

Abstract
Paenibacillus larvae is the causative agent of American foulbrood (AFB) disease which affects early larval stages during honeybee development. Due to its virulence, transmissibility, capacity to develop antibiotic resistance, and the inherent resilience of its endospores, Paenibacillus larvae is extremely difficult to eradicate from infected hives which often must be burned. AFB contributes to the worldwide decline of honeybee populations, which are crucial for pollination and the food supply. We have isolated a novel bacteriophage lysin, PlyPalA, from the genome of a novel Paenibacillus larvae bacteriophage originally extracted from an environmental sample. PlyPalA has an N-terminal N-acetylmuramoyl-L-alanine amidase catalytic domain and possesses lytic activity against infectious strains of Paenibacillus larvae without harming commensal bacteria known to compose the honeybee larval microbiota. A single dose of PlyPalA rescued 75% of larvae infected with endospores, showing that it represents a powerful tool for future treatment of AFB. This represents the first time that lysins have been tested for therapeutic use in invertebrates.
Keywords: enzyme, honeybee, larvae, lysin, Paenibacillus larvae
Introduction
American foulbrood (AFB) is a highly destructive bacterial disease affecting the honeybee, Apis mellifera; despite its name, it is distributed worldwide.1 The disease is caused by the endospores of the Gram-positive bacterium Paenibacillus larvae and only the first- or second-instar larval stages are susceptible.2 Infection occurs when honeybee larvae ingest endospores in contaminated food; after 12 hours have passed, P. larvae endospores germinate into vegetative cells that then proliferate and ultimately kill the honeybee larvae.2 The larvae are digested by secreted proteases and turned into a glue-like, ropy mass that then dries out and turns into a foulbrood scale.1 Vegetative cells then sporulate, forming billions of endospores that continue to spread through the hive or be transmitted to neighboring colonies.2 These spores are highly infectious; consumption of 10 or fewer endospores can lead to a fatal infection and endospores can remain infectious for several decades.1 Therefore, beekeepers are often forced to burn their equipment and infected hives in order to control the spread of AFB.2
Antibiotics are an ineffective treatment because they cannot destroy endospores, they may harm the honeybees themselves, render honey unfit for human consumption, and contribute to increasing antibiotic resistance among P. larvae strains.1 Indeed, due to long-term treatment of beehives with oxytetracycline in the United States, metagenomic screens have revealed that honeybee gut bacteria have accumulated resistance genes against tetracycline and oxytetracycline.3 This reservoir of resistance genes can then be used by pathogens to become more difficult to control.3
Bacteriophage lysins are modular enzymes containing a C-terminal cell-wall binding domain that determines strain and species specificity as well as one or more N-terminal catalytic domains that hydrolyze bonds within the bacterial peptidoglycan.4 Double-stranded DNA phages produce these enzymes during the late stage of the lytic cycle in order to disrupt the integrity of the cell wall, cause hypotonic lysis of the host cell, and release progeny virions into the environment.4 At least 4 different categories of lysins have been identified: endopeptidases and amidases that cleave peptide and amide bonds, respectively, as well as transglycosidases and lysozymes that cleave the glycosidic bonds.5
Most lysins lack signal sequences for secretion and therefore accumulate within the cytoplasm; thus they are dependent on small, hydrophobic, transmembrane proteins called holins in order to reach the peptidoglycan layer.5 Holins form large, non-selective pores in the inner bacterial membrane.5 This system of lysin and holin is known as the lambda paradigm.5 Lysins affecting Gram-positive bacteria are generally species-specific with rare exceptions such as the aminopeptidase PlySs2 which is active against the genera Streptococcus and Staphyloccocus.6 Because the peptidoglycan layer of Gram-positive bacteria is exposed, it is possible to kill unwanted species by exogenously applying lysin without need for the holin or the phage; thus, lysins are highly promising therapeutic agents.5
In this work, we present an amidase, PlyPalA, isolated from the genome of a novel double-stranded DNA bacteriophage from an environmental sample. We found that PlyPalA was highly lytic against pathogenic P. larvae strains, which makes it a promising treatment for AFB. Additionally, its specificity ensures that it would likely not harm the normal microbiota present in the gut of honeybee larvae. To our knowledge, this is the first lysin isolated with the express purpose of treating a disease in honeybees.
Results
Bioinformatic analysis of the plypalA gene
The genome of phage Xenia is 41,149 base pairs in length with 67 putative open reading frames (ORFs) predicted by annotation with the DNA Master program. Xenia_gp37, designated as PlyPalA, was identified as a putative phage lysin with an amino acid length of 224 residues and a predicted molecular mass of 25,943 Da. Analysis of the N-terminal region of the lysin using BLASTP 2.2.29+ showed similarity to peptidoglycan recognition protein (residues 21-142, E-value 2.55e-27), which is a conserved domain involved in peptidogylcan hydrolysis found in animals, phages, and bacteria,7 as well as N-acetylmuramoyl-L-alanine amidase (residues 22-136, E-value 6.08e-22). Critical Zn2+ binding residues known for the PGRP domain were successfully mapped onto the sequence as the following amino acids: His30, His130, and Cys138.8 Significant alignments were observed with previously-isolated Bacillus subtilis phage lysins CwlA (P24808.1, query coverage 75%, identity 53%, E-value 3e-53) and XlyA (P39800.1, query coverage 69%, identity 40%, E-value 5e-45).
Multiple Sequence Alignment of PlyPalA compared with other phage amidases
Several phage amidases from other bacterial species were bioinformatically compared with PlyPalA. Prophage amidase PlyPalP (ETK30343.1) was found within the fully sequenced genome of P. larvae DSM 25430 (CP003355.1).9 PlyL (ACQ48698.1) and PlyG (ABC40416.1) are phage amidases from the Bacillus anthracis genome.8 and from gamma phage10, respectively. Both lysins have been extensively characterized and serve as useful references for analysis of the PlyPalA sequence. LambdaBa01 (AIE36437.1) is a lysin from a prophage in the Bacillus thuringiensis genome and XlyA is a lysin found in a defective B. subtilis prophage.11 The amidase of prophage LambdaLm01 (EGJ23638.1) was found in the sequenced genome of Listeria monocytogenes strain Scott A.12 All of these lysins contain the same catalytic activity and thus should contain homologous N-terminal catalytic domains.
Amino acid residues participating in catalysis and residues participating in zinc-binding were initially identified after mutagenesis and structure determination of PlyL.8 Interestingly, these same amino acid residues are perfectly conserved among the Bacillus and P. larvae amidases that were compared. Catalytic residues for these phage amidases are marked with a red triangle and putative zinc-binding residues are marked with a blue triangle in Figure. 1 .
Figure 1.

Alignment of the amino acid sequence of PlyPalA with known and putative N-acetylmuramoyl-L-alanine amidases. Dots indicate the same amino acid as the top sequence. Zn2+ binding residues of PlyL have a blue triangle above and catalytic residues have a red triangle above. Software used was the CLC Main Workbench 7.5.1 (http://www.clcbio.com/).
According to the multiple sequence alignment (Fig. 1), there is considerable homology in the N-terminal domains of PlyL, PlyG, LambdaBa01, PlyPalP, PlyPalA, and, to a lesser extent, XlyA.
Purification of PlyPalA
With a predicted pI of 6.15 (http://web.expasy.org/compute_pi/), 6x His PlyPalA bound to the DEAE column at pH 7.6 and was eluted from the column at an ionic strength of 200 mM NaCl. Fractions containing lysin were applied to a Ni Sepharose column and eluted using a stepwise gradient of imidazole to a concentration of 250mM. The 6x His tag was then removed by overnight digestion with thrombin and the thrombin was removed using AEC. This was followed by SDS-PAGE to determine purity (Fig. 2) .
Figure 2.
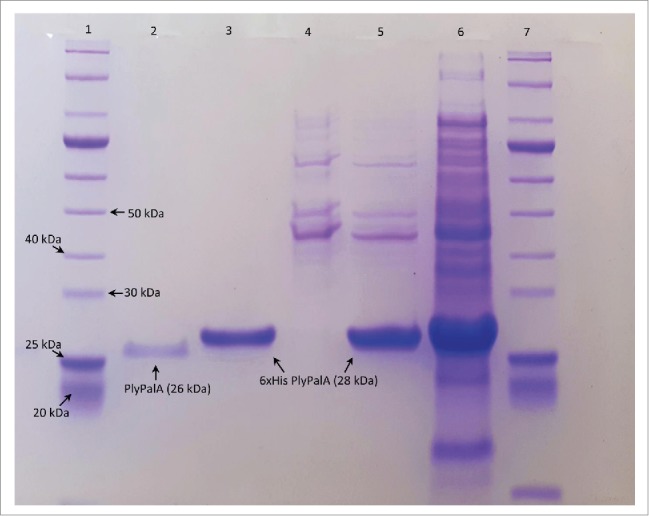
SDS-PAGE of lysin purification after AEC, IMAC, thrombin digest, and more AEC. The 10% acrylamide gel was run at 60V for 30 mins followed by 150V for 60 mins in buffer containing 50 mM Tris, 50 mM MOPS, 0.1% SDS, and 1 mM EDTA, pH 6.8. The lanes from left to right: (1) 10-250 kDa Protein Ladder (New England Biolabs), (2) 210 mM NaCl eluate from second AEC after thrombin digest, (3) 250 mM imidazole eluate from IMAC, (4) 25 mM imidazole eluate from IMAC, (5) 200 mM NaCl eluate from first AEC, (6) crude B-PER lysate of IPTG-induced E. coli, (7) 10-250 kDa Protein Ladder (New England Biolabs).
Biochemical characteristics
PlyPalA was tested at a range of pH and salinity values to determine which biochemical conditions enhance lytic activity. Buffers used were 20 mM 2-(N-morpholino)ethanesulfonic acid (MES) at pH of 5.5, 6.0, and 6.7 and 20 mM Tris at pH of 7.4, 8.0, and 9.0. Catalysis was present over the entire pH range tested although the amount of lysis significantly varied. The lysin was least active at an acidic pH in MES buffer and most active at a somewhat alkaline pH in Tris buffer (Fig. 3A). However, it did retain about 50% of its optimal activity at a mildly acidic pH; this is crucial because the midgut of a developing larva is slightly acidic.13
Figure 3.
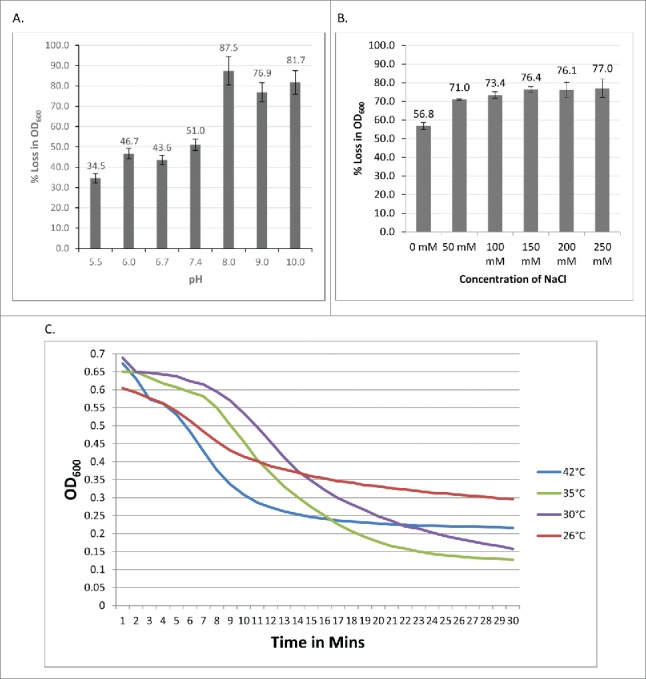
(A) PlyPalA activity at various pH values. Lysin was mixed with P. larvae B-3650 resuspended in buffers titrated to various pH and measured once per minute for 45 mins. Error bars represent the standard deviations of lysin-treated wells and buffer-treated wells (n = 3). (B) PlyPalA activity in various salinities. Readings were taken once per minute for 30 mins. Error bars represent standard deviations (n = 3). (C) PlyPalA activity at various temperatures. OD600 of 3 wells was taken once per minute for 30 mins.
Additionally, lysin activity was optimal when the buffer was supplemented with sodium chloride; the benefit from increased NaCl was maximal at a concentration of 150 mM and stayed essentially the same to 250 mM (Fig. 3B). This suggests that a cellular ionic environment may be necessary for proper PlyPalA folding and activity. Interestingly, lysin activity was best at 35°C, the same temperature honeybee larvae are incubated at in laboratory conditions in order to mimic conditions in a hive,14 and wells incubated at this temperature reached a lower OD 600 nm at equilibrium than those incubated at 42°C (Fig. 3C). Finally, like other known lysins, PlyPalA is inhibited by EDTA (Fig. 4A). Mg2+, Mn2+, Ca2+, and Co2+ all restored lytic activity after removal of EDTA by cutoff filter centrifugation (Fig. 4B). These biochemical conditions were then taken into account and used for further experiments to test the lysin against a variety of strains.
Figure 4.
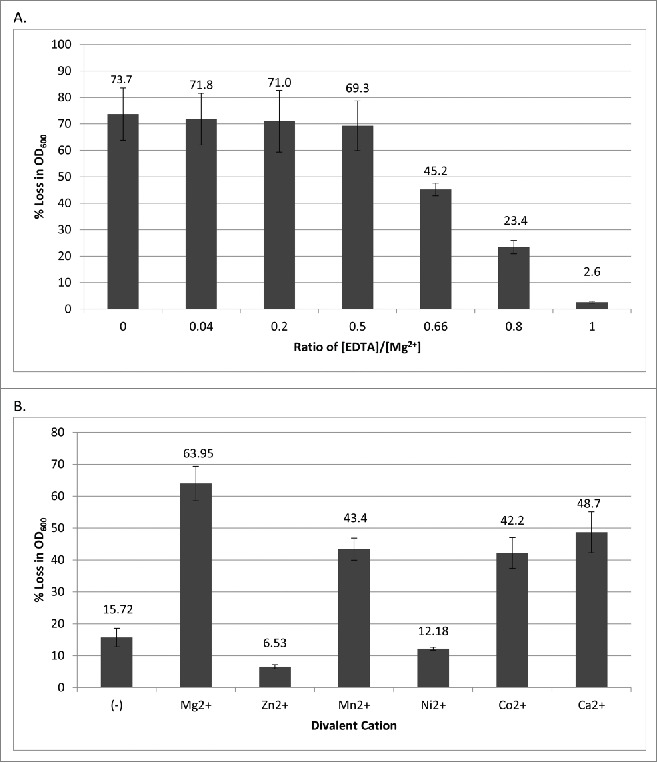
(A) PlyPalA inhibition by the metal cation chelator EDTA. After adding P. larvae and before adding lysin, EDTA was added to various concentrations as ratios of the known concentration of Mg2+ (5.0 mM). Error bars represent standard deviations (n = 3). (B) PlyPalA activity with Mn2+, Co2+, and Ca2+ cofactors. Cations were added to a concentration of 5.0 mM. The (-) control represents lysis with no cation added. Readings were taken once per minute for 30 mins. Error bars represent standard deviations (n = 3).
Finally, lysin activity seemed not to be affected by lyophilization. After being lyophilized overnight and resuspended in sterile water to its original concentration, the lysin caused a 66.87% ± 1.95% reduction in OD600 after 30 mins compared to a 64.17% ± 6.30% reduction caused by unlyophilized lysin at the same concentration when assayed on aliquots of the same cell suspension of P. larvae strain 3650. This difference is not significant (2-tailed 2 sample t-test, P = 0.5838).
Lytic activity against bacterial strains
PlyPalA was tested against 11 different strains of P. larvae and 14 other species of bacteria representing 9 different genera. ERIC genotypes were described previously15 according to amplification of repetitive DNA sequences present in P. larvae genomes. Strains 2188 and 2231, isolated from hive samples, showed colony morphology similar to genotype I and were confirmed by PCR to have signature bands of this genotype.
PlyPalA had lytic activity against P. larvae strains belonging to ERIC genotype I, reducing the OD600 of P. larvae B-3650 from 0.7 to 0.1 in 30 minutes at 35°C (Table 1). P. larvae strains corresponding to ERIC genotypes III/IV were affected much less (2-tailed 2 sample t-test, P = 0.005658). This indicates that ERIC genotypes I is significantly more susceptible to PlyPalA than genotypes III and IV. The lytic activity assay against bacteria other than P. larvae showed insignificant or nonexistent lysis for most strains tested (Table 2). Importantly, this includes Lactobacillus spp., Bacillus spp, Fructobacillus fructosus, and Bifidobacterium longum which correspond to genera that are abundant in the microbiota of honeybee larvae.13 Indeed, among the Gram-positive organisms present in the first through fifth larval instars, these genera represent the majority of bacterial diversity.13
Table 1.
P. larvae strain selectivity of PlyPalAa
| P. larvae strain | % Loss in OD600 | ERIC Genotype |
|---|---|---|
| NRRL B-3650 | 84.9% ± 0.6% | I |
| NRRL B-3554 | 78.0% ± 0.9% | I |
| ATCC 25748 | 72.8% ± 2.0% | I |
| NRRL B-2605 | 72.1% ± 1.2% | I |
| ATCC 25747 | 62.7% ± 1.4% | I |
| 2188 | 57.2% ± 2.6% | I |
| ATCC 25367 | 49.4% ± 1.6% | IV |
| 2231 | 42.4% ± 0.5% | I |
| ATCC 3688 | 23.3% ± 2.2% | III/IV |
| ATCC 25368 | 22.9% ± 3.3% | IV |
| ATCC 49843 | 12.2% ± 2.1% | IV |
| E. coli (-) | -1.6% ± 3.1% | N/A |
aPlyPalA is most effective against P. larvae strains corresponding to ERIC genotype I. Standard deviations were calculated from the ratio of OD600 of lysin-treated wells versus buffer treated wells (in triplicate). E. coli was used as a negative control. ERIC genotypes for NRRL B-2605 and ATCC 49843 were taken from a previous study17 and determined experimentally by ERIC PCR for other strains (data not shown).
Table 2.
Species and genus specificity of PlyPalAb
| Bacterial Genus/Species | Strain | % Loss in OD600 |
|---|---|---|
| Paenibacillus larvae (+) | NRRL B-3650 | 80.0% ± 0.9% |
| Paenibacillus polymyxa | DSM 36 | 26.2% ± 3.4% |
| Paenibacillus lentimorbus | ATCC 14707 | 20.0% ± 2.7% |
| Gluconobacter cerinus | ATCC 19441 | 10.4% ± 5.4% |
| Listeria innocua | ATCC 51742 | 8.0% ± 4.8% |
| Paenibacillus alvei | ATCC 6344 | 7.9% ± 6.2% |
| Staphylococcus aureus | ATCC 12600 | 8.2% ± 1.2% |
| Fructobacillus fructosus | ATCC 13162 | 6.7% ± 5.5% |
| Lactobacillus brevis | NRRL-1834 | 5.6% ± 2.8% |
| Bacillus subtilis | YB955 | 6.2% ± 6.9% |
| Bacillus megaterium | ATCC 12872 | 5.5% ± 1.8% |
| Paenibacillus popilliae | ATCC 14706 | 5.4% ± 7.2% |
| Lactobacillus gasseri | ATCC 3323 | 2.5% ± 1.4% |
| Bifidobacterium longum | ATCC BAA-999 | 1.3% ± 3.2% |
| E. coli (-) | 11303B | 0.58% ± 2.4% |
bPlyPalA is highly species-specific although it does exhibit some lysis against other Paenibacillus spp. Standard deviations were calculated from the ratio of OD600 of lysin-treated wells versus buffer treated wells in triplicate. E. coli was used as a negative control. P. larvae 3650 was used as a positive control.
Some lysis was observed for Paenibacillus polymyxa and Paenibacillus lentimorbus although not for Paenibacillus alvei or Paenibacillus popilliae, both of which exhibited a drop in OD600 even less than that of P. larvae ATCC 49843, the P. larvae strain least affected by PlyPalA. This may indicate that the cell walls of P. polymyxa and P. lentimorbus are somewhat similar to that of P. larvae ERIC genotypes III and IV and that P. alvei and P. popilliae cell walls lack the binding sites necessary for PlyPalA catalysis.
Bactericidal activity
PlyPalA was tested for the ability to kill selected bacterial strains, mostly P. larvae, which were chosen according to the OD600 decrease. These strains included 2 that were strongly affected in terms of OD600 (3650 and 25748), 2 that were moderately affected (2188 and 2231), and 2 that were not significantly lysed (49843 and E. coli 11303B). After 60 mins of exposure to 100 μg/mL PlyPalA, susceptible strains were reduced by just 1 to 2 logs, showing that PlyPalA is only mildly bactericidal (Fig. 5A). The concentration of PlyPalA had to be increased significantly, up to 700 μg/mL, in order to reduce viability by 4 logs (Fig. 5B).
Figure 5.
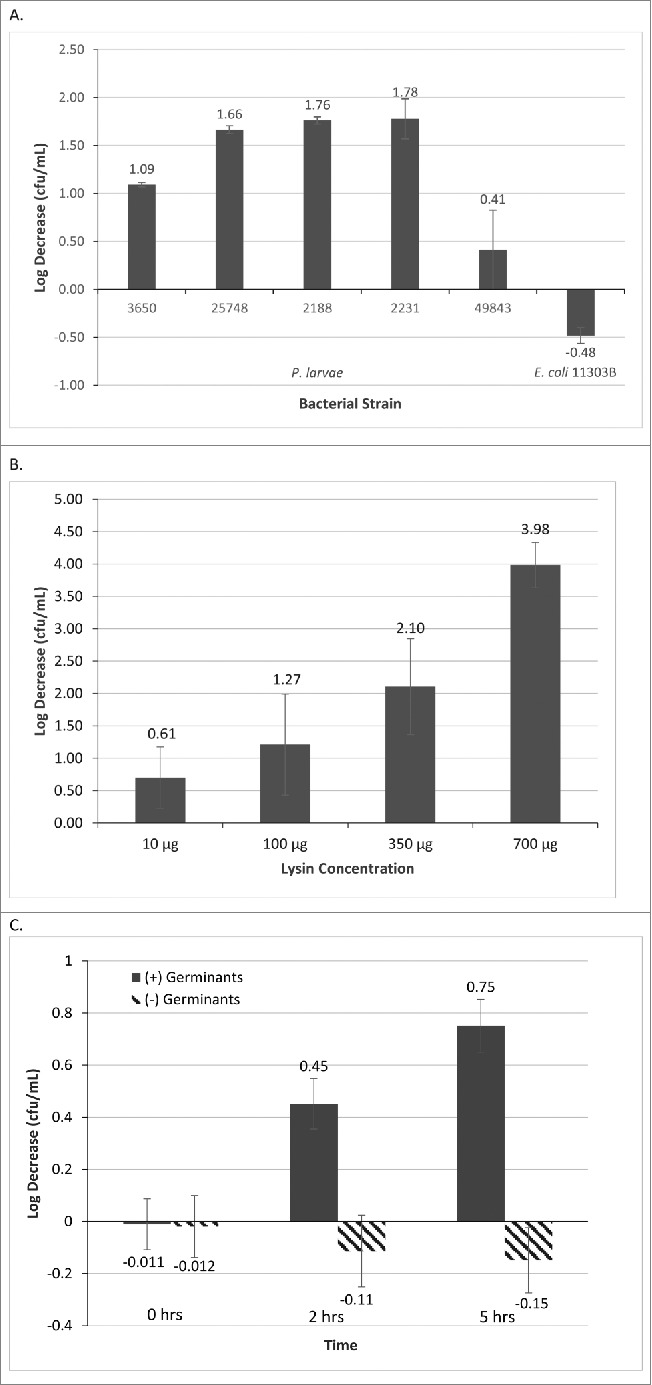
(A) PlyPalA bactericidal activity against selected P. larvae strains. Log-phase P. larvae were treated with 100 μg/ml PlyPalA for 60 mins, serially diluted, and plated onto MYPGP agar. Drop in viable cell count was calculated by comparing colonies formed before or after lysin treatment. E. coli was used as a negative control. Error bars represent standard deviations (n = 3). (B) PlyPalA dose response curve. Log-phase P. larvae 3650 was treated with varying concentrations of PlyPalA for 60 mins, serially diluted, and plated onto MYPGP agar. Drop in viable cell count was calculated by comparing colonies formed before or after lysin treatment. Error bars represent standard deviations (n=2). (C) PlyPalA sporicidal assay. P. larvae 3650 endospores were heat-treated to kill vegetative cells, then treated with or without germinants (MYPGP and 3 mM uric acid) and with or without 16 μg/mL lysin. They were incubated for various durations, serially diluted, and plated onto MYPGP agar. Drop in viable cell count was calculated by comparing lysin-treated spores with non-lysin-treated spores. Solid bars represented spores treated with germinants and dashed bars represent spores without germinants. Error bars represent standard deviations (n=3 for 2 hrs/5 hrs, n=6 for 0 hrs).
Sporicidal activity
PlyPalA was tested for its ability to kill P. larvae 3650 endospores or germinating cells. Endospores that were not induced to germinate yielded the same viable cell count whether or not they were treated with lysin, indicating that PlyPalA cannot kill endospores (Fig. 5C). Endospores that were exposed to germinants and lysin experienced a drop in cell viability; about a half of a log after 2 hours and 3-quarters of a log after 5 hours.
Treatment of infected honeybee larvae
Larvae less than 24 hrs post-hatching were transferred to sterile Petri dishes, fed, and kept at 35°C and 90% relative humidity. The following day, larvae were infected with P. larvae B-3650 spores and then treated with PlyPalA. Larval survival was compared to 2 groups, one where spores were given without treatment and one where only lysin buffer was administered. The buffer control represented a baseline mortality rate, which is ideally ≤ 15% for honeybee larvae in laboratory conditions.14
Larvae were treated by addition of lysin to food to a final concentration of 16 μg/mL, or approximately 160 ng/larva. Though no MIC or dose response curve was performed, this concentration was the maximum possible that could be administered without excessively diluting the food given the concentration of lysin stocks at the time. Lysin or spores were administered to larvae by feeding larvae directly on Petri dishes by pipette with food that had spores and/or lysin added immediately before. Survival was monitored over 8 d and the results for 2 separate experiments were combined and plotted as a Kaplan-Meier survival curve with standard errors indicated by bars (Fig. 6). Based on the log-rank test, the lysin treatment group showed a statistically significant difference (P < 0.0001) from the untreated, spore-infected group. Larval deaths were detected by morphological changes. Dying larvae lost bodily elasticity and segmentation patterns, stopped eating, and often became discolored, turning white, brown, or black and remaining much smaller in mass than their healthy counterparts (Fig. 7).15 Liquefaction often occurred, present as an outline of clear fluid around the larval body.
Figure 6.
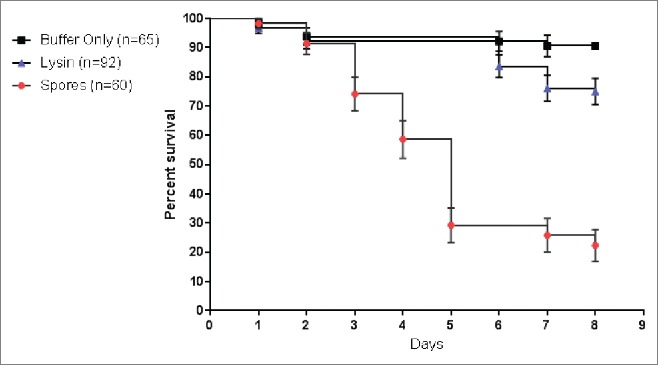
PlyPalA rescued honeybee larvae from American Foulbrood disease. Larvae were infected by addition of endospores to food immediately before feeding to a final concentration of 1×105 endospores/mL of food or approximately 1000 endospores per larva.
Figure 7.
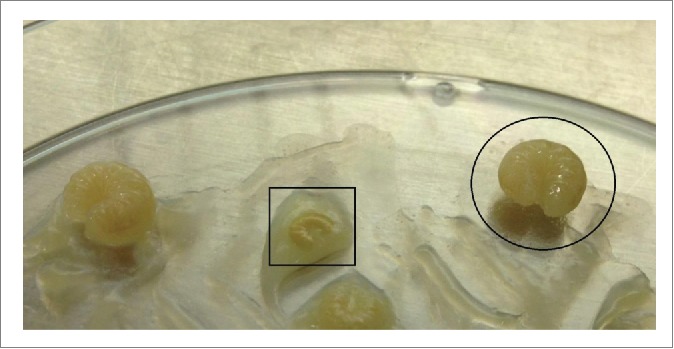
Comparison of healthy (circled) and dying larvae (box) on a petri dish where larvae were infected with spores and then treated with phage lysin.
Larvae infected with endospores alone experienced a survival rate of 23% (7/30) while the baseline survival rate without infection was 89% (58/65). Treatment with lysin alone boosted survival to 75% (69/92); if compared to the expected baseline survival rate of 89% simply due to larvae being in laboratory conditions, this represents a net survival of 84%. The majority of deaths in the spore-treated group occurred on D3 (12/60) and D5 (17/60). Notably, no deaths occurred in either lysin-treated group on D3; most of the deaths occurred on D6 (5/92) and D7 (7/92).
Discussion
A novel P. larvae phage lysin, PlyPalA, isolated from the novel phage Xenia, has shown lytic activity against many P. larvae strains in vitro and has successfully rescued honeybee larvae from P. larvae endospore infection in vivo. To our knowledge, although many lysins have been tested in animal models, this is the first time that lysins have been tested on an economically vital invertebrate.
PlyPalA shows specificity within the strains assayed. Its N-terminal domain, with putative N-acetylmuramoyl-L-alanine amidase activity, is homologous to that of many Bacillus spp. phage amidases yet it shows almost no lytic activity against Bacillus spp. Considering that they all have the same catalytic activity and that lysins are known to be modular, it is suggested that the the N-terminal domains of the Bacillus and Paenibacillus phage amidases analyzed in the alignment have a common ancestor that later diverged by adopting different C-terminal domains in order to adapt to different bacterial hosts. In contrast, the amidase of LambdaLm01 has a markedly different sequence and fewer conserved residues despite possessing the same predicted catalytic mechanism, so perhaps this is an example of convergent evolution rather than homology.
Of all the bacterial strains tested, only Paenibacillus polymyxa and Paenibacillus lentimorbus experienced more than an insignificant reduction in OD600. Commensal strains such as Fructobacillus fructosus and Lactobacillus spp., known to be present in the honeybee larval gut microbiota,13 were not affected. This indicates that PlyPalA would be much less harmful to the natural microbial community compared to a broad-spectrum antibiotic such as oxytetracycline. This is crucial because previous literature suggests that the honeybee gut bacteria are critical in defense against pathogens and neutralization of toxins.3 The lack of lysis in the commensal strains tested indicates that administering PlyPalA to honeybee larvae would likely not disturb the natural microbiota essential for larval immunity during the earliest stages of development.
The lytic specificity means it is not equally effective at lysing all infectious strains of P. larvae. Strains belonging to ERIC genotype II were not used in this study. Strains belonging to ERIC genotypes III and IV are only mildly affected, decreasing the efficacy of PlyPalA as a treatment tool for beehives contaminated with these particular strains. This implies that the cell wall ligand responsible PlyPalA binding is either expressed less in P. larvae ERIC genotypes III and IV, or that there are minor differences in ligand structure which account for less efficient binding and therefore less efficient lysis. The same situation may apply to Paenibacillus polymyxa and Paenibacillus lentimorbus which show similar susceptibility as P. larvae 3688 and 25368. Alternatively, minor differences in cell wall structure such as more extensive crosslinking may account for the difference in lysis. This specificity suggests that lysins may be useful as a rapid strain differentiation tool in the future, much like phages were used for phage typing in the past. Lysins would have the advantage of speed by measuring a decrease in turbidity over several minutes rather than waiting hours for phage infection and lysis to occur.
A previously characterized lysin, PlyG, has activity against endospores.14 PlyG, otherwise a typical phage lysin with a C terminus that binds to vegetative B. anthracis cells, contains a specific spore binding domain in its N-terminal region that recognizes B. anthracis spores.16 Unfortunately, PlyPalA does not have sporicidal activity. No significant drop in viability occurred when spores are treated with lysin, even after 5 hours of incubation. However, mild killing occurred when spores were co-incubated with both germinants and lysin, showing that as spores germinated and became vegetative, cell lysis followed thereafter. Therefore, it is likely that the mechanism of larval rescue involves co-ingestion of endospores with phage lysin, and then once the spores germinate in the midgut, the lysin destroys the vegetative cells before they can multiply and establish an infection.
The survival curves show that most of the deaths in the spore-infected group occurred on either D3 or D5 while most of the deaths in the lysin-treated group occurred in the later days. It is possible, then, that the lysin successfully killed spores germinating early on, but spores that stayed dormant for an extended period of time evaded the lysin. In certain larvae, these dormant spores were numerous enough to cause a lethal infection.
Despite its conserved Zn2+ –binding site, after chelation by EDTA PlyPalA activity is not restored by Zn2+. Many phage lysins can use Zn as a cofactor including Bacillus anthracis prophage lysin PlyL which was crystallized and found to coordinate a zinc ion in vitro.8 PlyPalA displays a strong preference for Mg2+ though it is also capable of using Mn2+, Co2+, and Ca2+, albeit less efficiently at the same concentration. Previously-characterized Bacillus phage lysins show similar but not identical dependence on divalent metal cations as cofactors. Somewhat similarly, LysB4 (AFF27501.1), active against Bacillus cereus, could utilize Zn2+, Mn2+, Ca2+, and Mg2+ as cofactors, although its catalytic domain is an endopeptidase, not an amidase.17 CwlV (BAA90649.1), a phage lysin active against Bacillus polymyxa, could utilize Zn2+, Mn2+, and Co2+ as cofactors, but not Mg2+ or Ca2+, despite having the same catalytic mechanism as PlyPalA, which is N-acetylmuramoyl-L-alanine amidase.18 Such data suggest that the particular cations enabling catalytic activity for phage lysins, while similar, are not perfectly predictable. They may be dependent on the exact geometry of the active site in complex with the peptidoglycan substrate.
Table 3.
Sources of bacterial strains and growth media used in their propagation
| Species | Strain | Medium | Source |
|---|---|---|---|
| Bacillus megaterium | ATCC 12872 | BHI | Dr. Ernesto Abel-Santos |
| Bacillus subtilis | YB955 | BHI | Dr. Eduardo Robleto |
| Bifidobacterium longum | ATCC BAA-999 | MRSC | Dr. Amber Howerton |
| Fructobacillus fructosus | ATCC 13162 | MRS | Agricultural Research Service, USDA |
| Gluconobacter cerinus | ATCC 19441 | MRS | Agricultural Research Service, USDA |
| Lactobacillus brevis | NRRL-1834 | MRS | Agricultural Research Service, USDA |
| Lactobacillus gasseri | ATCC 3323 | MRS | Dr. Amber Howerton |
| Listeria innocua | ATCC 51742 | MYPGP | American Type Culture Collection (ATCC) |
| Paenibacillus alvei | ATCC 6344 | MYPGP | ATCC |
| Paenibacillus lentimorbus | ATCC 14707 | MYPGP | ATCC |
| Paenibacillus polymyxa | GG49 | MYPGP | Ms. Diane Yost |
| Paenibacillus popilliae | ATCC 14706 | MYPGP | ATCC |
| Paenibacillus larvae | NRRL B-2605 | GmBHI | Agricultural Research Service, USDA |
| P. larvae | NRRL B-3650 | GmBHI | Agricultural Research Service, USDA |
| P. larvae | ATCC 25748 | GmBHI | ATCC |
| P. larvae | ATCC 25747 | GmBHI | ATCC |
| P. larvae | 2231 | GmBHI | Infected hive |
| P. larvae | 2188 | GmBHI | Infected hive |
| P. larvae | NRRL B-3544 | GmBHI | Agricultural Research Service, USDA |
| P. larvae | ATCC 3688 | GmBHI | ATCC |
| P. larvae | ATCC 25367 | GmBHI | ATCC |
| P. larvae | ATCC 25368 | GmBHI | ATCC |
| P. larvae | ATCC 49843 | GmBHI | ATCC |
| Staphylococcus aureus | ATCC 12600 | BHI | ATCC |
| E. coli | ATCC 11303B | LB | ATCC |
| E. coli | MON1 | TB | Monserate |
| E. coli | BL21 | TB | Dr. Ernesto Abel-Santos |
The function of the C-terminal domain in previously characterized phage lysins determined specificity.8 This agrees with our findings that although PlyPalA has a nearly identical catalytic domain to Bacillus spp. phage lysins, it does not cause lysis of Bacillus spp. The C-terminal domain has almost no similarity. Often called the cell-wall binding domain, this domain is previously known to possess strong and specific ligand affinity; the affinity is so strong that after lysis during the phage lytic cycle, the C-terminal domain keeps the lysin tethered to the peptidoglycan in order to prevent degradation of nearby hosts.5 Data from the turbidimetric assays suggest that this may also apply to PlyPalA. Figure 3C shows a reverse sigmoidal curve for drop in OD600 in which the turbidity dropped rapidly after an initial lag phase and then leveled off after an amount of time dependent on temperature. For example, at 42°C, OD600 stabilized after approximately 15 mins, quicker than at other temperatures; at 35°C, OD600 stabilized by about 30 mins. Perhaps both the tethering and the hydrolysis are accelerated by a higher temperature but only at the approximate temperature of a beehive does an optimal balance between the 2 occur.
PlyPalA at a concentration of 100 μg/mL was only mildly bactericidal, only reducing the viability in cfu/mL of susceptible P. larvae strains by 1.78 logs at best. This is markedly less than lysins previously examined in the literature such as PlySs2 which reduced the viability of pathogenic Listeria strains by over 5 logs and Staphylococcus strains by 2 to 5 logs at a similar concentration.6 Lysin concentration had to be septupled to a concentration of 700 μg/mL in the dose response curve in order to reduce viability by approximately 4 logs. Interestingly, there was not a direct correlation between OD600 decrease and decrease in viable cell count. P. larvae strain 3650 was the strain most strongly lysed in turbidimetric assays but its viability after incubation with PlyPalA was nearly one log better than strains 2188 or 2231. As expected, however, P. larvae 49843 was not strongly affected by the lysin, further suggesting that other treatments should be recommended for AFB caused by ERIC IV strains.
One important consideration is how to apply this lysin in the field to prevent infection by P. larvae. Although no experiments on actual beehives were conducted in this investigation, the fact that lyophilization did not reduce lysin activity at all suggests that the dust method may be a possibility. This method is currently used for the antibiotic tylosin; essentially, tylosin is mixed with confectioner's sugar and applied regularly to frames in the beehive on weekly intervals.19 Such a method showed a significant reduction in diseased hives after 65 days;19 therefore, a similar approach may be adapted for the lysin in future tests by lyophilizing it, mixing it with confectioner's sugar, and sprinkling it on top of frames.
In summary, PlyPalA represents a promising breakthrough in the ongoing struggle against pathogenic P. larvae and the decreasing honeybee populations worldwide. It maintains activity in a wide range of biochemical conditions, protects larvae in vivo from lethal infection, does not harm commensal bacteria known to be present in the larval gut, and is readily produced and purified as a recombinant protein in E. coli. These characteristics support PlyPalA as a lysin worthy of further characterization and eventual application in the field as a treatment for American Foulbrood disease.
Materials and Methods
Bacterial growth and preservation
Media were obtained from Difco Laboratories, Inc., and chemicals were obtained from Sigma-Aldrich unless otherwise stated. Paenibacillus, Listeria, and Brevibacterium strains were cultivated in MYPGP medium consisting of Mueller Hinton broth (Sigma Aldrich, 70192), yeast extract (BD Biosciences, 211930), sodium pyruvate, glucose (J.T. Baker, 1919-05), and KH2PO4 as described previously.20 Bacillus and Staphylococcus strains were cultivated in Brain Heart Infusion (BHI) broth (BD Biosciences, 237300). P. larvae strains were cultivated in BHI broth supplemented with an additional 0.4% glucose, 1 mM CaCl2, and 1 mM MgCl2, henceforth referred to as GmBHI. Lactobacillus, Fructobacillus, and Gluconobacter strains were cultivated in de Man, Rogosa, and Sharpe (MRS) broth (EMD Millipore, 110661). Bifidobacterium strains were cultivated anaerobically in MRSC broth supplemented with 5 mg/L resazurin (Sigma Aldrich, R7017) as a redox indicator and 0.5 g/L L-cysteine (Sigma Aldrich, 168149) as a reducing agent. E. coli strains were cultivated either in Tartoff-Hobbs broth.21 (TB) or in LB.22
Bacteria were grown at 37°C in an environmental shaker (Barnstead LabLine MaxQ 4000, SHKE4000-5) at 150 rpm except for Gluconobacter cerinus which was cultivated at 30°C. All bacterial strains were preserved in 15% glycerol at −80°C.
Phage genomic DNA extraction and sequencing
Phage Xenia, a dsDNA siphophage, was originally isolated from an infected scale enriched with P. larvae strain NRRL B-2605. After three transfers of single plaques, phage Xenia was amplified overnight on P. larvae strain ATCC 25748 in flasks at a low multiplicity of infection (∼0.1). Cellular debris was pelleted by centrifugation at 3220 x g for 15 mins (Eppendorf Centrifuge 5810) then the supernatant was vacuum filtered using 0.2 μm cellulose acetate membrane filters with a pore size of (VWR, 28145-477). Titers were determined by soft agar overlays23 and lysates containing a phage concentration greater than 1 × 109 pfu/mL were selected for DNA extraction.
To the phage lysate, DNase I (New England Biolabs, M0303S) and RNase A (Sigma-Aldrich, R4875) were added at a final concentration of 10 U/mL and 2.5 μg/mL and incubated at 37°C for 30 mins. Following heat inactivation, the phages were pelleted by centrifugation with PEG-6000 and NaCl as previously described.24 Phage pellets were resuspended in 200 μL of buffer containing 10 mM Tris, 5 mM MgSO4, 0.5 mM EDTA, and 0.5 mM CaCl2, adjusted to pH 7.6. Proteinase K (New England Biolabs, P8107S) and SDS (Research Organics, Inc., 3306262) were then added to final concentrations of 40 U/mL and 0.45%, respectively, and the sample was incubated at 55°C for 45 mins.25
Samples were then cooled to room temperature and DNA extraction based on phenol was then carried out as previously described,26 except that vortexing of the sample was only carried out for 15s in order to minimize shearing of genomic DNA. The presence of genomic DNA was verified on an agarose gel. The concentration of phage DNA was determined by a PicoGreen (Life Technologies, P7589) fluorescence assay and then sequenced with the Nextera XT DNA Sample Preparation Kit (Illumina Inc., FC-131-1024) according to manufacturer's instructions. These libraries were sequenced on a MiSeq Desktop Sequencer at the Environmental Genetics and Genomics Laboratory (EnGGen) at Northern Arizona University.
Phage genomic analysis and cloning of PlyPalA
Using the program Geneious, overlapping reads were assembled into the complete genome of phage Xenia yielding an average coverage depth of 123× and a minimum coverage depth of 41× (http://www.geneious.com/). Identification and annotation of open reading frames was performed using DNA Master (http://cobamide2.bio.pitt.edu).
A putative lysin gene, PlyPalA, was amplified from phage genomic DNA using Phusion High-Fidelity DNA Polymerase (New England Biolabs, M0530S) under the following conditions: 98°C for 30 seconds, 30 cycles of 98°C for 7 seconds, 60°C for 25 seconds, and 72°C for 30 seconds, and a final extension step of 72°C for 5 mins. The PCR used the following primers: 5′-AAGACCCCATATGATGGAAATCAGAGAAATGC-3′ (forward) and 5′-TGAACTCGAGCTACTACAGGCTACTCGCTAA-3' (reverse) (Integrated DNA Technologies, Inc.). The bolded nucleotides signify restriction sites for the endonucleases NdeI (New England Biolabs, R0146S) and XhoI (New England Biolabs, R0111S), respectively. The 700 bp amplicon was verified by agarose gel electrophoresis.
Both the amplicon and the plasmid vector pET-28a(+) (Novagen), containing a 6x His tag, a gene for kanamycin resistance, and a T7 promoter, were cut with NdeI and XhoI.27 Then, the gene and plasmid were ligated using T4 DNA ligase (New England Biolabs) overnight at 16°C. After heat inactivation, the ligation product, pET-28a(+)_PlyPalA, was transformed into E. coli MON1 cells according to manufacturer's instructions (Monserate Biotechnology Group, 5001). E. coli transformants were plated onto TB agar supplemented with 50 μg/mL kanamycin and grown overnight at 37°C. Random colonies were picked and marked on the plate and DNA was extracted from single colonies using NaOH as previously described.28
PCR amplification to confirm the size and presence of the insert was then carried out with the following T7 primers: 5′-TAATACGACTCACTATAG-3′ (forward) and 5′-GCTAGTTATTGCTCAGCGG-3′ (reverse) (https://dnasu.org/DNASU/VectorPrimers.jsp). Thermocycle conditions were 98°C for 30 seconds, 30 cycles of 98°C for 7 seconds, 52°C for 20 seconds, and 72°C for 30 seconds, and a final extension step of 72°C for 2 mins. The exact gene sequence was verified by sequencing on an ABI 3130 Genetic Analyzer (Applied Biosystems). Transformants corresponding to the correct sequence were propagated in flasks containing TB with kanamycin. The plasmid was extracted by a miniprep procedure using the Mini Plus Plasmid Extraction System kit (Viogene, GF2001) according to manufacturer's instructions. This plasmid was then transformed into E. coli strain BL21 (Novagen) according to manufacturer's instructions. The PlyPalA gene was submitted to the GenBank database under the accession number KT167538.
Recombinant expression and purification of PlyPalA
After transformation with pET-28a(+)_PlyPalA, an E. coli BL21 colony was picked and propagated in TB with kanamycin at 37°C with shaking at 150 rpm overnight. This starter culture was used to inoculate fresh TB with kanamycin. Expression of PlyPalA was induced by adding IPTG (Gold Biotechnology, I2481C5) to a final concentration of 1 mM to a log-phase culture with an OD600 of approximately 0.85. The flask was then transferred to 30°C and incubated for 3 hours with shaking at 200 rpm to allow expression. The cells were then chilled on ice, pelleted by centrifugation at 3,220 × g for 15 mins (Eppendorf Centrifuge 5810), and frozen at -80°C.
Proteins were extracted from cell pellets using the B-PER Complete Reagent according to the manufacturer's instructions (Thermo Scientific, 90084). The crude protein extracts were then directly applied to a HiTrap DEAE Sepharose Fast Flow anion exchange column (AEC) equilibrated with 20 mM Tris, pH 7.6, and eluted using a stepwise gradient of NaCl according to manufacturer's instructions (GE Healthcare, 17-5055-01).
Fractions containing lysin were then supplemented with imidazole to a final concentration of 25 mM to the column and applied to a HisTrap FF Crude immobilized metal ion affinity chromatography (IMAC) column (GE Healthcare, 11-0004-58). The column was washed extensively with buffer containing 20 mM Tris, 25 mM imidazole and 500 mM NaCl, pH 7.6, and then eluted with a stepwise gradient of imidazole (Sigma Aldrich, I5513) from 50 mM to a concentration of 250 mM.
The lysin was changed into buffer containing 20 mM Tris and 100 mM NaCl, pH 8.0 using PD-10 desalting columns (General Electric, 17-0851-01). Finally, the 6x His tag was removed by incubating the tagged protein with 20 U of thrombin (Sigma Aldrich, T4648) overnight at room temperature. Thrombin was separated from the lysin using AEC. The resulting lysin was dialyzed into lysin buffer (20 mM Tris, 10 mM MgCl2, pH 8.0). Protein concentrations were quantified by a bicinchoninic acid (BCA) assay (Bio-World, 20831001-1) according to manufacturer's instructions. Lysin was then stored in 1 mL aliquots in lysin buffer at −80°C.
In vitro characterization of PlyPalA
The optimal biochemical conditions for enzymatic activity and strain/species specificity were determined using a spectrophotometric method. Frozen pellets of log-phase bacterial cultures were resuspended in 180 μL of lysin buffer in 96-well microtiter plates (Falcon, 353910). Then, 20 μL of either PlyPalA to a final concentration of 10 μg/mL or lysin buffer was added to each well in triplicate. Spectrophotometric measurements at a wavelength of 600 nm (OD600) were taken using an Infinite M1000 plate reader (Tecan) every minute over 30 mins at 35°C, or 45 mins for the pH range assay. The percentage loss of turbidity was calculated as follows: 1-(average OD600 for wells containing lysin/average OD600 for wells containing buffer)*100%.
For the divalent cation assay, special precautions were taken to remove contaminating cations. Both the resuspended cells and lysin were incubated with 5.0 mM EDTA at room temperature for 20 mins. EDTA was removed from the cells by centrifugation at 6,000 × g at 4°C for 5 mins followed by resuspension in buffer containing 20 mM MES and 2.5 mM EDTA adjusted to pH 6.7. EDTA was removed from the lysin by centrifugation at 14,000 x g at room temperature for 25 mins using 3 KDa cutoff centrifugal filters (Amicon, Z677094). The cell resuspension was then supplemented with chloride (Mg2+, Mn2+, Ni2+, Co2+, Ca2+) or sulfate (Zn2+) salts of various divalent cations to a final concentration of 5.0 mM.
To test lysin viability after lyophilization, purified lysin in lysin buffer at a concentration of 100 μg/mL was lyophilized overnight using a FreeZone Freeze Dry System (Labconco, 7740040) and then resuspended the following day in sterile water. The resuspended lysin was then assayed for turbidity reduction of P. larvae strain 3650 in 96-well plates as described above compared to lysin of the same concentration that had never been lyophilized.
Bactericidal activity
Bacteria from overnight cultures were diluted into fresh media and grown to mid-log phase, then centrifuged and resuspended in microfuge tubes using buffer containing lysin at a concentration of 100 μg/mL to an OD600 of 0.100 (approximately 108 cfu/mL; the actual cell count was determined by serially diluting and plating untreated cells on MYPGP agar). These were incubated at 35°C for one hr with occasional inversion. After incubation, the cells were serially diluted and plated on MYPGP agar. The loss of viability was calculated as follows: log(number of viable cells without lysin treatment) – log(number of viable cells with lysin treatment). The dose response curve was carried out identically except lysin was diluted as needed using buffer.
Sporicidal activity
P. larvae strain 3650 endospores were diluted to approximately 106 cfu/mL and vegetative cells were killed by heating the spores at 68°C for 15 min.29 Then, spores were diluted to 105 cfu/mL and separated into 4 treatment groups. Spores were treated with or without germinants as well as with or without lysin. P. larvae requires tyrosine and uric acid for germination.30 In order to mimic germination conditions in the honeybee larval gut, MYPGP was used as a source of tyrosine (as well as other amino acids, to ensure growth of cells) and uric acid was added to the mix to a concentration of 3 mM.30 For groups without germinants, sterile ddH2O was added to the same volume instead. Lysin was added to a concentration of 16 μg/mL to ensure that the ratio of spores:lysin per mL would be the same as in the in vivo experiments (1000 spores:160 ng of lysin). For groups without lysin, lysin buffer was added instead. All four treatment groups were incubated for different time points (t = 0, 2, and 5 hrs), serially diluted, and plated on MYPGP agar. The loss of viability was calculated as follows: log(number of viable cells from spores not treated with lysin) – log(number of viable cells from spores treated with lysin). Each time point compared loss of viability with germinants versus without germinants side by side to compare killing of spores with germinating cells.
Rearing of honeybee larvae
Methods for rearing honeybee larvae were adapted from standard methods14 as follows. Larvae were obtained from colonies with no clinical symptoms of disease. Queens were caged in order to confine them to a particular region on the comb and observed for oviposition.14 Larvae were grafted from the frames less than 24 hrs after hatching. Larvae were kept in sterile Petri dishes, stored in plastic containers containing a solution of 10% glycerol to maintain an internal humidity of 90% in a 34-35°C SHEL LAB Digital Laboratory Incubator. A pan of H2O was placed at the bottom shelf to maintain 80% humidity in the incubator itself. Food was prepared as a mix of royal jelly, glucose, fructose, yeast extract, and water as previously described.31
Larvae were fed an increasing amount of food each day, beginning with 10 μL on D0 and D1, according to amounts specified previously14 except that feeding continued for an additional 2 days (50 μL on D7, then 60 μL on D8) in order to monitor mortality until pupation. Additionally, 800 μL of food was diluted with 200 μL GmBHI, vortexed to ensure homogeneity, and warmed to 34-35°C immediately before feeding.
On Day 1 after grafting, larvae were infected by administering food containing a spore concentration of approximately 1×105 P. larvae B-3650 endospores/mL which equals 1000 endospores per larva. One group of larvae was administered food containing 16 μg/mL of lysin. Treatments and spores were simultaneously administered. A control group was fed food diluted with lysin buffer (800 μL plus 200 μL) alone to determine the baseline survival rate. Each experimental group had an n = 30 for the controls and n = 45 for the lysin group and experiments were repeated twice.
Survival rates for each experimental group were monitored each day immediately before feeding. Larvae were marked as dead if they lost bodily segmentation or changed color to brown, black, or completely white.15 Dead larvae were carefully removed using sterile swabs. The survival rate data were statistically analyzed using Kaplan-Meier survival curves with standard deviations, P values, and 95% confidence intervals calculated using the Prism software program (GraphPad Software).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed
Acknowledgments
For technical assistance with SDS-PAGE and plate readers, we thank Chris Yip. We thank Dr. Richard Gomer from Texas A&M for his generous gift of 3 kDa cutoff filters. We thank Dr. Ernesto Abel-Santos for E. coli cloning strain BL21 and plasmid pET-28a(+). We also thank Dr. Gary Kleiger's lab for the PD-10 desalting column. Sequencing of the lysin gene construct was performed by Casey Hall from the UNLV Genomics Core. Sequencing of the novel phage was done by Andrew Krohn from Northern Arizona University. Finally, we thank Israel Alvarado for the generous gift of P. larvae 3650 endospores as well as technical assistance.
Funding
Funding for this research was provided by United States Department of Agriculture grant NEVR-2010-03755, National Science Foundation EPSCoR II Award IIA-1301726, and the 2014 American Society for Microbiology Undergraduate Research Fellowship.
References
- 1. Genersch E. American foulbrood in honeybees and its causative agent, Paenibacillus larvae. J Invertebr Pathol 2009; 103:S10–9; PMID:19909971; http://dx.doi.org/ 10.1016/j.jip.2009.06.015 [DOI] [PubMed] [Google Scholar]
- 2. de Graaf DC, Alippi AM, Antúnez K, Aronstein KA, Budge G, De Koker D, De Smet L, Dingman D, Evans JD, Foster LJ, et al. . Standard methods for American foulbrood research. J Apicult Res 2012; 52(1):1–26 [Google Scholar]
- 3. Tian B, Fadhil NH, Powell JE, Kwong WK, Moran NA. Long-term exposure to antibiotics has caused accumulation of resistance determinants in the gut microbiota of honeybees. MBio 2012; 3(6):e00377-12; PMID:23111871; http://dx.doi.org/ 10.1128/mBio.00377-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fischetti VA. Bacteriophage lysins as effective antibacterials. Curr Opin Microbiol 2008; 11(5):393–400; PMID:18824123; http://dx.doi.org/ 10.1016/j.mib.2008.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Oliveira H, Melo LDR, Santos SB, Nobrega FL, Ferreira EC, Cerca N, Azeredo J, Kluskens LD. Molecular aspects and comparative genomics of bacteriophage endolysins. J Virol 2013; 87(8):4558–70; PMID:23408602; http://dx.doi.org/ 10.1128/JVI.03277-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gilmer DB, Schmitz JE, Euler CW, Fischetti VA. Novel bacteriophage lysin with broad lytic activity protects against mixed infection by Streptococcus pyogenes and methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 2013; 57(6):2743–50; PMID:23571534; http://dx.doi.org/ 10.1128/AAC.02526-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dziarski R, Gupta D. The peptidoglycan recognition proteins. Genome Biol 2006; 7(8):232; PMID:16930467; http://dx.doi.org/ 10.1186/gb-2006-7-8-232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Low LY, Yang C, Perego M, Osterman A, Liddington RC. Structure and lytic acivity of a Bacillus anthracis prophage endolysin. J Biol Chem 2005; 280:35433–9; PMID:16103125; http://dx.doi.org/ 10.1074/jbc.M502723200 [DOI] [PubMed] [Google Scholar]
- 9. Djukic M, Brzuszkiewicz E, Fünfhaus A, Voss J, Gollnow K, Poppinga L, Liesegang H, Garcia-Gonzalez E, Genersch E, Daniel R. How to kill the honey bee larva: genomic potential and virulence mechanisms of Paenibacillus larvae. PLoS One 2014; 9(3):e90914; PMID:24599066; http://dx.doi.org/ 10.1371/journal.pone.0090914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schuch R, Nelson D, Fischetti VA. A bacteriolytic agent that detects and kills Bacillus anthracis. Nature 2002; 418:884–9; PMID:12192412; http://dx.doi.org/ 10.1038/nature01026 [DOI] [PubMed] [Google Scholar]
- 11. Longchamp PF, Mauël C, Karamata D. Lytic enzymes associated with defective prophages of Bacillus subtilis: sequencing and characterization of the region comprising the N-acetylmuramoyl-L-alanine amidase gene of prophage PBSX. Microbiology 1994; 140:1855–67; PMID:7921239; http://dx.doi.org/ 10.1099/13500872-140-8-1855 [DOI] [PubMed] [Google Scholar]
- 12. Briers Y, Klumpp J, Schuppler M, Loessner MJ. Genome sequence of Listeria monocytogenes Scott A, a clinical isolate from a food-borne listeriosis outbreak. J Bacteriol 2011; 193(16):4284–5; PMID:21685277; http://dx.doi.org/ 10.1128/JB.05328-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vojvodic S, Rehan SM, Anderson KE. Microbial gut diversity of Africanized and European honeybee larval instars. PLoS One 2013; 8(8):e72106; PMID:23991051; http://dx.doi.org/ 10.1371/journal.pone.0072106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Crailsheim K, Brodschneider R, Aupinel P, Behrens D, Genersch E, Vollmann J, Riessberger-Gallé U. Standard methods for artificial rearing of Apis mellifera larvae. J Apicult Res 2012; 52(1):1–16 [Google Scholar]
- 15. Genersch E, Forsgren E, Pentikäinen J, Ashiralieva A, Rauch S, Kilwinski J, Fries I. Reclassification of Paenibacillus larvae subsp. pulvifaciens and Paenibacillus larvae subsp. larvae as Paenibacillus larvae without subspecies differentiation. Int J Syst Evol Microbiol 2006; 56:501–11; PMID:16514018; http://dx.doi.org/ 10.1099/ijs.0.63928-0 [DOI] [PubMed] [Google Scholar]
- 16. Yang H, Wang DB, Dong Q, Zhang Z, Cui Z, Deng J, Yu J, Zhang X, Wei H. Existence of Separate Domains in Lysin PlyG for Recognizing Bacillus anthracis Spores and Vegetative Cells. Antimicrob Agents Chemother 2012; 56(10):5031–9; PMID:22802245; http://dx.doi.org/ 10.1128/AAC.00891-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Son B, Yun J, Lim J, Shin H, Heu S, Ryu S. Characterization of LysB4, an endolysin from the Bacillus cereus-infecting bacteriophage B4. BMC Microbiol 2012; 12:3; PMID:22235902; http://dx.doi.org/ 10.1186/1471-2180-12-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shida T, Hattori H, Ise F, Sekiguchi J. Mutational analysis of catalytic sites of the cell wall lytic N-acetylmuramoyl-L-alanine amidases CwlC and CwlV. J Biol Chem 2001; 276:28140–6; PMID:11375403; http://dx.doi.org/ 10.1074/jbc.M103903200 [DOI] [PubMed] [Google Scholar]
- 19. Elzen PJ, Westervelt D, Causey D, Ellis J, Hepburn HR, Neumann P. Method of application of tylosin, an antibiotic for American foulbrood control, with effects on small hive beetle (Coleoptera: Nitidulidae) populations. J Econ Entomol 2002; 95(6):1119–22; PMID:12539820; http://dx.doi.org/ 10.1603/0022-0493-95.6.1119 [DOI] [PubMed] [Google Scholar]
- 20. Dingman DW, Stahly DP. Medium promoting sporulation of Bacillus larvae and metabolism of medium components. Appl Environ Microbiol 1983; 46:860–9; PMID:16346399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tartoff KD, Hobbs CA. Improved media for growing plasmid and plasmid clones. Bethesda Research Laboratories Focus 1987; 9(12):2 [Google Scholar]
- 22. Green MR, Sambrook J. Molecular cloning: A laboratory manual. 4th ed. New York: Cold Spring Harbor Laboratory Press; 2012. 2028 p [Google Scholar]
- 23. Hurst CJ, Reynolds KA. Chapter 48: Sampling viruses from soil. In: Manual of environmental microbiology. 2nd ed. Hurst CJ, Crawford RL, Knudsen GR, McInerney MJ, Stetzenbach L, editors. Washington, DC: American Society for Microbiology Press; 2002. 527-534 p [Google Scholar]
- 24. Yamamoto KR, Alberts BM. Rapid bacteriophage sedimentation in the presence of polyethylene glycol and its application to large-scale virus purification. Virol 1970; 40(3):734–44; http://dx.doi.org/ 10.1016/0042-6822(70)90218-7 [DOI] [PubMed] [Google Scholar]
- 25. Sambrook J, Russell DW. Extraction of bacteriophage λ DNA from large-scale cultures using proteinase K and SDS. CSH Protoc 2006; 2006(1):1484-94. [DOI] [PubMed] [Google Scholar]
- 26. Cheng HR, Jiang N. Extremely rapid extraction of DNA from bacteria and yeasts. Biotechnol Lett 2006; 28(1):55–9; PMID:16369876; http://dx.doi.org/ 10.1007/s10529-005-4688-z [DOI] [PubMed] [Google Scholar]
- 27. Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 1995; 177(14):4121–30; PMID:7608087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schuch R, Fischetti VA, Nelson DC. A genetic screen to identify bacteriophage lysins. Methods Mol Biol 2009; 502:307–19; PMID:19082564; http://dx.doi.org/ 10.1007/978-1-60327-565-1_18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Alvarado I, Elekonich MM, Abel-Santos E, Wing EJ. Comparison of in vitro methods for the production of Paenibacillus larvae endospores. J Microbiol Methods 2015; 116:30–2; PMID:26130193; http://dx.doi.org/ 10.1016/j.mimet.2015.06.011 [DOI] [PubMed] [Google Scholar]
- 30. Alvarado I, Phui A, Elekonich MM, Abel-Santos E. Requirements for in vitro germination of Paenibacillus larvae spores. J Bacteriol 2012; 195(5):1005; PMID:23264573; http://dx.doi.org/ 10.1128/JB.01958-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Peng YSC, Mussen E, Fong A, Montague MA, Tyler T. Effects of chlortetracycline on honey-bee worker larvae reared in vitro. J Invertebr Pathol 1992; 60:127–33; http://dx.doi.org/ 10.1016/0022-2011(92)90085-I [DOI] [Google Scholar]