
FIGURE 1.
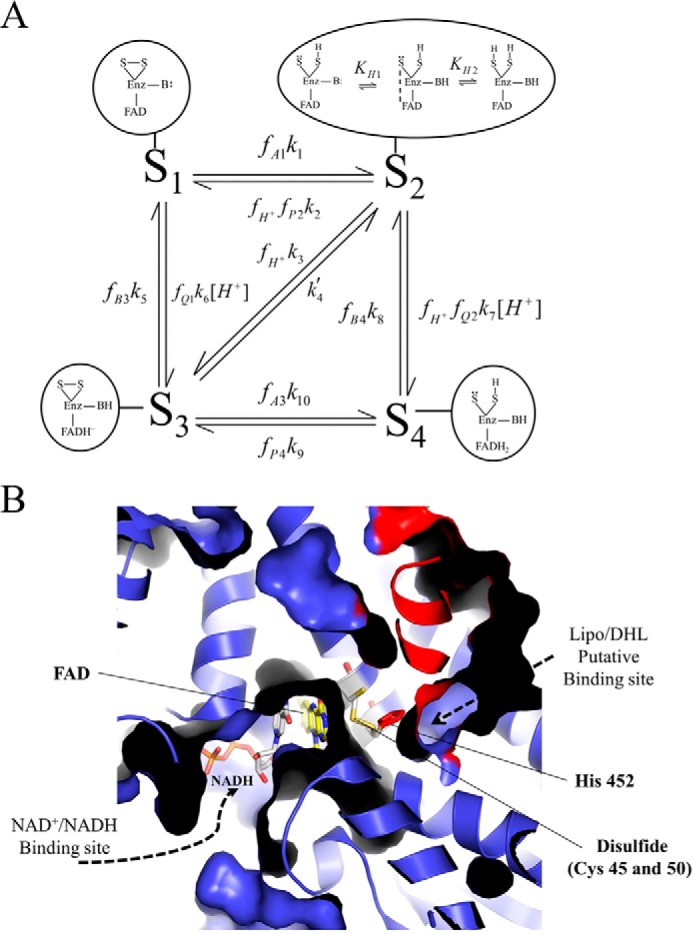
Mammalian E3 kinetic model and substrate/product-binding sites. A, pig heart E3 kinetic model consists of four redox states as follows: oxidized (S1), hydride-reduced disulfide (S2), hydride-reduced FAD (S3), and a hydride-reduced disulfide and FAD state (S4). Each enzyme redox state is depicted in the corresponding bubble captions, which illustrate the disulfide (S-S), flavin (FAD), and active site base (B:) chemical forms as part of the E3 enzyme (Enz). In the model, S2 has the ability to undergo (de)protonation, where the middle schematic in the bubble caption represents the charge transfer complex required for hydride transfer between the thiolate redox center and FAD cofactor to advance the mechanism between S2 and S3 enzyme states (3). Dihydrolipoamide, NAD+, lipoamide, NADH, and protons are represented by A, B, P, Q, and H+, respectively. Fractional occupancies, the fraction of a substrate or product bound to a given enzyme state, are represented by f, and subscripts indicate the substrate/product bound to a specific state (S1 through S4). Substrates and products are considered to bind randomly within each redox state and in rapid equilibrium compared with chemical steps. Our assumption of rapid equilibrium binding is supported by previous studies (3, 43, 46). B, x-ray structure of human E3 (Protein Data Bank code 1ZMD) (4) illustrates the general substrate/product binding situation common to E3s, where the re face of the FAD cofactor is exposed, and the si face is guarded by an active site disulfide. In the human E3 structure (4), NAD+ and NADH were shown to bind the re face of the FAD cofactor, whereas the si face does not bind either NAD redox state. There are available structures with bound NAD+ (Protein Data Bank code 1ZMC) and NADH (Protein Data Bank code 1ZMD). We chose to show the NADH-bound structure, which has a different binding mode than NAD+ where the nicotinamide ring of NADH is oriented toward the FAD cofactor. Structurally homologous enzymes in the flavin disulfide reductase family (69), molecular dynamics simulations (70), and structures of bound lipoamide inhibitors reveal the lipoamide binding cavity (71). The E3 human structure (4) is shown in a cutaway view so that the binding pockets, located in the protein interior, can be viewed. The structure (a dimer) is colored so that one monomer is blue and the other is red. Active site components are annotated accordingly with the bound NADH (gray) and FAD (yellow) cofactors shown in stick representation. The active site disulfide (Cys-45 and -50 in humans) and histidine (residue 452 in humans) are shown as yellow and red sticks, respectively. This figure was made using PyMOL (72) and Protein Data Bank code 1ZMD.