) patients had EZH2 mutations with one each of L378Kfs*6, Y731C and C565Y.
Taken together, among the 94 patients examined for all the six genes of epigenetic regulators, 66.0% of patients had at least one gene mutation; 62.1% (59/95) involved in DNA methylation including DNMT3A, TET2, IDH1, and IDH2 mutations. Mutations in genes of histone modifiers, ASXL1 and EZH2, were rare (7/94, 7.4%) in MLL-PTD AML.
Other concomitant gene mutations in MLL-PTD AML
FLT3-ITD and FLT3-TKD mutations were detected in 44.9% (44/98) and 13.3% (13/98) of patients, respectively. Other mutations of class I signaling pathways occurred less frequently, with the frequencies of 6.1% for PTPN11, 5.1% for NRAS and 2.2% for CSF1R. None had JAK2V617F, KRAS or CKIT mutations. For class II mutations of transcription pathway, RUNX1 mutations were detected in 23.5% (23/98) patients, including 16 frame-shift mutations leading to premature stop codon, three missense mutations, and four nonsense mutations. Single CEBPA mutations were found in four patients but none had double CEBPA mutations. No NPM1 mutation was detected. Among tumor suppressor genes, WT1 mutations were detected in 10.2% (10/98), and none was positive for TP53 mutation. Considering the functional classes of the genes, mutations in class I, class II and tumor suppressor genes occurred in 63.3%, 23.5% and 10.2% of patients, respectively. Together, 91.8% (90/98) of patients had at least one additional gene mutations examined in MLL-PTD AML patients.
Correlations between gene mutations and clinical parameters
The clinical parameters including age, sex, hemoglobin level, platelet counts, white blood cell (WBC) counts, percentage of circulating blasts or marrow blasts, and cytogenetic risk groups were correlated with gene mutation status of DNMT3A, TET2, IDH1/IDH2, RUNX1 and FLT3-ITD. TET2 mutated patients were significantly older (median 71 vs. 54 years, P = 0.004), and the presence of mutations of DNMT3A (61 vs. 54 years, P = 0.053) had a trend of association with older age. IDH1/IDH2 mutations were associated with a lower WBC counts (11.3 × 109/L and 41.5 × 109/L, P = 0.059). MLL-PTD AML patients with FLT3-ITD had significantly higher WBC counts (63.5 vs. 14.9 × 109/L, P = 0.001), circulating blasts (71.8 vs. 61.7%, P = 0.036) and marrow blasts (82.9 vs. 69.3%, P = 0.001) than patients without FLT3-ITD. No significant differences were found with regard to other clinical characteristics according to the mutation status of each gene (Supplementary Table S4).
Survival analysis
Outcome analyses were restricted to patients receiving standard therapy (55/98, 56.1%). We assessed the impact of age (>50 vs <= 50 years), FLT3-ITD, IDH1/IDH2, RUNX1, DNMT3A, and TET2 mutations on EFS and OS. Patients more than 50 years old had significantly shorter EFS (median 0 vs. 9.9 months, P < 0.001) and OS (median 6.7 vs. 11.7 months, P = 0.001) (Figures 2A–2B). MLL-PTD AML patients with DNMT3A mutations had inferior EFS (median 0 vs. 6.8 months, P = 0.026) and OS (median 6.0 vs. 11.5 months, P = 0.032) compared to those without DNMT3A mutations (Figure 2C–2D). The mutational status of FLT3-ITD, RUNX1, IDH1/IDH2, TET2, or ASXL1 did not have significant influence on EFS and OS (Supplementary Figure S1).
Figure 2. Survivals of patients with MLL-PTD AML.
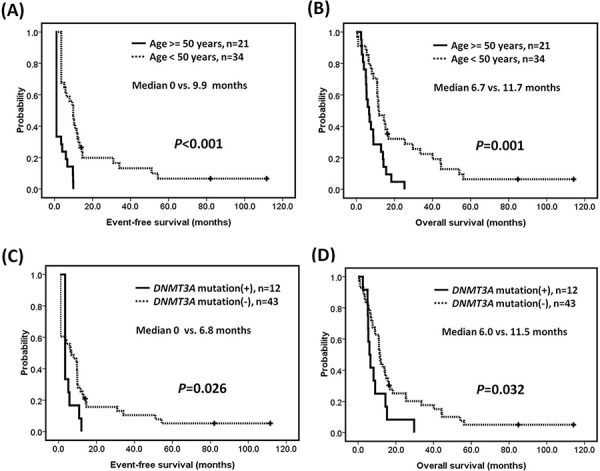
Kaplan-Meier estimates of A. event-free survival and B. overall survival according to age, and C. event-free survival and D. overall survival according to the mutation status of DNMT3A in patients with MLL-PTD associated AML.
DISCUSSION
It is still unclear how MLL-PTD contributes to AML. A MllPTD/WT mice model was reported not to develop leukemia, but characterized by a proliferative advantage, abnormal self-renewal capability, and myeloid differentiation blockage in hematopoietic stem/progenitor cells [19]. Because MLL-PTD alone does not generate leukemia, acquisition of additional cooperating mutations is required for the development of AML. To understand the genetic background and determine whether the prognosis of MLL-PTD is influenced by any additional genetic aberration, we comprehensively investigated the pathobiologically important genetic aberrations known to be involved in myeloid neoplasms including DNA methylation, chromatin modifiers, activating signaling, myeloid transcription factors, and tumor suppressors in a large cohort of patients with MLL-PTD associated AML. We observed that MLL-PTD rarely occurred alone in AML. More than 90% of AML patients with MLL-PTD had at least one additional gene mutation. The most common concomitant gene mutations in MLL-PTD associated AML were FLT3-ITD, followed by DNMT3A mutation, RUNX1 mutation, IDH1/IDH2 mutation, and TET2 mutation.
Of the class I mutation involving activated signaling pathways, FLT3 mutations were the most common concomitant gene mutations in MLL-PTD associated AML, with a frequency of 45% for FLT3-ITD, and up to 58% if FLT3-TKD was included. Other class I mutations, such as PTPN11, NRAS, KRAS, and CSF1R mutations, were less frequently observed. Of the class II mutation involving hematopoietic transcription factors, we observed that RUNX1 mutations were the most frequent co-existed gene mutation. Other class II mutations, such as double CEBPA and NPM1 mutation, were absent in MLL-PTD associated AML. It has been reported that RUNX1 mutations were significantly associated with MLL-PTD [20, 21]. The high frequency of RUNX1 mutation and absent of NPM1 or double CEBPA mutations might contribute in the poor outcome of patients with MLL-PTD AML. Gene mutations involving DNA methylation were common, with DNMT3A, IDH1/IDH2 and TET2 mutations accounting for 62% of patients with MLL-PTD associated AML. The high frequency of FLT3-ITD and mutations in epigenetic and/or chromatin remodeling-associated genes were generally in line with other cohorts with smaller patient numbers [10, 22, 23]. Another study including 225 MLL-PTD AML patients examined 12 genes published as an abstract, in which the frequency of DNMT3A, FLT3-ITD, IDH1, IDH2, and RUNX1 mutations were 44.7%, 33.3%, 14.5%, 29.4%, and 25.8% [24]. However, gene mutations in DNMT3A, NRAS, KRAS, and TP53 were only examined in half of their patients, and TET2, EZH2, PTPN11, and CSF1R mutations were not analyzed.
Taking the functional classification of genes into consideration, the most frequent concurrent gene mutations were genes of activating signaling pathways and epigenetic modifiers. The cooperative functions of MLL-PTD with FLT3-ITD mutations in the leukemogenesis of AML have been studied by Zorko et al, who demonstrated that the MllPTD/wt:Flt3ITD/wt double knock-in mice spontaneously developed AML and recapitulated the cytogenetic and molecular characteristics of human MLL-PTD and FLT3-ITD AML [25]. It is recently found that RUNX1 physically and functionally interacts with MLL, thereby regulating the epigenetic status of critical cis-regulatory elements for hematopoietic genes [26, 27]. In addition to the prevalent FLT3-ITD and RUNX1 mutations in our study, MLL-PTD and gene mutations regulating DNA methylation frequently coexisted in our MLLI-PTD AML patients. As MLL-PTD is a regulator of histone modifications and frequently associated with other gene mutations involving DNA methylation, it is suggested that mechanistic interaction or crosstalk between MLL and other epigenetic regulators, especially DNMT3A, TET2 and IDH1/IDH2, may exist for the regulation of multiple hematopoietic related genes. However, the mechanism of interaction between MLL and DNMT3A, TET2 or IDH1/IDH2 was unclear, which significantly veiled the roles of epigenetic regulators in leukemogenesis, and restrict the development of new therapies. The underlying mechanism of aberrant DNA methylation or chromatin modulation in the leukemogenesis of MLL-PTD associated AML merits further investigation.
The present study is one of the largest series to specifically investigate the impact of additional genetic aberrations in MLL-PTD associated AML. We found that in addition to older age, DNMT3A mutations conferred an inferior OS in MLL-PTD AML patients. Multivariate analysis for age and DNMT3A mutation was not performed because the proportional hazard assumption was not met. Our findings suggest that the detection of DNMT3A mutations in MLL-PTD associated AML can therefore be recommended to identify the subgroup of patients with extremely poor outcome. In addition, the detection of gene mutations involving epigenetic regulators may also provide potential molecular targets or tailored treatment. Several promising treatment strategies were recently reported, including the in vivo benefit of targeting aberrant epigenetics in MLL-PTD associated AML patients [28], the anti-leukemic activity of liposomal bortezomib delivered as a single agent in the MllPTD/wt:Flt3ITD/wt double knock-in mice through the pharmacologic increase in miR-29b [29], and dose intensification of anthracycline in induction therapy to improve the survival of AML patients with DNMT3A mutations and/or MLL-rearrangement [30]. Very recently, Traina et al. reported that DNMT3A and/or TET2 mutations were the independent predictors for a favorable response to DNMT inhibitors of 5-azacytidine and/or decitabine in myelodysplastic syndromes and related neoplasms [31]. Moreover, the preliminary data of an ongoing clinical trial with decitabine with or without bortezomib showed DNMT3A mutations conferred a better response to the hypomethylating agents in AML patients [32]. IDH is another promising target. Chaturvedi et al. recently reported that an inhibitor targeting mutant IDH1 induced apoptosis and decreased colony formation in methylcellulose of IDH1-mutant human primary bone marrow cells [33]. Wang F et al. also demonstrated that treatment with IDH2/R140Q inhibitor induced differentiation in TF-1 erythroleukemia and primary human AML cells in vitro [34]. FLT3 mutations were common in patients with MLL-PTD AML. FLT3 inhibitor to target FLT3 receptor tyrosine kinase is a potential treatment strategy which is under development or clinical trials [35–37]. In addition, FLT3-ITD can be monitored for minimal residual disease by using next generation sequencing to detect outgrowth of variable FLT3-ITD mutant clones during relapse in a single test [38, 39]. In the future, tailored, molecular directed combination therapy will hold promise to improve the outcome of patients with MLL-PTD AML who have high frequency of gene mutations in epigenetic regulators and activated signaling pathways.
In summary, we analyzed gene mutations in 98 patients with de novo MLL-PTD associated AML. We found that nearly two thirds of AML patients with MLL-PTD had at least one gene mutation involving in epigenetic regulators or activating signaling pathways. DNMT3A, IDH1/IDH2 and TET2 mutations were most common and DNMT3A mutation was associated with a poor outcome. We also confirmed that FLT3-ITD and RUNX1 mutations frequently coexisted with MLL-PTD in AML patients. Our findings suggested that MLL-PTD, cooperated with other gene mutations involving epigenetic regulators, might result in deregulated gene expression and the leukemogenesis of AML. The cooperative biological function of MLL-PTD and other mutations in epigenetic regulators warrants further study. As AML patients with MLL-PTD even received intensive therapy such as stem cell transplantation still died of the disease, novel target therapy and tailored treatment according to their concomitant genetic aberrations will hold promise to improve the outcome of patients with MLL-PTD associated AML.
MATERIALS AND METHODS
Patient samples
Analyses of diagnostic bone marrow samples were carried out in 98 patients with de novo AML harboring MLL-PTD in Chang Gung Memorial Hospital and Mackay Memorial Hospital. G-banding method was used for karyotypic analysis. Cytogenetic categorization of favorable-, intermediate- and adverse-risk groups followed the criteria recommended by ELN guidelines [15]. Mutational analyses for MLL-PTD, DNMT3A, IDH1 and IDH2 were performed for all patients (n = 98), and TET2, ASXL1 and EZH2 were performed for 94 patients. Data of other molecular markers, including gene mutations in class I of activated signaling pathways (FLT3-ITD, FLT3-TKD, JAK2V617F, NRAS, KRAS, CKIT, CSF1R, PTPN11), class II of transcription pathways (RUNX1, NPM1, CEBPA), and tumor suppressor genes (WT1, TP53) were available for all patients. The study was approved by the Institutional Review Board of Chang Gung Memorial Hospital and Mackay Memorial Hospital.
Detection of gene mutations
Cell fractionation, DNA or RNA extraction, and cDNA preparation were performed as previously described [16]. MLL-PTD was screened by reverse transcriptase (RT)-PCR and confirmed by real-time quantitative RT-PCR TaqMan assay using the primer sequences described in details in Supplementary Tables S1–S2. The 30 normal subjects had a normalized copy number (NCN) of 0.00008–0.0012 for e9e3 and 0.00029–0.00073 for e11e3, respectively. Sixty-nine patients had repetition of e9e3 with median NCN of 1.25 (range 0.21–9.78), and 29 patients had repetition of e11e3 with median NCN of 1.04 (range 0.44–5.78). The genomic DNA-PCR or reverse transcriptase-PCR assays for the detection of FLT3-ITD, FLT3-TKD, CKIT and CSF1R mutations, point mutations at codons 12, 13, and 61 in exons 1 and 2 of NRAS and KRAS genes, whole coding sequences (exons 3–8) of RUNX1 mutations, mutated genes of CEBPA, PTPN11, JAK2V617F, NPM1, exons 4–10 of TP53, exons 1–3 and exons 7–9 of WT1, whole exons of DNMT3A and TET2, exon 4 of IDH1 and IDH2, DNMT3A, and exon 12 of ASXL1 were carried out as previously described [17]. For the detection of EZH2 mutation, we used denaturing high performance liquid chromatography as the screening assay followed by direct sequencing for abnormal PCR products as described before [16].
Statistical analysis
Fisher's exact test, the χ2 analysis, and Wilcoxon's rank-sum test were used whenever appropriate to make comparisons between groups. Estimates of survival were calculated according to the Kaplan-Meier method. Comparisons of estimated survival curves were analyzed by the log-rank test. In all analysis, the P-values were two-sided and considered statistically significant when values lower than 0.05. Statistical analysis was carried out by SPSS version 17.0 software (SPSS Inc, Chicago, IL).
SUPPLEMENTARY FIGURES AND TABLES
Acknowledgments
The authors thank Ms Ting-Yu Huang for secretarial assistance.
Footnotes
CONFLICTS OF INTEREST
None.
FUNDING
This work was supported by grant from National Health Research Institute, Taiwan (NHRI-EX93-9011SL), grant from National Science Council, Taipei, Taiwan (NSC 97-2314-B-182-011-MY3), grant from the Ministry of Health and Welfare, Taiwan (MOHW103-TD-B-111-09), grant from Mackay Memorial Hospital, Taipei, Taiwan (MMH-E-101-09), and grants from Chang Gung Memorial Hospital, Taipei, Taiwan (OMRPG380031, OMRPG3C0021).
Author contributions
LYS and DCL designed the research. HWK and LYS analyzed the data and wrote the paper. LYS, DCL, MCK JHW, PD, PNW, and TLL provided patient samples and clinical data. YSS, STL, THL, CYL, and CHL performed the laboratory work.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
- 1.Basecke J, Whelan JT, Griesinger F, Bertrand FE. The MLL partial tandem duplication in acute myeloid leukaemia. Br J Haematol. 2006;135:438–449. doi: 10.1111/j.1365-2141.2006.06301.x. [DOI] [PubMed] [Google Scholar]
- 2.Yu BD, Hess JL, Horning SE, Brown GAJ, Korsmeyer SJ. Altered Hox expression and segmental identity in Mll-mutant mice. Nature. 1995;378:505–508. doi: 10.1038/378505a0. [DOI] [PubMed] [Google Scholar]
- 3.Huret JL. KMT2A (myeloid/lymphoid or mixed lineage leukemia) Atlas Genet Cytogenet Oncol Haematol. Available from: http://atlasgeneticsoncology.org//Genes/MLLID13.html.
- 4.Caligiuri MA, Schichman SA, Strout MP, Mrózek K, Baer MR, Frankel SR, Barcos M, Herzig GP, Croce CM, Bloomfield CD. Molecular rearrangement of the ALL-1 gene in acute myeloid leukemia without cytogenetic evidence of 11q23 chromosomal translocations. Cancer Res. 1994;54:370–373. [PubMed] [Google Scholar]
- 5.Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, Hess JL. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10:1107–1117. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- 6.Whitman SP, Hackanson B, Liyanarachchi S, Liu S, Rush LJ, Maharry K, Margeson D, Davuluri R, Wen J, Witte T, Yu L, Liu C, Bloomfield CD, et al. DNA hypermethylation and epigenetic silencing of the tumor suppressor gene, SLC5A8, in acute myeloid leukemia with the MLL partial tandem duplication. Blood. 2008;112:2013–2016. doi: 10.1182/blood-2008-01-128595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steudel C, Wermke M, Schaich M, Schäkel U, Illmer T, Ehninger G, Thiede C. Comparative analysis of MLL partial tandem duplication and FLT3 internal tandem duplication mutations in 956 adult patients with acute myeloid leukemia. Genes Chromosomes Cancer. 2003;37:237–251. doi: 10.1002/gcc.10219. [DOI] [PubMed] [Google Scholar]
- 8.Shih LY, Liang DC, Fu JF, Wu JH, Wang PN, Lin TL, Dunn P, Kuo MC, Tang TC, Lin TH, Lai CL. Characterization of fusion partner genes in 114 patients with de novo acute myeloid leukemia and MLL rearrangement. Leukemia. 2006;20:218–223. doi: 10.1038/sj.leu.2404024. [DOI] [PubMed] [Google Scholar]
- 9.Döhner K, Tobis K, Ulrich R, Fröhling S, Benner A, Schlenk RF, Döhner H. Prognostic significance of partial tandem duplications of the MLL gene in adult patients 16 to 60 years old with acute myeloid leukemia and normal cytogenetics: a study of the Acute Myeloid Leukemia Study Group Ulm. J Clin Oncol. 2002;20:3254–3261. doi: 10.1200/JCO.2002.09.088. [DOI] [PubMed] [Google Scholar]
- 10.Whitman SP, Caligiuri MA, Maharry K, Radmacher MD, Kohlschmidt J, Becker H, Mrózek K, Wu YZ, Schwind S, Metzeler KH, Mendler JH, Wen J, Baer MR, et al. The MLL partial tandem duplication in adults aged 60 years and older with de novo cytogenetically normal acute myeloid leukemia. Leukemia. 2012;26:1713–1717. doi: 10.1038/leu.2012.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schnittger S, Kinkelin U, Schoch C, Heinecke A, Haase D, Haferlach T, Büchner T, Wörmann B, Hiddemann W, Griesinger F. Screening for MLL tandem duplication in 387 unselected patients with AML identify a prognostically unfavorable subset of AML. Leukemia. 2000;14:796–804. doi: 10.1038/sj.leu.2401773. [DOI] [PubMed] [Google Scholar]
- 12.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, Harris CC, Lichti CF, Townsend RR, et al. DNMT3A Mutations in Acute Myeloid Leukemia. N Engl J Med. 2010;363:2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrózek K, Margeson D, Holland KB, Whitman SP, Becker H, Schwind S, Metzeler KH, Powell BL, Carter TH, et al. IDH1 and IDH2 Gene Mutations Identify Novel Molecular Subsets Within De Novo Cytogenetically Normal Acute Myeloid Leukemia: A Cancer and Leukemia Group B Study. J Clin Oncol. 2010;28:2348–2355. doi: 10.1200/JCO.2009.27.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Massé A, Kosmider O, Le Couedic JP, Robert F, Alberdi A, Lécluse Y, Plo I, Dreyfus FJ, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360:2289–2301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 15.Döhner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA, Lo-Coco F, Naoe T, Niederwieser D, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115:453–474. doi: 10.1182/blood-2009-07-235358. [DOI] [PubMed] [Google Scholar]
- 16.Kao HW, Liang DC, Wu JH, Kuo MC, Wang PN, Yang CP, Shih YS, Lin TH, Huang YH, Shih LY. Gene Mutation Patterns in Patients with Minimally Differentiated Acute Myeloid Leukemia. Neoplasia. 2014;16:481–488. doi: 10.1016/j.neo.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang DC, Liu HC, Yang CP, Jaing TH, Hung IJ, Yeh TC, Chen SH, Hou JY, Huang YJ, Shih YS, Huang YH, Lin TH, Shih LY. Cooperating gene mutations in childhood acute myeloid leukemia with special reference on mutations of ASXL1, TET2, IDH1, IDH2, and DNMT3A. Blood. 2013;121:2988–2995. doi: 10.1182/blood-2012-06-436782. [DOI] [PubMed] [Google Scholar]
- 18.Liang DC, Chang TT, Lin KH, Lin DT, Lu MY, Chen SH, Liu HC, Lin MT, Lee MT, Shu SG, Chang TK, Chen JS, Hsiao CC, et al. Improved treatment results for childhood acute myeloid leukemia in Taiwan. Leukemia. 2006;20:136–141. doi: 10.1038/sj.leu.2403979. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Yan X, Sashida G, Zhao X, Rao Y, Goyama S, Whitman SP, Zorko N, Bernot K, Conway RM, Witte D, Wang QF, Tenen DG, et al. Stress hematopoiesis reveals abnormal control of self-renewal, lineage bias, and myeloid differentiation in Mll partial tandem duplication (Mll-PTD) hematopoietic stem/progenitor cells. Blood. 2012;120:1118–1129. doi: 10.1182/blood-2012-02-412379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gaidzik VI, Bullinger L, Schlenk RF, Zimmermann AS, Röck J, Paschka P, Corbacioglu A, Krauter J, Schlegelberger B, Ganser A, Späth D, Kündgen A, Schmidt-Wolf IG, et al. RUNX1 mutations in acute myeloid leukemia: results from a comprehensive genetic and clinical analysis from the AML study group. J Clin Oncol. 2011;29:1364–1372. doi: 10.1200/JCO.2010.30.7926. [DOI] [PubMed] [Google Scholar]
- 21.Schnittger S, Dicker F, Kern W, Wendland N, Sundermann J, Alpermann T, Haferlach C, Haferlach T. RUNX1 mutations are frequent in de novo AML with noncomplex karyotype and confer an unfavorable prognosis. Blood. 2011;117:2348–2357. doi: 10.1182/blood-2009-11-255976. [DOI] [PubMed] [Google Scholar]
- 22.Whitman SP, Ruppert AS, Marcucci G, Mrózek K, Paschka P, Langer C, Baldus CD, Wen J, Vukosavljevic T, Powell BL, Carroll AJ, Kolitz JE, Larson RA, et al. Long-term disease-free survivors with cytogenetically normal acute myeloid leukemia and MLL partial tandem duplication: a Cancer and Leukemia Group B study. Blood. 2007;109:5164–5167. doi: 10.1182/blood-2007-01-069831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schlenk RF, Döhner K, Krauter J, Fröhling S, Corbacioglu A, Bullinger L, Habdank M, Späth D, Morgan M, Benner A, Schlegelberger B, Heil G, Ganser A, et al. Mutations and Treatment Outcome in Cytogenetically Normal Acute Myeloid Leukemia. N Engl J Med. 2008;358:1909–1918. doi: 10.1056/NEJMoa074306. [DOI] [PubMed] [Google Scholar]
- 24.Haferlach C, Kohlmann A, Kern W, Halferlach T, Schnittger S. Cytogenetic and molecular genetic characterization of MLL-PTD positive AML in comparison to MLL-translocated AML. Blood. 2013;122:2557. [Google Scholar]
- 25.Zorko NA, Bernot KM, Whitman SP, Siebenaler RF, Ahmed EH, Marcucci GG, Yanes DA, McConnell KK, Mao C, Kalu C, Zhang X, Jarjoura D, Dorrance AM, et al. Mll partial tandem duplication and Flt3 internal tandem duplication in a double knock-in mouse recapitulates features of counterpart human acute myeloid leukemias. Blood. 2012;120:1130–1136. doi: 10.1182/blood-2012-03-415067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang G, Zhao X, Wang L, Elf S, Xu H, Zhao X, Sashida G, Zhang Y, Liu Y, Lee J, Menendez S, Yang Y, Yan X, et al. The ability of MLL to bind RUNX1 and methylate H3K4 at PU.1 regulatory regions is impaired by MDS/AML-associated RUNX1/AML1 mutations. Blood. 2011;118:6544–6552. doi: 10.1182/blood-2010-11-317909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koh CP, Wang CQ, Ng CE, Ito Y, Araki M, Tergaonkar V, Huang G, Osato M. RUNX1 meets MLL: epigenetic regulation of hematopoiesis by two leukemia genes. Leukemia. 2013;27:1793–1802. doi: 10.1038/leu.2013.200. [DOI] [PubMed] [Google Scholar]
- 28.Bernot KM, Siebenaler RF, Whitman SP, Zorko NA, Marcucci GG, Santhanam R, Ahmed EH, Ngangana M, McConnell KK, Nemer JS, Brook DL, Kulp SK, Chen CS, et al. Toward personalized therapy in AML: in vivo benefit of targeting aberrant epigenetics in MLL-PTD-associated AML. Leukemia. 2013;27:2379–2382. doi: 10.1038/leu.2013.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bernot KM, Nemer JS, Santhanam R, Liu S, Zorko NA, Whitman SP, Dickerson KE, Zhang M, Yang X, McConnell KK, Ahmed EH, Muñoz MR, Siebenaler RF, et al. Eradicating acute myeloid leukemia in a MllPTD/wt:Flt3ITD/wt murine model: a path to novel therapeutic approaches for human disease. Blood. 2013;122:3778–3783. doi: 10.1182/blood-2013-06-507426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Van VP, Dolgalev I, Thomas S, Aminova O, Huberman K, Cheng J, Viale A, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–1089. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Traina F, Visconte V, Elson P, Tabarroki A, Jankowska AM, Hasrouni E, Sugimoto Y, Szpurka H, Makishima H, O'Keefe CL, Sekeres MA, Advani AS, Kalaycio M, et al. Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia. 2014;28:78–87. doi: 10.1038/leu.2013.269. [DOI] [PubMed] [Google Scholar]
- 32.Metzeler KH, Walker A, Geyer S, Garzon R, Klisovic RB, Bloomfield CD, Blum W, Marcucci G. DNMT3A mutations and response to the hypomethylating agent decitabine in acute myeloid leukemia. Leukemia. 2012;26:1106–1107. doi: 10.1038/leu.2011.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chaturvedi A, Araujo Cruz MM, Jyotsana N, Sharma A, Yun H, Görlich K, Wichmann M, Schwarzer A, Preller M, Thol F, Meyer J, Haemmerle R, Struys EA, et al. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood. 2013;122:2877–2887. doi: 10.1182/blood-2013-03-491571. [DOI] [PubMed] [Google Scholar]
- 34.Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, Yang H, Gross S, Artin E, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340:622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- 35.Lee-Sherick AB, Zhang W, Menachof KK, Hill AA, Rinella S, Kirkpatrick G, Page LS, Stashko MA, Jordan CT, Wei Q, Liu J, Zhang D, DeRyckere D, et al. Efficacy of a Mer and Flt3 tyrosine kinase small molecule inhibitor, UNC1666, in acute myeloid leukemia. Oncotarget. 2015;6:6722–6736. doi: 10.18632/oncotarget.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sammons SL, Pratz KW, Smith BD, Karp JE, Emadi A. Sorafenib is tolerable and improves clinical outcomes in patients with FLT3-ITD acute myeloid leukemia prior to stem cell transplant and after relapse post-transplant. Am J Hematol. 2014;89:936–938. doi: 10.1002/ajh.23782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liegel J, Courville E, Sachs Z, Ustun C. Use of sorafenib for post-transplant relapse in FLT3/ITD-positive acute myelogenous leukemia: maturation induction and cytotoxic effect. Haematologica. 2014;99:e222–224. doi: 10.3324/haematol.2014.109975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shih LY, Huang CF, Wu JH, Lin TL, Dunn P, Wang PN, Kuo MC, Lai CL, Hsu HC. Internal tandem duplication of FLT3 in relapsed acute myeloid leukemia: a comparative analysis of bone marrow samples from 108 adult patients at diagnosis and relapse. Blood. 2002;100:2387–2392. doi: 10.1182/blood-2002-01-0195. [DOI] [PubMed] [Google Scholar]
- 39.Bibault JE, Figeac M, Hélevaut N, Rodriguez C, Quief S, Sebda S, Renneville A, Nibourel O, Rousselot P, Gruson B, Dombret H, Castaigne S, Preudhomme C. Next-generation sequencing of FLT3 internal tandem duplications for minimal residual disease monitoring in acute myeloid leukemia. Oncotarget. 2015 doi: 10.18632/oncotarget.4333. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.