

Abstract
Today, Fmoc SPPS is the method of choice for peptide synthesis. Very‐high‐quality Fmoc building blocks are available at low cost because of the economies of scale arising from current multiton production of therapeutic peptides by Fmoc SPPS. Many modified derivatives are commercially available as Fmoc building blocks, making synthetic access to a broad range of peptide derivatives straightforward. The number of synthetic peptides entering clinical trials has grown continuously over the last decade, and recent advances in the Fmoc SPPS technology are a response to the growing demand from medicinal chemistry and pharmacology. Improvements are being continually reported for peptide quality, synthesis time and novel synthetic targets. Topical peptide research has contributed to a continuous improvement and expansion of Fmoc SPPS applications. Copyright © 2015 European Peptide Society and John Wiley & Sons, Ltd.
Keywords: solid‐phase peptide synthesis, Fmoc/tBu, aspartimide, peptide thioester, post‐translational modification, protecting group, racemisation
Abbreviations
- Bzl
benzyl
- Boc
t‐butyloxycarbonyl
- DIC
diisopropyl carbodiimide
- EDT
ethanedithiol
- Far
farnesyl
- GC
gas chromatography
- Mob
4‐methoxybenzyl
- MPAA
dynamic contrast enhanced
- DSC
4‐mercaptophenylacetic acid
- Mpe
3‐methyl‐3‐pentyl
- Oxyma
ethyl cyano(hydroxyimino) acetate
- PTM
post‐translational modification
- Pbf
2,2,4,6,7‐pentamethyldihydrobenzofuran‐5‐sulfonyl
- Pmc
2,2,5,7,8‐pentamethylchromanyl‐6‐sulfonyl
- TIS
triisopropylsilane
- Tmob
2,4,6‐trimethoxybenzyl
- Trt
triphenylmethyl.
Introduction
The success of peptide drugs, notably glucagon‐like peptide 1 receptor agonists, and a promising pipeline of peptide drugs has renewed interest in synthetic peptides 1, 2. Additionally, the rapidly emerging field of peptide‐based biomaterials has further stimulated demand 3. The majority of synthetic peptides are now prepared by Fmoc solid‐phase peptide synthesis (SPPS) 4. Classical t‐butyloxycarbonyl (Boc) SPPS is now generally only used for specialist applications. Initially, the success of the Fmoc chemistry was due to its rapid adoption by non‐chemists as biologists realised they could quickly prepare peptides suitable for antibody production using inexpensive machines and avoid the use of anhydrous hydrogen fluoride (HF) 5. Fmoc SPPS was easy to automate because there was no need for corrosive TFA in the synthetic cycles and because deprotection released a fluorene group with strong UV absorption properties that gave a useful indicator of synthesis success 6. For peptide chemists themselves, Fmoc chemistry provided a solution to the previously limiting conditions of the Boc method as the deprotection conditions were compatible with modified peptides, such as phosphorylated and glycosylated peptides and for peptide libraries 7. The concern with the Boc technique had always been the lack of complete differentiation in the reaction conditions for cleavage of the Boc group and semipermanent side‐chain protection. The iterative use of TFA could cleave small amounts of the side‐chain protecting groups at each cycle and cause progressive loss of peptide from the polymer support. In contrast, Fmoc SPPS provided an orthogonal combination of temporary and permanent protecting groups.
Fmoc belongs to a set of urethane protecting groups including the benzyl carbamate (benzyloxycarbonyl) and Boc protecting groups that suppress racemisation during activation and coupling. Carpino and Han introduced the Fmoc group for solution chemistry, but it proved unsuitable 8, 9. The initial cleavage product, dibenzofulvene, is reactive and can reattach to the liberated amine or be potentially difficult to separate from the product, in contrast to Boc where the deprotection product, butylene, is volatile. It was notable for its exceptional lability to bases, particularly secondary amines. When screened alongside several other base‐labile candidates for its application to solid phase, the Fmoc group found its métier, as on the solid support, the dibenzofulvene and any associated adducts could be simply washed away 10, 11. Furthermore, the release of the Fmoc group gave a unique method to monitor deprotection 12.
There have been considerable advances in the length of peptides synthesised. Partly, this is a consequence of the improvements in purity of the Fmoc building blocks. Mostly, however, it has been due to the success of applying pseudoprolines 13, 14 and backbone protection 15 to the synthesis of long peptides overcoming the difficult sequence problem 16. Although the figure of around 50 amino acids is often given in publications as the average target that can be routinely synthesised, in practice, this figure is meaningless as many much shorter sequences are extremely problematic and synthetic success is not guaranteed.
Whilst the combination of side‐chain protecting groups used by peptide chemists for routine Fmoc/tert‐butyl (tBu) chemistry has remained largely unchanged for more than 15 years (Table 1), many recognise that for some amino acids, particularly arginine, asparagine, aspartic acid, histidine and cysteine, the choice is suboptimal. However, the adoption of new and superior protecting groups by peptide chemists has been slow mainly because the standard ones are produced cheaply and ultrapure in industrial scales for good manufacturing practice peptide production. In this review, we will highlight some new developments, with the hope that by bringing their benefits to a wider audience, we will encourage their take‐up by peptide chemists and help stimulate innovation in the development of basic peptide synthesis reagents. For further information on the origins of the methodology and current practice, a number of excellent reviews are available 7, 17, 18, 19, 20, 21.
Table 1.
Standard TFA‐labile protecting groups for Fmoc SPPS [Fmoc‐Xaa(P)‐OH]
Developments in Nα‐Fmoc amino acid derivatives
Purity of Nα‐Fmoc amino acids
The industrialisation and regulation of Fmoc‐protected amino acid derivatives have led to a significant improvement in the quality of the 20 standard Fmoc‐protected amino acid building blocks 29. Most Fmoc amino acids are now available in remarkably high RP‐HPLC purity of >99% although a number of well‐documented side reactions can occur during the introduction of the Fmoc group to the Nα‐amine of an amino acid. The most frequently encountered is the Lossen‐type rearrangement, which leads to the formation of Fmoc‐β‐Ala‐OH and Fmoc‐β‐Ala‐Xaa‐OH using 9‐fluorenylmethyloxycabonyl N‐hydroxysuccinimide 30, 31, and the unwanted carboxyl activation, which generates the Fmoc‐Xaa‐Xaa‐OH dipeptide using 9‐fluorenylmethyl chloroformate 32, 33. These impurities will be incorporated into the growing peptide chain. Therefore, it is important when using HPLC analysis for quality control of the Fmoc building block that these impurities do not co‐elute.
To prevent this, an intermediate silylation with chlorotrimethylsilane has been proposed to protect the carboxylic acid and prevent amino acid oligomerisation during Fmoc protection 34. The requirements for improved oxime base reagents have been developed for the clean introduction of the Nα‐Fmoc protecting group aiming to replace the N‐hydroxysuccinimide activation 35, 36.
The International Conference on Harmonisation for standards of active pharmaceutical ingredient production (Q11) requires optical purity, acetic acid content and free amine content to be specified of the amino acids 37. Enantiomeric purity can be quantified greater than 99.9% by gas chromatography (GC)‐MS 38.
The presence of acetic acid in Fmoc‐amino acid derivatives is a serious problem, as it cannot be detected by RP‐HPLC and causes permanent capping. Some commercial preparation of trifunctional amino acids like Fmoc‐Arg[2,2,4,6,7‐pentamethyldihydrobenzofuran‐5‐sulfonyl (Pbf)]‐OH and Fmoc‐Asn/Gln[triphenylmethyl (Trt)]‐OH can contain significant amounts. As acetic acid has an Mr of only 60, negligible amounts lead to significant chain terminations during peptide assembly. Levels <0.02% are required for a clean SPPS.
Another consideration is the content of free amino acid in an Fmoc‐amino acid derivative. This can result in incorporation of multiple copies of the target amino acid into the peptide chain. Furthermore, free amino acid can compromise long‐term storage as traces of the free amine promote autocatalytic Fmoc cleavage. However, quantification of the free amine today is problematic. Suppliers provide either a GC‐based method with a limit of detection of 0.2% or a semiquantitative TLC‐ninhydrin assay.
Advances in side‐chain protection
Aspartic acid
The most serious side reaction during Fmoc chemistry is aspartimide formation (Scheme 1) 39. It is caused by exposure of the peptide sequence containing aspartic acid to strong base. Aspartimide formation is therefore a major problem for the synthesis of long peptides and sequences containing multiple aspartic acid residues. Aspartimide formation is particularly pernicious as it can lead to the formation of nine different by‐products, some of which will co‐elute with the target peptide. Attack by water yields the undesired d/l‐β‐aspartyl peptides in a ratio of 3 : 1 to the α‐aspartyl peptide 40, 41, 42. Ring opening by piperidine gives a mixture of d/l‐α‐piperidides and d/l‐β‐piperidides. Ring opening by amino groups leads to the formation of dipeptides or cyclic peptides 43. In most cases, α‐piperidides and β‐piperidides are easily separated from the target peptide by RP‐HPLC; however, resolution of the epimerised α‐aspartyl peptide is very difficult or impossible 44. The extent of aspartimide formation is highly dependent on the nature of the amino acid following the aspartyl residue. Sequences that are particularly aspartimide prone are ‐Asp‐Gly‐, ‐Asp‐Asp‐, ‐Asp‐Asn‐, ‐Asp‐Arg‐, Asp‐Thr‐ and ‐Asp‐Cys‐ 45, 46 with Asp‐Gly being the worst case (Table 2) 47. Ser and Thr need to be side chain protected 45. The importance of the aspartyl side‐chain protection has been demonstrated in many studies 48. Aspartimide formation was exacerbated by the use of less bulky protecting groups: ODie 1 (Figure 1) 49 > OMpe 50 > OtBu 51 > O‐1‐adamantyl 52, trityl‐based 51 > OBzl, OAll 48, 51 and O‐phenacyl 53.
Scheme 1.
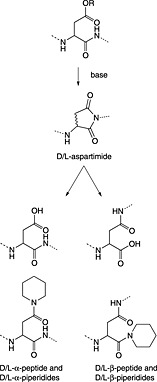
Aspartimide formation.
Table 2.
Aspartimide sensitive sequences, Asp‐Xaa
| Xaa | Degree of aspartimide formation |
|---|---|
| Gly | +++++ |
| Asn(Trt) | +++ |
| Asp(OtBu) | ++ |
| Arg(Pbf) | ++ |
| Ser/Thr | ++ |
| Cys(Acm) | ++ |
| Cys(Trt) | + |
| Thr(tBu) | + |
| Ala | + |
From highly sensitive (+++++) to weakly sensitive (+).
Figure 1.
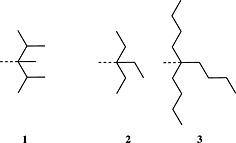
Structures of novel aspartate protections Die (1,1‐diisopropyl‐ethyl) 1, Epe (3‐ethyl‐3‐pentyl) 2 and Bno (5‐butyl‐5‐nonyl) 3.
Moreover, aspartimide formation also depends on the peptide conformation 54. The influence of conformation on aspartimide formation has been exploited by the use of pseudoproline dipeptides immediately before an aspartimide‐prone Asp(OAll) residue that enabled the synthesis of N‐glycopeptides containing the Asn‐Xaa‐Ser/Thr sequence 55, 56.
Recently, novel derivatives have been developed to directly address aspartimide formation 57. These derivatives incorporate trialkylcarbinol‐based esters Epe 2 and Bno 3 (Figure 1) which appear to provide excellent protection against this side reaction, as illustrated by the data presented in Table 3.
Table 3.
Composition of crude products obtained from peptide resins VKDXYI after treatment with 20% piperidine in DMF at room temperature
| Asp(OR) R | Aspartimide per cycle for X = Asna , b (%) | d‐Asp for X = Asn (%) | Aspartimide per cycle for X = Arga , b (%) | d‐Asp for X = Arg (%) |
|---|---|---|---|---|
| tBu | 1.65 | 9.1 | 1.24 | 25.1 |
| Mpe | 0.49 | 4.2 | 0.4 | 11.0 |
| Epe | 0.19 | 2.2 | 0.13 | 3.1 |
| Bno | 0.06 | 0.9 | 0.06 | 1.4 |
Calculation by first order decay.
Based on 10‐min treatments 57.
The addition of acidic modifiers has long been known to reduce the problem of aspartimide formation in Fmoc SPPS 42. Recently, Subirós‐Funosas et al. 58 have shown that the addition of 1 M ethyl cyano(hydroxyimino) acetate (Oxyma) Pure in 20% piperidine in dimethylformamide (DMF) reduces the levels of aspartimide‐related impurities. In the case of the Asp(OtBu)‐Gly‐containing test peptide, fragment 1‐6 scorpion toxin II, impurities are reduced from 44% to 15% for a 6 + 6‐h treatment 58.
The only strategy, however, that currently offers complete protection from aspartimide formation is backbone protection of the aspartyl α‐carboxyamide bond 47, 48, 59, 60, 61. However, this comes with its own associated problems because of difficulties with acylating the secondary amine formed by introduction of the backbone protection and is generally only routinely used for the synthesis of peptides containing Asp‐Gly, for which a number of building blocks are commercially available: Fmoc‐Asp(OtBu)‐(Dmb)Gly‐OH (Dmb 11), Fmoc‐(Dmb)Gly‐OH and Fmoc‐(FmocHmb)Gly‐OH (Hmb 12) 15, 62 (Table 5).
Table 5.
Backbone amide protection in use in Fmoc SPPS
| Introduction | Acylation | Removal | Safety catch | Reference | |
|---|---|---|---|---|---|
| Dmb 11
|
Automated SPPS | Standard coupling to DmbGly; all others are very sterically hindered | TFA | No | Weygand et al., 1966 102; Blaakmeer et al., 1991 105; Cardona et al., 2008 15 |
| FmocDmbGly building block | |||||
| Automated SPPS | |||||
| Dipeptide building block FmocXaaDmbGly | |||||
| Hmb 12
|
Automated SPPS FmocHmbXaa; building block | Standard coupling to HmbGly; for all others, FmocAA symmetric anhydride in DCM onto HmbAA unless both are beta‐branched | TFA | Yes, acetylated Hmb is TFA resistant 107 | Johnson et al., 1993 106; Ede et al., 1996 112 |
| On‐resin reduction | |||||
| Pseudoproline 13
|
Automated SPPS | Standard coupling | TFA | No | Wöhr et al., 1995 14, 113 |
| Dipeptide building block; limited to XaaSer or XaaThr | |||||
| Hmsb 14
|
Building block | Coupling with standard conditions to HmsbAla and HmsbLeu demonstrated | NH4I/TFA; TmsBr/EDT/TFA/thioanisole | Yes sulfone/sulfide safety catch | Offer et al., 1997 114; Abdel‐Aal et al., 2014 115 |
| Automated on‐resin reduction | |||||
| Hnb 15
|
On‐resin reduction | Intramolecular acyl transfer assists subsequent acylation; good tolerance for a range of residues | hν | Yes, UV irradiation orthogonal to acidolysis | Miranda et al., 2000 116 |
| Mmsb 16
|
Fmoc building block | Repeat couplings required to quantitatively acylate | NH4I/TFA | Yes, sulfone/sulfide safety catch | Paradis‐Bas et al., 2014 117 |
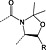
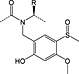
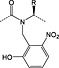
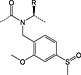
Arginine
4‐Methoxy‐2,3,6‐methylbenzenesulfonyl was initially the standard protecting group for arginine but frequently required overnight or longer deprotection with TFA/thioanisole cocktails. The 2,2,5,7,8‐pentamethylchromanyl‐6‐sulfonyl (Pmc) protecting group, introduced in 1987, showed a dramatic reduction in deprotection time 63, 64. The unpatented but conceptually similar Pbf has subsequently become the standard protection; it was introduced in 1993 and was reported to be slightly more labile than Pmc 22. Removal of Pbf is usually complete within 1–2 h; however, with peptides containing multiple arginine residues, extended cleavage times are still required. A new derivative, Fmoc‐Arg(MIS)‐OH, with improved deprotection kinetics compared with Fmoc‐Arg(Pbf)‐OH has been introduced. The MIS 4 group (1,2‐dimethylindole‐3‐sulfonyl), (Figure 2) was completely cleaved from a model peptide with 1 : 1 TFA/DCM in 30 min compared with 4% of the peptide protected with Pbf 65. The apparent drawback of this derivative is the release of dimethylindole‐3‐sulfonic acid co‐precipitates in ether with the product peptide when water is used in the scavenger cocktail.
Figure 2.
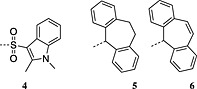
Structures of the novel arginine protecting groups MIS (1,2‐dimethylindole‐3‐sulphonyl) 4, and dibenzosuberyl 5, dibenzosuberenyl 6.
Surprisingly, dibenzosuberyl 5 and dibenzosuberenyl 6 have never found broad application 66 despite being removed under the mildest reported conditions whilst reducing δ‐lactam 67, 68 and ornithine formation 69 (Figure 2).
Cysteine
Cysteine racemisation occurs with base‐mediated activation methods, such as those using phosphonium or uronium reagents 70, 71. It is particularly high with microwave heating and preactivation 72. It can be avoided in all these cases by using carbodiimide activation 73, 74.
Several alternatives to the standard trityl protecting group have been reexamined in studies to overcome cysteine racemisation 75 using the model peptide H‐Gly‐Cys‐Phe‐NH2 (Table 4). Couplings were performed under basic conditions using HCTU/6‐Cl‐HOBt/DIPEA (4/4/8) activation with 1‐min preactivation, at both room and elevated temperatures. Reduction of cysteine racemisation correlated with a decrease of the steric bulk and an increase of the electron density at the sulfur. MBom 9, (Figure 3) was most effective, reducing the formation of d‐Cys to 0.4%. Unfortunately, it is associated with a number of side reactions due to the formaldehyde and methoxybenzyl cation released during cleavage 76, 77. Complete suppression of these side products was not possible 78.
Table 4.
Influence of cysteine side‐chain protecting groups on cysteine racemisation using basic activation conditions and 1‐min preactivation 75
| Conditions | Racemisation [%] (d‐Cys/l‐Cys peptide) | ||||||
|---|---|---|---|---|---|---|---|
| Trt | Dpm | Ddm | Bzl | Mob | Tmob | MBom | |
| Conventional SPPS | 8.0 | 1.2 | 0.8 | 5.3 | 1.7 | 0.6 | 0.4 |
| 50°C | 10.9 | 3.0 | 1.8 | n.d. | n.d. | n.d. | 0.8 |
| 80°C | 26.6 | 4.5 | 2.5 | n.d. | n.d. | n.d. | 1.3 |
Figure 3.
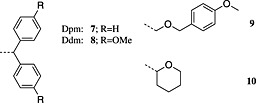
Structures of the novel cysteine sulfhydryl protecting groups Dpm (diphenylmethane) 7, Ddm (4,4′‐dimethoxydiphenylmethane) 8, MBom (4‐methoxybenzyl‐oxymethyl) 9 and THP (2‐tetrahydropyranyl) 10.
Tetrahydropyranyl (THP 10) (Figure 3) sulfhydryl protection would seem an ideal solution. THP promises similar benefits of suppressed racemisation of MBom but without its associated deprotection artefacts and an inexpensive starting material 79.
Epimerisation and β‐elimination of C‐terminal cysteine residues anchored to a resin via a benzyl ester remain problematic. The influence of S protecting groups on these side reactions has been investigated 80. Bz‐Ser(tBu)‐Cys(Trt)‐NovaSynTGT resin treated with 20% piperidine over 6 h formed 23% d‐Cys(Trt), an extraordinarily large amount given that trityl‐based linkers are believed to help prevent this problem. Under the same conditions, the corresponding peptide made with Fmoc‐Cys(MBom)‐OH gave 6% d‐Cys.
Histidine
Historically, histidine has been a notoriously racemisation‐prone residue. Histidine is conventionally introduced using Fmoc‐His(1‐Trt)‐OH without special precautions. However, this residue can undergo significant racemisation during coupling, particularly when base‐mediated couplings are used or the reaction is slow. The racemisation‐prone character of histidine is caused by the imidazole π‐nitrogen promoting the enolisation of histidine active esters 81, 82, 83, 84. Therefore, the most effective approach to preserving the chiral integrity of histidine is to employ imidazole π‐nitrogen protection. Jones and co‐workers were the first to describe such protection introducing the Nπ‐benzyloxymethyl (Bom) group, which is used in Boc chemistry 85. Later, they introduced the analogous derivative, Fmoc‐His(Bum)‐OH, for Fmoc SPPS based on the TFA‐labile t‐butoxymethyl (Bum) group 86. Others also described Nπ‐1‐adamantyloxymethyl protection 87.
This topic has been revisited with the introduction of Fmoc‐His(MBom)‐OH 78, 88. Loss of histidine chiral integrity was compared for Fmoc‐His(Trt)‐OH and Fmoc‐His(MBom)‐OH using HCTU/6‐Cl‐HOBt/DIPEA (4/4/8) activation. In the case of Fmoc‐His(Trt)‐OH, the level of racemisation increased with the preactivation time from 1% without preactivation to 7.8% with 5 min of preactivation. In contrast Fmoc‐His(MBom)‐OH reduced epimerisation to 0.3% with 5‐min preactivation. Microwave heating at 80°C and Nτ‐Trt protection gave racemisation of 16.6% whereas the Nπ‐MBom group gave 0.8%. Nevertheless, the preparation of Fmoc‐His(MBom)‐OH remains expensive, and as previously mentioned, the MBom group is associated with a number of undesirable side reactions.
Generally, acidic coupling conditions, such as diisopropyl carbodiimide (DIC)/HOBt at ambient temperature, are sufficient to maintain histidine stereochemistry even with Fmoc‐His(Trt)‐OH 74. However, histidine racemisation will always be a risk with the current range of Nτ‐protected derivatives. The search of a cost‐efficient solution for this problem remains unresolved.
Enhancing Fmoc SPPS efficiency
The greatest problem of peptide chemistry is peptide insolubility, of either unprotected peptides in aqueous buffer 89, 90, 91, 92 or fully protected peptides in organic solvents 89, 93, 94. This obstacle has prevented the development of many areas of peptide chemistry: for example, a general method for the assembly of protected peptide fragments.
Poor solubility was recognised from the very beginning of SPPS to be the direct cause of synthetic problems 95. Furthermore, it can complicate or prevent chemical characterisation. Although poor solubility can often be anticipated, as it is predominantly associated with hydrophobic sequences, prediction remains difficult 96, 97. Chemists have dealt with poor peptide solubility by further functionalising the peptide with hydrophilic groups such as polyarginine or polylysine to enhance solubility. Notable results have been achieved with this approach including the synthesis of membrane proteins 98, 99 and the notoriously insoluble insulin A‐chain 100.
The presence of N‐alkylated amino acids such as sarcosine and proline at regular intervals along a peptide sequence has long been known to guarantee high peptide solubility 90, 96. This improvement in peptide solubility is directly related to maintaining an open‐chain, disordered conformation by removing hydrogen bonds. The effect occurs on the solid phase where proline residues, evenly spaced throughout a sequence, assist peptide synthesis, presumably again by maintaining a fully solvated, disordered conformation 101. Modifying the peptide backbone promises a more systematic approach to peptide solubility. However, development of this was slow because many peptide chemists reasoned that adding hydrophobic groups to the backbone would decrease solubility of the peptide, whereas the opposite effect, a dramatic increase is observed.
The possibility of using a reversible group (Table 5) for temporarily increasing peptide solubility was first suggested by Weygand and co‐workers using 2,4‐dimethoxybenzyl (Dmb 11) 102. The optimal distancing of the backbone substitution (every six residues) was identified by Narita and co‐workers 89, 103. Sheppard proposed using backbone protection as a strategy for overcoming difficult sequences 104 as the difficult sequence phenomenon could be considered as the reappearance of poor peptide solubility on resin 16. Therefore, installing backbone protection before the onset of aggregation and thereafter at every six residues would prevent the onset of difficult sequences. Adding a substituent to the amide bond would prevent its participation in intermolecular hydrogen bonding and prevent interchain aggregation 96. The use of Dmb introduced a new problem: the problematic acylation of hindered secondary amines 105. Nevertheless, DmbGly dipeptide building blocks are commercially available and have found widespread use against difficult sequences and aspartimide formation 15. The Sheppard group overcame the obstacle of steric hindrance with the introduction of Hmb 12. Acylation could occur at the accessible 2‐hydroxy position, and subsequent intramolecular transfer assists subsequent acylation of the secondary amine 106. Hmb protection entered widespread use; however, it had a major drawback: the ease of acylation of Hmb‐protected amines differs between residues, and the reaction requires DCM as solvent with lengthy (overnight) coupling times. However, an unintended consequence of using Hmb, but one that gave it a great advantage over alternative methods of backbone protection, was its stability to TFA treatment when the 2‐hydroxyl group was acetylated 107. This was found useful for the purification on HPLC of β‐amyloid, as the deprotected peptide retaining backbone protection was much easier to handle 108, 109. A wide variety of other backbone protections have been proposed including dicyclopropylmethyl 110 and other structures 111.
The problem of Hmb is the lengthy time required to effect complete O,N‐acyl transfer. This can be accelerated by introducing electron‐withdrawing groups to the benzene ring. In one approach (Scheme 2, Hmsb 14), a sulfoxide group was positioned para to the 2‐hydroxyl position, which generates an active ester when acylated, that is favourably positioned for intramolecular acyl transfer 114, 115, 118. Mild reduction to the sulfide makes the group labile to acidolysis, an approach also employed in the Mmsb group 16 117. Alewood and co‐workers achieved similar results with a photolabile group, Hnb 15. Acylation generates an activated nitrophenol ester in situ and accelerates acyl transfer. Hmsb, Mmsb and Hnb backbone protection could be retained on the side‐chain‐deprotected peptides for their solubilising properties 116.
Scheme 2.
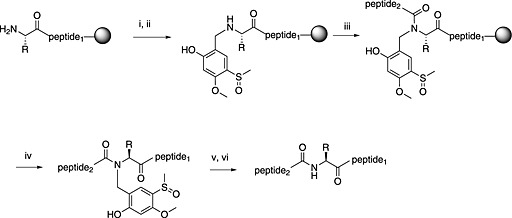
Principle of the safety‐catch backbone protecting groups using Hmsb: (i) 1.1 eq. of the corresponding salicylaldehyde in DMF; (ii) NaBH4, DMF; (iii) continuing SPPS; (iv) standard TFA cleavage; (v) NH4I/DMS reduction; (vi) standard TFA cleavage.
An important advance was the demonstration of on‐resin reduction of Hmb onto the peptide resin 112. The reductive amination was quantitative with a single equivalent of the salicylaldehyde precursor. This was also successfully applied to Hnb and Hmsb, simplifying the introduction of the backbone protection and enabling automation.
One limitation of the benzyl‐type protection is the formation of long‐lived benzyl cations during deprotection that can react with the peptide, a particular concern with tryptophan. Additionally, on some long peptides and near‐basic residues, prolonged acidolysis is required for removal.
The most popular approach to date is unrelated to classical benzyl‐type amide substitution but instead exploits dimethyloxazolidine derivatives of serine or threonine, referred to as pseudoproline dipeptides 13. The related cysteine equivalent, the thiazolidine had been used by the Kemp group as a protecting group for ligation 119 and had been identified as being particularly resilient to epimerisation 120. Mutter used a different rationale from that used for Hmb and hypothesised that by ‘kinking’ the peptide, they broke structure and made the peptides more soluble. The key to their popularity is that no special conditions are required for installation of the pseudoproline dipeptide, two residues are incorporated in a single step and TFA deprotection is uncomplicated 14, 113. Unsurprisingly, many of the applications of pseudoprolines mirror those of Dmb‐derived backbone protection, such as improved cyclisation, aspartimide suppression and epimerisation‐free segment coupling. However, they are limited to sequences containing serine and threonine at convenient positions. Impressively long peptides have been synthesised using pseudoprolines, notably fas 13, ubiquitin 62 and D2 domain of vascular endothelial growth factor receptor 1 121.
Another approach for overcoming chain association is the O‐acyl isopeptide method (Scheme 3) 122, 123, 124. The desired peptide is first synthesised as a depsipeptide derived from a serine or threonine residue. Such depsipeptide analogues of aggregation‐prone peptides are more soluble and consequently more easily purified. Once purified, the depsipeptide is converted to the native form by adjusting the pH to 7.4, when spontaneous O‐acyl to N‐acyl migration occurs (Scheme 3) 125, 126.
Scheme 3.
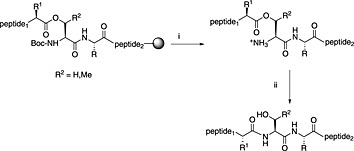
O,N‐acyl shift following the use of isoacyl dipeptide secondary amino acid surrogates. (i) Standard global peptide TFA cleavage; (ii) pH 7.4.
It has been predicted that the future of synthetic protein chemistry would come from a combination of native chemical ligation and backbone protection 127. Recent work supports this. The first application of backbone protection in ligation was reported for the synthesis of bovine pancreatic trypsin inhibitor with the unprotected peptide thioester fragment possessing an Hmb 128. Liu and co‐workers reported the synthesis of the M2 ion channel using a version of Hmb showing greatly improved solubility in aqueous buffer 129. Noteworthy in this context is the Ag+‐promoted thioester ligation of Tim‐3 using the related O‐acyl isopeptide approach to improve segment solubility 130. In contrast to pseudoprolines, the acyl isopeptide analogues can be retained after side‐chain deprotection. Any general method of backbone protection must be easy to install, easy to acylate and simple to remove; it is also desirable that it can be retained after the rest of the peptide has been deprotected. The most important consideration for it to be practically useful and widely used is, however, cost.
Post‐translational modifications
Understanding the role of protein post‐translational modifications (PTMs) in cell signalling, gene expression, and protein processing and translocation is of enormous interest 131, 132, 133, 134. Ready access to peptides containing post‐translational modified amino acid residues, for use as probes, for use as inhibitors or for raising antibodies against protein PTMs, has been crucial for advances in these areas.
Fmoc SPPS is generally the method of choice for the synthesis of such modified peptides because many of the most important PTMs, such as glycosylation and phosphorylation, are not stable to HF cleavage conditions. The milder chemistry of the Fmoc method allows almost all PTMs to be introduced during chain elongation using the appropriate preformed protected amino acid building blocks. The most notable exceptions are ubiquitinylation and farnesylation, which will be discussed in the following.
Table 6 contains the most frequently utilised building blocks for the synthesis of peptides bearing the most commonly encountered PTMs.
Table 6.
Commercially available building blocking blocks for introduction of principle PTMs
| PTM | Introduction | Comments | Reference |
|---|---|---|---|
| Phosphorylation Ser/Thr | |||
|
|
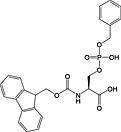
Fmoc‐Ser(PO(OBzl)OH)‐OH 17 | Best coupled using imminium‐based reagents 135 | Wakamiya et al., 1994 136 |
|
|
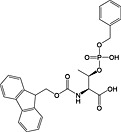
Fmoc‐Thr(PO(OBzl)OH)‐OH 18 | Best coupled using imminium‐based reagents 135 | White and Beythien, 1996 137 |
|
|
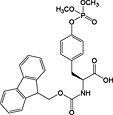
Fmoc‐Tyr(PO(OMe)2)‐OH 19 | Compatible with all coupling methods; monodemethylated by piperidine; requires TMSBr/TFA for side‐chain deprotection | Kitas et al., 1989 138 |
|
|
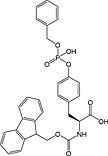
Fmoc‐Tyr(PO(OBzl)OH)‐OH 20 | Best coupled using imminium‐based reagents 154 | White and Beythien, 1996 137 |
|
|
Fmoc‐Tyr(PO(OBzl)2)‐OH 21 | Compatible with all coupling methods; monodebenzylated by piperidine | Perich and Reynolds, 1991 139 |
|
|
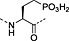
Fmoc‐Tyr(PO3H2)‐OH 22 | Best coupled using imminium‐based reagents; issues with pyrophosphate formation 143 | Ottinger et al., 1993 140 |
|
|
Fmoc‐Tyr(PO(NMe2)2)‐OH 23 | Compatible with all coupling methods; deprotected with TFA/water (9 : 1) | Chao et al., 1995 141 |
|
|
Fmoc‐Tyr(PO(OMDPSE)2)‐OH 24 | Compatible with all coupling methods; MDPSE groups removed with TFA | Chao et al., 1994 142 |
|
|
Fmoc‐Ppa(Bzl)‐OH 25 | Best coupled using imminium‐based reagents | Chauhan et al., 2007 143 |
|
|
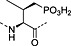
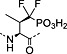
Fmoc‐Pmp‐OH 26 | Best introduced with HATU/DIPEA coupling | Marseigne et al., 1988 144 |
|
|
Fmoc‐F2Pmp‐OH 27 | Best introduced with HATU/DIPEA coupling | Gordeev et al., 1994 145 |
| Sulfation Tyr | |||
|
|
Fmoc‐Tyr(SO3nP)‐OH 28 | Neopentyl ester is stable to TFA; cleaved with sodium azide/DMSO or aq. ammonium acetate | Simpson and Widlanski, 2006 [146, 147] |
|
|
Fmoc‐Tyr(SO3DCV)‐OH 29 | DCV ester stable to TFA; DCV cleaved by Zn/AcOH reduction | Ali and Taylor, 2009 148, 149 |
| Methylation Arg | |||
|
|
Fmoc‐Arg(Me,Pbf)‐OH 30 | For introduction of monomethyl arginine | White, 2006 150 |
|
|
Fmoc‐ADMA(Pbf)‐OH 31 | For introduction of asymmetric dimethylarginine | White et al., 2006 150 |
|
|
Fmoc‐SDMA(Boc2)‐ONa 32 | For introduction of symmetric dimethylarginine | White et al., 2006 150 |
| Methylation Lys | |||
|
|
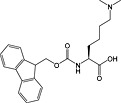
Fmoc‐Lys(Me,Boc)‐OH 33 | For introduction of monomethyl lysine | |
|
|
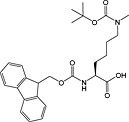
Fmoc‐Lys(Me2)‐OH 34 | For introduction of dimethyl lysine, basic side chain can promote Fmoc loss and double insertions during synthesis 151 | |
|
|
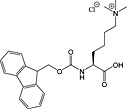
Fmoc‐Lys(Me3Cl)‐OH 35 | For introduction of trimethyl lysine | |
| Citrullation | |||
|
|
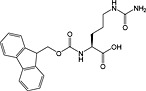
Fmoc‐citrulline‐OH 36 | For introduction of citrullation | |
| Glycosylation Asn | |||
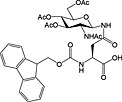
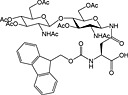
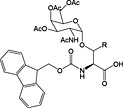
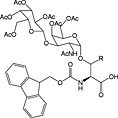
|
|
Fmoc‐Asn(β‐d‐GlcNAc(Ac)3)‐OH 37 | Building block for introduction of monosaccharide fragment of N‐linked glycoproteins | Meldal and Bock, 1990 152 |
|
|
Fmoc‐Asn(β‐d‐GlcNAc(Ac)3‐(1‐4)‐β‐d‐GlcNAc(Ac)2)‐OH 38 | Building block for introduction of chitobiose fragment of N‐linked glycoproteins | Meinjohanns et al., 1998 153 |
| Glycosylation Ser (R = H)/Thr (R = Me) | |||
|
|
Fmoc‐Ser/Thr(α‐d‐GlnNAc(Ac)3)‐OH 39 | Building block for introduction of Tn antigen oligosaccharide fragment | Paulsen and Adermann, 1989 154 |
|
|
Fmoc‐Ser/Thr(β‐d‐Gal(Ac)4‐(1‐3) α‐d‐GlnNAc(Ac)2)‐OH 40 TF antigen | Building block for introduction of TF antigen oligosaccharide fragment | Irazoqui et al., 1999 155 |
|
|
Fmoc‐Ser/Thr(sialylOMe(Ac)4‐(1‐6)‐α‐d‐GlnNAc(Ac)2)‐OH 41 STn antigen | Building block for introduction of STn antigen oligosaccharide fragment | Liebe and Kunz, 1997 156 |
|
|
Fmoc‐Ser/Thr(sialylOMe(Ac)4‐(1‐3)‐β‐d‐Gal(Ac)3‐(1‐3) α‐d‐GlnNAc(Ac)2)‐OH 42 STF antigen | Building block for introduction of STn antigen oligosaccharide fragment | Komba et al., 1999 157 |
|
|
Fmoc‐Ser/Thr(β‐d‐GlcNAc(Ac)3)‐OH 43 | Building block for introduction of β‐GlcNAc modification; building blocks tend to racemise 158 | Arsequell et al., 1994 159 |
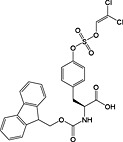
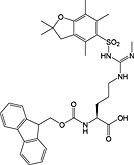
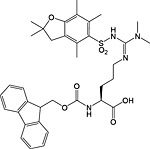
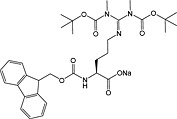
Phosphorylation
Reversible phosphorylation of proteins is involved in signal transduction and on/off control of enzymes 160. In eukaryotes, phosphorylation occurs on serine, threonine, tyrosine and histidine 161 residues, whereas in prokaryotes, it is also observed on lysine and arginine 162.
The synthesis of peptides containing phosphorylated serine, threonine and tyrosine is well established and has been reviewed 163. There are two approaches: postsynthetic global phosphorylation and introduction of a preformed phosphoamino acid building block. Global phosphorylation (Scheme 4) involves selective phosphitylation of the appropriate hydroxyamino acid on the solid phase, with a protected phosphoramidite, followed by oxidation of the resultant P(III) triester to the P(V) triester. The favoured approach utilises dibenzyl‐N,N‐diisopropylphosphoramidite with oxidation using anhydrous t‐butyl hydroperoxide 164.
Scheme 4.
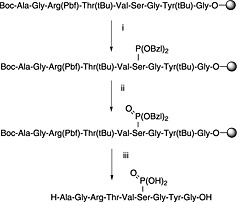
Global phosphorylation strategy (phosphitylation–oxidation method): (i) (BzlO)2‐PN(i‐Pr)2/tetrazole; (ii) tBuOOH; (iii) standard global TFA cleavage.
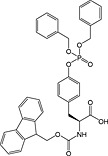
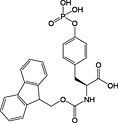
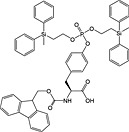
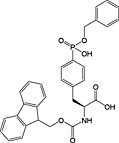
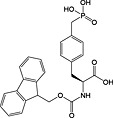
This method has largely been superseded by the building block approach, owing to its greater convenience 165. For introduction of phosphotyrosine, free phosphate, phosphodiester and triester, phosphoamidate‐based building blocks are available (Table 6). Fmoc‐Tyr(PO3H2)‐OH 22 140 is seldom used owing to issues with pyrophosphate formation between adjacent Tyr(PO3H2) residues 166, 167. Fmoc‐Tyr(PO(OBzl)OH)‐OH 20 137 is the most frequently employed reagent; however, the acidic hydroxyl group causes problems during coupling reactions 135, as piperidine counterion to the phosphate consumes activated amino acid derivative, necessitating an additional equivalent of amino acid to be employed for every monobenzyl phosphate introduced.
In contrast, Fmoc‐Tyr(PO3Bzl2)‐OH 21 138, 168, 169 couples smoothly but is converted into Fmoc‐Tyr(PO(OBzl)OH)‐OH by piperidine and so suffers from the same issues as the monoprotected derivative during subsequent chain extension 170.
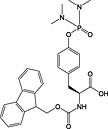
Fmoc‐Tyr(PO(NMe2)2)‐OH 23 141, 171 presents no problems during peptide assembly; however, regeneration of phosphotyrosine from the phosphodiamidate requires overnight treatment with TFA containing 10% water 141.
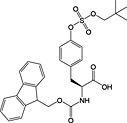
With phosphoserine and phosphothreonine, the situation is further complicated by the propensity of their phosphotriesters to undergo β‐elimination under basic conditions 172. For these amino acids, it is necessary to use a partially protected phosphodiester for their introduction. The favoured derivatives are monobenzyl esters, Fmoc‐Ser(PO(OBzl)OH)‐OH 17 136, 173 and Fmoc‐Thr(PO(OBzl)OH)‐OH 18 137, 174. The acidic phosphate is thought to become deprotonated during Fmoc deprotection, thereby inhibiting deprotonation of the amino acid α‐proton and subsequent β‐elimination. In practice, these derivatives do not offer complete protection from elimination, particularly in the case of phosphoserine and during microwave‐accelerated synthesis 173, 175.
Problems can occur with these benzyl‐protected derivatives during the TFA cleavage reaction as a result of alkylation of sensitive residues by the released benzyl carbocation.
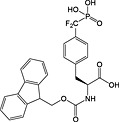
Nonhydrolysable analogues of all three O‐phosphoamino acids have been developed (Table 7), to aid in the preparation of phosphopeptide tools for use as phosphatase inhibitors and as antigens for raising antibodies. However, at present, only analogues of phosphotyrosine are commercially available: Fmoc‐Pmp‐OH 26 144, Fmoc‐F2Pmp‐OH 27 145, 176 and Fmoc‐Ppa(benzyl)‐OH 25 143.
Table 7.
Nonhydrolysable analogues of phosphoamino acid residues.
| Structure | Name | Reference |
|---|---|---|
|
|
Phosphonomethylalanine (Pma) 44 | Engel, 1977 177 |
|
|
Difluorophosphonomethylalanine (F2Pma) 45 | Berkowitz et al., 1994 178 |
|
|
β‐(Phosphonomethyl)aminobutyric acid (PmAbu) 46 | Ruiz et al., 1994 179 |
|
|
(Difluorophosphonomethyl)aminobutyric acid (F2PmAbu) 47 | Berkowitz et al., 1996 180 |
|
|
Phosphonomethyl phenylalanine (Pmp) 48 | Marseigne et al., 1988 144 |
|
|
Hydroxyphosphonomethyl phenylalanine (HPmp) 49 | Burke et al., 1993 181 |
|
|
Fluorophosphonomethyl phenylalanine (FPmp) 50 | Burke et al., 1993 181 |
|
|
Difluorophosphonomethyl phenylalanine (F2Pmp) 51 | Burke et al., 1993 182 |
|
|
Phosphonophenylalanine Ppa 52 | Lui et al., 2002 183 |
|
|
2‐Phospho‐4‐furylalanine 53 | Schenkels et al., 1999 184 |
|
|
1‐(2‐Phosphonoethyl)‐1H‐1,2,3‐triazol‐4‐ylalanine 54 | Yang et al., 2011 185 |
|
|
4‐Phosphonotriazolylalanine 55 | Kee et al., 2010 186 |
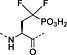
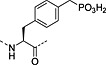
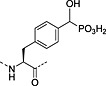
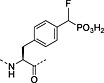
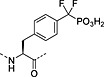
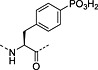
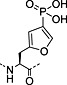
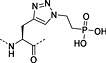
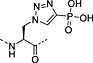
The former isostere appears to be a poor substitute for phosphotyrosine, as its use often leads to a significant reduction in biological activity, which is interpreted as being caused by the lack of the H‐bond acceptor phenyl oxygen and incomplete ionisation of the phosphonic acid at neutral pH (Pmp 48, pKa2 7.72, vs pTyr, pKa2 6.22) 187 F2Pmp 51 in contrast has a pKa2 of 5.71 and is therefore fully ionised at neutral pH, and the methylene fluorine atoms can undergo H‐bonding. Peptides substituted with F2Pmp exhibit higher binding affinities to SH2 domains than Pmp analogues 176. Enhancements of 1000‐fold in affinities of F2Pmp‐containing peptide compared with those containing Pmp are reported 188, 189.
Unfortunately, for peptides containing phosphorylated basic amino acids, the building block approach is not appropriate as N‐phosphates are not stable to TFA 161, 190. For phosphohistidine, nonhydrolysable analogues have been developed to overcome this problem, a phosphofurylalanine 53 analogue 184 and phospho triazolyl alanine analogues 54 and 55 185, 186. The Fmoc‐protected derivative of 55 191, 192, 193 shows particular promise because peptides containing this moiety have been able to elicit antiphosphohistidine antibodies 194, 195. Recently, the synthesis of pLys‐containing peptides has been reported using a strategy involving postassembly N‐phosphorylation followed by saponification from a base‐labile resin (Scheme 5) 196.
Scheme 5.
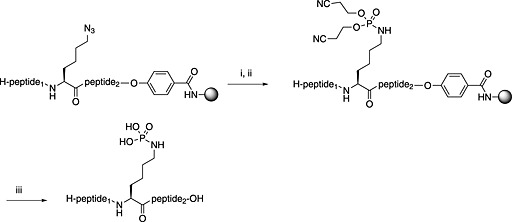
On‐resin synthesis of phospholysine peptides by the use of a base‐labile hydroxymethylbenzoic acid resin and tris(cyanoethyl)phosphite: (i) TFA/TIS (95:5), 2 h; (ii) 4 × 5 eq. P(OCH2CH2CN)3 in DMF, 45°C, 48 h; (iii) 0.25 M NaOH in dioxane, 0°C, 20 min and immediate neutralisation; advantage one‐step chromatographic purification under basic condition 196.
Recently, a method for pyrophosphorylation of pSer has been reported 197, which enables the investigation of pyrophosphorylation 198.
Sulfation
It is believed that up to 1% of all protein tyrosine residues in eukaryotes are sulfated; however, the biological role of tyrosine sulfation is poorly understood 199, 200. Sulfation is thought to be involved in the modulation of the extracellular protein–protein interactions of secreted and transmembrane proteins 201, 202. It is also an essential requirement for maintaining the biological activity of a number of peptide hormones such as gastrin II, cholecystokinin and caerulein 203, 204.
One of the principal hurdles to studying tyrosine sulfation is the difficulty in obtaining site‐specifically sulfated peptides for use as biological probes or antigens for raising antibodies. This is because tyrosine sulfate esters are rapidly degraded in acid and fragment during mass spectrometry, making their synthesis and characterisation highly problematic. The basic principles for the synthesis of sulfated peptides have been elaborated by methods in solution and then transferred to SPPS. For an exhaustive review, see 205.
Recent advances were achieved by protecting the sulfate, which stabilises it during the TFA cleavage, enabling standard reaction conditions to be used without significant loss of the sulfate. The use of four protecting groups has been examined in detail: azidomethyl 206, trichloroethyl 149, 207, dichlorovinyl (DCV) 148 and neopentyl (nP) 146. Of these, nP protection appears to offer particular promise as the group is stable to piperidine and TFA but can be removed postcleavage with either sodium azide or ammonium acetate 147. Fmoc‐sulfotyrosine building blocks are available, bearing DCV and nP protecting groups: Fmoc‐Tyr(SO3DCV)‐OH 29 and Fmoc‐Tyr(SO3nP)‐OH 28.
Farnesylation
S‐Farnesylation of protein C‐terminal cysteinyl residues is thought to be involved in modulating protein–membrane and protein–protein interactions 208, 209. Stepwise synthesis of farnesylated peptide probes is challenging as the unsaturated farnesyl group is subject to addition reactions during TFA cleavage. Therefore, the S‐farnesyl group is generally introduced by treating cysteinyl peptides that are either protected with base‐labile groups or fully deprotected, with farnesyl bromide in solution 210 or on resin 211. For an exhaustive review, see 212.
In the context of Fmoc SPPS, solution and solid‐phase approaches to farnesylation are nicely exemplified by the following reported syntheses of yeast mating pheromone a‐factor. The synthesis of this peptide is complicated by the fact that it contains not only a farnesyl group but also a C‐terminal cysteine methyl ester.
Scheme 6 shows a yeast mating pheromone a‐factor involving farnesylation in solution 213. Note the use of TMS‐diazomethane for the conversion of the easily epimerised C‐terminal cysteinyl residue to its methyl ester.
Scheme 6.
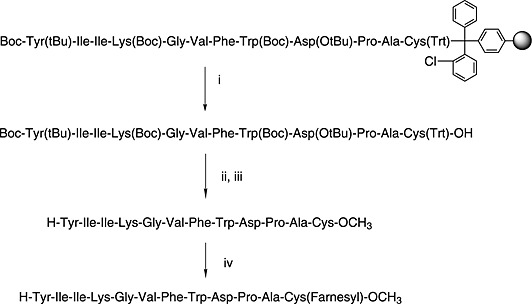
Synthesis of S‐farnesylated peptide methyl esters by Fmoc SPPS. (i) 1% TFA in DCM; (ii) TMS‐diazomethane in DCM, methanol 9 : 1; (iii) TFA/TIS/EDT/H2O (92.5 : 2.5 : 2.5 : 2.5); (iv) Far‐Br in DMSO/DMF/acetonitrile (3 : 3 : 1) 213.
In the solid‐phase approach shown in Scheme 7 214, the peptide is prepared on the acid‐stable hydrazinobenzoyl resin 215, the side‐chain protection is removed with TFA and the Cys residue is farnesylated. Oxidation followed by methanolysis releases the desired peptide methyl ester. Approaches to the peptidyl cysteine methyl ester involving side‐chain anchoring of the C‐terminal cysteine methyl ester 216 are not recommended as cysteine esters are not optically stable in the presence of piperidine 80.
Scheme 7.
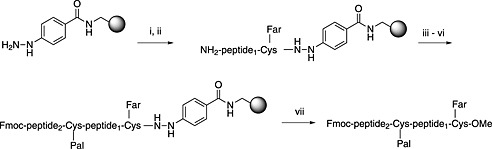
Synthesis of S‐palmitoylated and S‐farnesylated peptide methyl esters by Fmoc SPPS. (i) Fmoc‐Cys(Far)‐OH (4 eq.), DIC/HOBt; (ii) SPPS; (iii) ) Fmoc‐Cys(Pal)‐OH (4 eq.), DIC/HOBt; (iv) 1% 1,8‐diazabicyclo [5.4.0]undec‐7‐ene in DMF 2 × 30 s; (v) Fmoc‐AA‐OH (5 eq.), HATU (5 eq.), DIPEA (20 eq.), DCM/DMF (4 : 1); (vi) SPPS; (vii) Cu(OAc)2, pyridine, acetic acid, methanol, DCM, oxygen atmosphere, 3 h.
Methylation
Protein methylation occurs predominantly on lysine and arginine residues of cytosolic and nuclear proteins and is involved in protein targeting and signalling and in the epigenetic control of gene expression 134, 217, 218, 219, 220. Methylation of arginine is mediated by peptidylarginine methyltransferases (PRMTs), giving monomethyl, symmetric dimethyl and asymmetric dimethyl modifications. Lysine methyltransferases effect methylation of lysine, producing monomethyl, dimethyl and trimethyl modifications 221.
Building blocks are available for the introduction during Fmoc SPPS of all biologically relevant methylated lysine [Fmoc‐Lys(Me,Boc)‐OH 33, Fmoc‐Lys(Me2)‐OH 34 and Fmoc‐Lys(Me3Cl)‐OH 35] and arginine residues [Fmoc‐Arg(Me,Pbf)‐OH 30, Fmoc‐ADMA(Pbf)‐OH 31, Fmoc‐SDMA(Boc2)‐ONa 32] (Table 6). In the case of the methylated lysine, the only point of note is that there is anecdotal evidence to suggest the basic side chain of dimethyllysine can promote Fmoc cleavage during subsequent coupling reactions, leading to double additions 151.
For dimethylarginine, side‐chain‐protected 150 (31 and 32) and unprotected derivatives are available. However, the use of the latter are not recommended because of low reactivity resulting from γ‐lactam formation and the potential for ornithine formation.
Citrullination
Conversion of arginine residues in proteins to citrulline is performed by enzymes known as peptidylarginine deiminases. Antibodies against citrullinated fibrin and fibrinogen are associated with rheumatoid arthritis and other autoimmune diseases, and citrullination is involved in epigenetic gene control by modification of histones 161, 222. For chemical synthesis, the introduction of citrulline into synthetic peptides is usually performed using a side‐chain‐unprotected building block. Coupling is slow, presumably as a result of competing γ‐lactam formation. The Pbf side‐chain‐protected derivative Fmoc‐citrulline(Pbf)‐OH has been described and should provide a more robust approach 151.
Glycosylation
Most secreted proteins are glycosylated, conferring heterogeneity to the glycoprotein by the type of sugar occupying the glycosylation site (glycoform) and the site occupancy. Only extremely rarely are proteins naturally expressed as a single glycoform. The dissection of the roles of the oligosaccharide is an area of great interest and has demanded a source of defined, chemically homogeneous glycoproteins.
A great deal of this demand has been met by breakthroughs in the expression of single‐glycoform proteins, for example, the use of the glycosidase inhibitors such as kifunensine in mammalian expression systems or the use of engineered yeast or cell lines 223. Nevertheless, important work in dissecting the role of glycosylation continues to be performed with synthetic glycopeptides, for example, its role in stabilisation of protein folds 224.
The synthesis of glycoproteins is one of the grand challenges of organic chemistry and continues to stretch the frontiers of organic synthesis. Notable achievements have been the synthesis of ribonuclease C 225 and synthesis of part of gp120 226. The synthesis of glycoproteins has been reviewed 227, 228, 229, 230, 231.
Glycoproteins are made by native chemical ligation. These glycoproteins bear either the desired side‐chain oligosaccharide or a truncated oligosaccharide that can be enzymatically extended or transglycosylated postsynthesis.
The methods used to produce the smaller glycopeptide components of the glycoproteins depend on whether the sugar is linked to the peptide chain by oxygen (O‐linked via serine and threonine) or nitrogen (N‐linked via asparagine). The synthesis of simple glycopeptides, with detailed practical protocols, has been described 7.
The most important class of O‐linked glycosides has a 2‐acetamido‐2‐deoxy‐α‐d‐galactopyranosyl (GalNAc) unit attached to serine or threonine. Such O‐glycosides are found in a wide range of proteins, such as mucin secreted from epithelial cells, the tumor‐associated sialyl‐T, sialyl‐Tn‐antigen and gp120 from HIV 232. These O‐linked glycopeptides are generally prepared using preformed Fmoc‐protected glycoamino acid building blocks, and a number are commercially available including those for the introduction of sialyl‐T and sialyl‐Tn antigens (Table 5, 39, 40, 41 and 42). These are stable to TFA and piperidine and are hence compatible with Fmoc SPPS methods 233. Sequential glycosylation has been employed to elaborate synthetic glycopeptides bearing this core O‐GalNAc unit, using the appropriate glycosyl transferases and nucleotide 234, 235.
Modification of serine or threonine residues with 2‐acetamido‐2‐deoxy‐β‐d‐glucopyranosyl (GlcNAc) is functionally more akin to phosphorylation than glycosylation 236. The addition and removal of O‐AcNH‐β‐Glc is a dynamic process controlled by a transferase, UDPGlcNAc polypeptide transferase, and removed by a β‐N‐acetylglucosaminidase. Only a single sugar is added, and the carbohydrate is not further extended. O‐AcNH‐β‐Glc glycosylation processes are thought to be involved in transcription, signal transduction, apoptosis and glucose homeostasis. Fmoc‐Ser(Ac3AcNH‐β‐Glc)‐OH and Fmoc‐Thr(Ac3AcNH‐β‐Glc)‐OH building blocks 43 are commercially available.
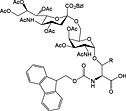
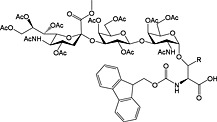
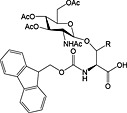
Most N‐linked glycans possess a trimannose‐di‐N‐acetylchitobiose pentasaccharide core linked via a b(1 → Nb) linkage to Asn. Like O‐linked glycopeptides, N‐linked glycopeptides can be prepared by the introduction of glycosylated building blocks during SPPS (37 and 38) 152, 237, 238. They can also be introduced convergently by acylation of glycosylamines with a peptide aspartyl side chain either in solution by Lansbury aspartylation 239 or convergent assembly on solid phase 48, 55, 61, 240.
Peptide thioesters by Fmoc SPPS
In the Boc method, peptide thioesters can be prepared directly 241, 242, whereas for the Fmoc method, the use of piperidine at each cycle is not compatible with a thioester at the C‐terminus 243, 244. Several indirect methods have therefore been proposed. But none so far match the Boc method for simplicity and yield. The use of HF in the last step of Boc SPPS precludes the preparation of peptide thioesters bearing acid‐sensitive PTMs and has largely driven the development of an Fmoc method. The approaches used to prepare peptide thioesters with Fmoc protocols can be classified into two types: those that use a safety‐catch approach or those that exploit an intramolecular O,S or N,S acyl transfer step to transform a bond stable to Fmoc synthesis into a thioester. Currently, the safety catch approach is more prevalent.
The first preparation of Fmoc peptide thioesters used a safety‐catch sulfamylbutyryl resin. Following chain assembly, the sulfonamide linker is activated by alkylation usually by treatment with iodoacetonitrile or TMS‐diazomethane. The fully protected peptide is cleaved from the resin by sodium thiophenolate in DMF, and the resulting protected peptide thioester is treated with acid to remove side‐chain protection (Scheme 8) 244. Numerous variations on this approach have since been developed 245.
Scheme 8.
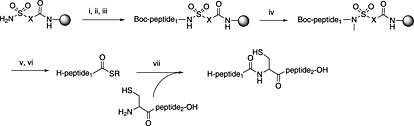
Principle of the sulfamyl safety‐catch linker for peptide thioester Fmoc SPPS, X = butyl or phenyl. (i) Fmoc‐AA (4 eq.), PyBOP, DIPEA (8 eq.); (ii) SPPS; (iii) Boc2O; (iv) TMS‐diazomethane; (v) R‐SH, NaSPh; (vi) standard TFA cleavage; (vii) 6 M guanidine, phosphate buffer pH 7.8, 1% thiophenol.
The sulfamylbutyryl approach has been used to synthesise impressive targets, including long peptide thioesters 246, glycoproteins 225, 247 and phosphoproteins 248. However, the thiolysis step can sometimes be problematic because of poor solvation of the resin‐bound peptide and difficulties with peptide recovery from DMF. It has been demonstrated that the thiolysis step is unnecessary and the methyl sulfamylbutyryl linker itself in the presence of mercaptophenylacetic acid additive can perform ligation directly in the ligation buffer 249.
Another widely used safety‐catch linker was adapted 250 from the N‐acyl urea safety catch 251, 252. The ortho‐di‐aniline system 3,4‐diaminobenzoic acid is activated after chain assembly with p‐nitrophenyl chloroformate (Scheme 9). This linker approach has been successfully used to give PTM‐modified protein precursors, including glycoproteins 230. Recently, a variation of this approach has been described based on 3‐amino‐4‐methylaminobenzoic acid 253. This modification improves regioselectively during acylation of the linker.
Scheme 9.
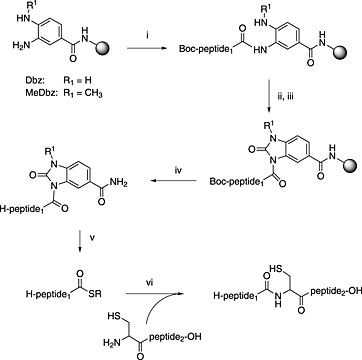
Use of diaminobenzoic acid linker (Dbz) as safety‐catch linker for peptide thioester Fmoc SPPS. (i) SPPS; (ii) 4‐nitrochloroformate; (iii) DIPEA; (iv) standard TFA cleavage; (v) R‐SH, NaSPh; (vi) 6 M guanidine, phosphate buffer pH 7.8, 1% thiophenol.
Latterly, there has been much success revisiting the classical hydrazide/azide 254 as a route to peptide thioesters for their application in ligation 255, 256, 257. Side reactions have been observed including intramolecular acylation of an adjacent lysine side chain 258. In a notable study, biologically expressed ubiquitin bearing a C‐terminal cysteine has been successfully converted to a hydrazide and subsequently converted to an azide and ligated 259. The reaction is dependent on the ability of a C‐terminal cysteine to transiently rearrange to a thioester 260. This rearrangement has become the basis for the other major classes of approaches to the synthesis of peptide thioesters: those that use an intramolecular acyl transfer to give the peptide thioester.
The N,S‐acyl transfer approach (Scheme 10) developed from the related area of ligation auxiliaries 261. Whilst attempting to remove a ligation auxiliary, 2‐mercapto‐4,5‐dimethoxybenzyl, from a peptide with TFA, the Aimoto group observed the appearance of a compound with a different retention time by HPLC but with the same mass, assigned as the thioester 262. Interestingly, this migration had been previously observed 263. Furthermore, the Aimoto group demonstrated that this could be exchanged with another thiol and used for the synthesis of several proteins in good yields 264. N‐alkylated cysteine also underwent this reaction 265, and this observation was extended to other tertiary amides bearing a C‐terminal δ‐ or ε‐thio akyl or aryl group 266. Many of these linkers are converted to a thioester in a two‐step reaction, rearrangement occurs irreversibly under strong acidic conditions followed by exchange to give a peptide thioester suitable for use in ligation. Later, it was observed that most of these linkers undergo acyl transfer under neutral conditions and can therefore be used directly in ligation.
Scheme 10.
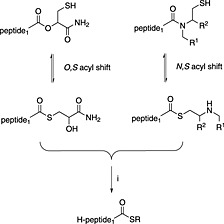
O,S‐acyl 267 and N,S‐acyl 265 shift for peptide thioester preparation, (i) R‐SH, NaSPh.
Botti and co‐workers pioneered the idea of an O,S shift on a linker derivatised from cysteine 267. They reported the successful synthesis of a protein, NNY‐Rantes (1–68), but cautioned that the method was prone to hydrolysis. This work led others to consider the possibility that the amide bond existed in equilibrium with a thioester and could be shifted further to the thioester form by the addition of thiol additives (Scheme 10). Evidence emerged that under some conditions amides with an adjacent δ‐thiol or ε‐thiol functionality could participate directly in native chemical ligation. The introduction of 4‐mercaptophenylacetic acid (MPAA) as a water‐soluble thiol additive to ligation reactions had a major impact 268, enabling many of the amide systems that showed N,S‐acyl transfer in acid to be used directly in ligation. The application of MPAA to ligation using N‐alkylated amides as a latent thioester was first demonstrated with the bis(sulfanylethyl)amido (SEA) linker 269. Transfer and ligation are performed in a single step, and they have reported the synthesis of many impressive targets with this approach (Scheme 11). The SEA linker has been used for the synthesis of 76mer SUMO, and this has in turn been conjugated 270. The Otaka group first reported the N,S‐acyl shift properties of sulfanylethylanilide in acid 271. Later, they were able to demonstrate the use of this linker directly in ligation using MPAA 272. They have since reported the synthesis of a glycoprotein in four pieces with their approach 273. Both linkers, SEA and sulfanylethylanilide, are difficult to acylate.
Scheme 11.
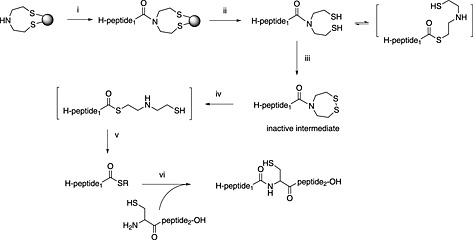
Use of the SEA‐linker for peptide thioester Fmoc SPPS. (i) SPPS; (ii) standard TFA cleavage; (iii) iodine oxidation; (iv) N,S‐acyl shift: 0.2 M tris(2‐carboxyethyl)phosphine/MPAA; (v) R‐SH, NaSPh; (vi) 6 M guanidine, phosphate buffer pH 7.8, 1% thiophenol.
These N,S rearrangements used N‐substituted amides; however, cysteine itself has been demonstrated to be in equilibrium between an amide and a thioester, and this has been exploited for the synthesis of cyclic peptides 274. Substitution at the Cα of cysteine to give α‐methylcysteine further promoted N,S‐acyl transfer by slightly twisting the amide bond and increasing its reactivity 274 so that it could participate in intermolecular ligation reactions (Scheme 12) 128. One advantage of using α‐methylcysteine is that it is stable to many postsynthetic manipulations including hydrazine treatment that makes it compatible with the use of Hmb backbone protection and other protecting group strategies. Its lack of N‐substitution made it compatible with standard linkers as it was less susceptible to diketopiperazine formation 128.
Scheme 12.

α‐Methylcysteine as a thioester surrogate.
However, these N,S‐acyl transfer methods are currently limited by slow‐ligation kinetics in comparison with safety‐catch or Boc methods and require careful control of pH and reaction conditions to achieve good results. This will undoubtedly change as this is a fast‐moving area of study 275. It is remarkable, however, that proteins are now being routinely synthesised using an amide bond as the active group. This approach has already generated interest for the preparation of dynamic peptide libraries 276. Nevertheless, the search for a straightforward, low‐cost Fmoc alternative for the preparation of thioester peptides continues because of the wealth of potential applications.
The use of heating in Fmoc SPPS
Since the inception of solid‐phase synthesis, heat has been used to speed up peptide assembly 277. A programmable heating block was incorporated into one of the first Fmoc continuous‐flow peptide synthesisers, LKB Biolynx, concomitant with introduction of Fmoc SPPS in the 1980s. The use of heating has been recently reviewed 18, 278.
Heating can be applied conventionally or by microwave or infrared irradiation. There are, however, associated problems exacerbated by heating: loss of peptide loading during peptide assembly 279, 280, cysteine and histidine racemisation 72, 281, 282, 283, 284, aspartimide formation 72, 281, 285 and elevated levels of β‐elimination in serine building blocks like pSer 175 and Ser‐glycan moieties 278.
There is a trend to return to simple coupling reagents in conjunction with heating. A carbodiimide/hydroxybenzotriazole‐based coupling protocol at 86°C accelerated the synthesis of β‐amyloid (1–42) 282, 283. The group of Jensen also found that DIC/Oxyma performed very well 284, 286. This topic has been comprehensively reviewed 18, 278.
Applying heat to Fmoc SPPS should reduce synthesis time and potentially suppress chain aggregation. The use of microwave heating in combination with backbone protection has been applied to the synthesis of aggregation‐prone sequences like the islet amyloid polypeptide 287, 288 and the influenza virus haemagglutinin 115. A comparison of the use of peptide backbone protection and microwave heating in the synthesis of difficult sequences showed that backbone protection was more effective in preventing aggregation than heating 115.
The major concern when using heating during Fmoc deprotection is aspartimide formation. This has been partially addressed by the substitution of piperidine with piperazine and the incorporation of acidic modifiers into the deprotection mixture 74, 285. However, despite these measures, aspartimide formation is still evident 57, 285.
Conclusion
Fmoc SPPS is very widely used and effective; however, it is still far from meeting its potential. It is generally considered that, as SPPS is a stepwise process with errors compounded throughout the synthesis, it cannot ever compete with the templated process of expression. Nevertheless, as we have outlined in this review, the constant improvement of side‐chain protection strategies and increasing purity of the building blocks have made previously unobtainable, lengthy targets accessible.
From the increasing number of long targets synthesised using commercially available backbone protection, there is a growing realisation that the major obstacle to peptide synthesis is peptide aggregation from interchain association on the growing peptide resin. Pseudoprolines are a good answer to address this problem and are used routinely to prevent association. They are limited by the dipeptide building blocks available, but as more researchers realise the benefits of backbone protection, there is a need for a more general backbone protection that can also be retained after synthesis to improve peptide handling properties.
Nonspecialists were responsible for the success of the Fmoc method and will be again for the future of Fmoc. The strongest uptake of Fmoc chemistry has been for the study of new materials. Chemical synthesis has the advantage of complete atom‐by‐atom control over the whole peptide, and multiple nonnatural or post‐translationally modified residues can be added. With the advances in backbone protection, heating and improved reliability of automation, we are on the threshold of machines suitable for the nonspecialist researcher to routinely reach synthetic peptides of lengths comparable with those for chemical nucleotide synthesis of above 100 residues.
Biographies
Raymond obtained his Ph.D. under the guidance of Prof. L. Moroder at the Technical University of Munich in 2000 and continued his work as a postdoctoral fellow at the Max‐Planck Institute of Biochemistry in Martinsried in Professor Moroder's group. He then took over the position of laboratory head at the Bachem Holding AG in Bubendorf, before moving to Merck & Cie, Schaffhausen, in 2009 as a principal scientist. His research interests are focused on the development and application of peptide chemistry tools and techniques supporting Fmoc SPPS for the Novabiochem® brand.

Peter obtained his Ph.D. under the guidance of Dr Brian Ridge at the University of Exeter in 1985. He worked as a postdoctoral fellow at the University of Bath from 1984 to 1986 under Prof. Malcolm Campbell, working on the structure–activity relationship of peptides related to melanin concentration hormone. In 1986, he joined LKB and was involved in the development of continuous‐flow peptide synthesisers. He moved to Novabiochem in 1988 as head of research and development, where he has worked since in various roles. His research interests have ranged from combinatorial chemistry, solid‐phase organic synthesis and peptide chemistry. However, his main interest has always been the development of chemical tools to facilitate Fmoc SPPS.

John was the last member of the peptide laboratory at the laboratory of Molecular Biology Cambridge before it closed, where Fmoc SPPS was developed by Atherton and Sheppard. John went on to postdoctoral work at The Scripps Research Institute with Phil Dawson where he translated work on backbone protection from the Sheppard lab into auxiliaries for extending native chemical ligation. He was made deputy director of the Scripps Oxford laboratory before moving to the National Institute for Medical Research Mill Hill in 2009, now part of the Francis Crick Institute.

Behrendt, R. , White, P. , and Offer, J. (2016) Advances in Fmoc solid‐phase peptide synthesis. J. Pept. Sci., 22: 4–27. doi: 10.1002/psc.2836.
References
- 1. Fosgerau K, Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discov. Today 2015; 20: 122. [DOI] [PubMed] [Google Scholar]
- 2. Kaspar AA, Reichert JM. Future directions for peptide therapeutics development. Drug Discov. Today 2013; 18: 807. [DOI] [PubMed] [Google Scholar]
- 3. Boyle AL, Woolfson DN. De novo designed peptides for biological applications. Chem. Soc. Rev. 2011; 40: 4295. [DOI] [PubMed] [Google Scholar]
- 4. Sheppard R. The fluorenylmethoxycarbonyl group in solid phase synthesis. J. Pept. Sci. 2003; 9: 545. [DOI] [PubMed] [Google Scholar]
- 5. Walter G. Production and use of antibodies against synthetic peptides. J. Immun. Meth. 1986; 88: 149. [DOI] [PubMed] [Google Scholar]
- 6. Dryland A, Sheppard RC. Peptide synthesis. Part 8. A system for solid‐phase synthesis under low pressure flow conditions. J. Chem. Soc., Perkin Trans. 1 1986; 125. [Google Scholar]
- 7. Chan WC, White PD. Fmoc Solid Phase Peptide Synthesis, Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- 8. Carpino LA, Han GY. 9‐Fluorenylmethoxycarbonyl amino‐protecting group. J. Org. Chem. 1972; 37: 3404. [Google Scholar]
- 9. Bodanszky M, Deshmane SS, Martinez J. Side reactions in peptide synthesis. 11. Possible removal of the 9‐fluorenylmethyloxycarbonyl group by the amino components during coupling. J. Org. Chem. 1979; 44: 1622. [Google Scholar]
- 10. Atherton E, Logan CJ, Sheppard RC. Peptide synthesis. Part 2. Procedures for solid‐phase synthesis using N α‐fluorenylmethoxycarbonylamino‐acids on polyamide supports. Synthesis of substance P and of acyl carrier protein 65–74 decapeptide. J. Chem. Soc., Perkin Trans. 1 1981; 538. [Google Scholar]
- 11. Chang CD, Meienhofer J. Solid phase peptide synthesis using mild base cleavage of Nalpha‐fluorenylmethyloxycarbonylamino acids, exemplified by a synthesis of dihydrosomatostatin. Int. J. Pept. Protein Res. 1978; 11: 246. [DOI] [PubMed] [Google Scholar]
- 12. Cameron L, Meldal M, Sheppard RC. Feedback control in organic synthesis. A system for solid phase peptide synthesis with true automation. J. Chem. Soc., Chem. Commun. 1987; 4: 270. [Google Scholar]
- 13. White P, Keyte JW, Bailey K, Bloomberg G. Expediting the Fmoc solid phase synthesis of long peptides through the application of dimethyloxazolidine dipeptides. J. Pept. Sci. 2004; 10: 18. [DOI] [PubMed] [Google Scholar]
- 14. Wöhr T, Mutter M. Pseudo‐prolines in peptide synthesis: direct insertion of serine and threonine derived oxazolidines in dipeptides. Tetrahedron Lett. 1995; 36: 3847. [Google Scholar]
- 15. Cardona V, Eberle I, Barthelemy S, Beythien J, Doerner B, Schneeberger P, Keyte J, White PD. Application of DMB‐dipeptides in the Fmoc SPPS of difficult and aspartimide‐prone sequences. Int. J. Pept. Res. Ther. 2008; 14: 285. [Google Scholar]
- 16. Kent SBH, in Peptides, structure and function, Proceedings of the 9th American Peptide Symposium (Eds.: Deber CM, Hruby VJ, Kopple KD), Pierce Chemical Company, Rockford, 1985, pp. 407.
- 17. Zompra AA, Galanis AS, Werbitzky O, Albericio F. Manufacturing peptides as active pharmaceutical ingredients. Future Med. Chem. 2009; 1: 361. [DOI] [PubMed] [Google Scholar]
- 18. Sabatino G, Papini AM. Advances in automatic, manual and microwave‐assisted solid‐phase peptide synthesis. Curr. Opin. Drug. Discovery Dev. 2008; 11: 762. [PubMed] [Google Scholar]
- 19. Pires DAT, Bemquerer MP, do Nascimento CJ. Some mechanistic aspects on Fmoc solid phase peptide synthesis. Int. J. Pept. Res. Ther. 2014; 20: 53. [Google Scholar]
- 20. Fields GB. Molecular Biomethods Handbook, Humana: Totowa, NJ, 1998; 527. [Google Scholar]
- 21. Isidro‐Llobet A, Alvarez M, Albericio F. Amino acid‐protecting groups. Chem. Rev. 2009; 109: 2455. [DOI] [PubMed] [Google Scholar]
- 22. Carpino LA, Shroff H, Triolo SA, Mansour E‐SM, Wenschuh H, Albericio F. The 2,2,4,6,7‐pentamethyldihydrobenzofuran‐5‐sulfonyl group (Pbf) as arginine side chain protectant. Tetrahedron Lett. 1993; 34: 7829. [Google Scholar]
- 23. Sieber P, Riniker R. Protection of carboxamide functions by the trityl residue: application to peptide synthesis. Tetrahedron Lett. 1991; 32: 739. [Google Scholar]
- 24. Photaki J, Taylor‐Papadimitriou J, Sakarellos C, Mazarakis P, Zervas L. On cysteine and cystine peptides. V. S‐trityl and S‐diphenylmethyl‐cysteine and ‐cysteine peptides. J. Chem. Soc., Perkin 1 1970; 19: 2683. [DOI] [PubMed] [Google Scholar]
- 25. Sieber P, Riniker B. Protection of histidine in peptide synthesis: a reassessment of the trityl group. Tetrahedron Lett. 1987; 28: 6031. [Google Scholar]
- 26. Anderson GW, Callahan M. tert‐Butyl esters of amino‐acids and peptides and their use in peptide synthesis. J. Am. Chem. Soc. 1960; 82: 3359. [Google Scholar]
- 27. Beyermann HC, Bontekoe JS. The t‐butoxy group, a novel hydroxyl‐protecting group for use in peptide synthesis with hydroxy‐amino acids. Recl. Trav. Chim. Pays‐Bas 1961; 81: 691. [Google Scholar]
- 28. White P, in Peptides, chemistry & biology. Proceedings of the 12th American Peptide Symposium (Eds.: Smith JA, Rivier JE), ESCOM, Leiden, 1992, pp. 537.
- 29. Eggen I, Gregg B, Rode H, Swietlow A, Verlander M, Szajek A. Control strategies for synthetic therapeutic peptide APIs part II: raw material considerations. Bio. Pharm. International. 2014; 27: 24. [Google Scholar]
- 30. Obkircher M, Stähelin C, Dick F. Formation of Fmoc‐β‐alanine during Fmoc‐protections with Fmoc‐OSu. J. Pept. Sci. 2008; 14: 763. [DOI] [PubMed] [Google Scholar]
- 31. Hlebowicz E, Andersen A, Andersson L, Moss B. Identification of Fmoc‐β‐Ala‐OH and Fmoc‐β‐Ala‐amino acid‐OH as new impurities in Fmoc‐protected amino acid derivatives. J. Pept. Res. 2005; 65: 90. [DOI] [PubMed] [Google Scholar]
- 32. Sigler GF, Fuller WD, Chaturvedi NC, Goodman M, Verlander M. Formation of oligopeptides during the synthesis of 9‐fluorenylmethyloxycarbonyl amino acid derivatives. Biopolymers 1983; 22: 2157. [Google Scholar]
- 33. Tessier M, Albericio F, Pedroso E, Grandas A, Eritja R, Giralt E, Granier C, Rietschoten J. Amino‐acids condensations in the preparation of Nα‐9‐fluorenylrnethyloxycarbonylamino‐acids with 9‐fluorenylmethylchloroformate. Int. J. Pept. Protein Res. 1983; 22: 125. [DOI] [PubMed] [Google Scholar]
- 34. Bolin DR, Sytwu I‐I, Humiec F, Meienhofer J. Preparation of oligomer‐free Nα‐Fmoc and Nα‐urethane amino acids. Int. J. Pept. Protein Res. 1989; 33: 353. [Google Scholar]
- 35. Khattab SN, Subirós‐Funosas R, El‐Faham A, Albericio F. Oxime carbonates: novel reagents for the introduction of fmoc and Alloc protecting groups, free of side reactions. Eur. J. Org. Chem. 2010; 2010: 3275. [Google Scholar]
- 36. Khattab SN, Subiros‐Funosas R, El‐Faham A, Albericio F. Cyanoacetamide‐based oxime carbonates: an efficient, simple alternative for the introduction of Fmoc with minimal dipeptide formation. Tetrahedron 2012; 68: 3056. [Google Scholar]
- 37. The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), http://www.ich.org [DOI] [PMC free article] [PubMed]
- 38. Gerhardt J, Nicholson GJ, in Peptides 2000, Proceedings of the 26th European Peptide Symposium (Eds.: Martinez J, Fehrentz A), Editions EDK, Paris, 2001, pp. 563.
- 39. Subirós‐Funosas R, El‐Faham A, Albericio F. Aspartimide formation in peptide chemistry: occurrence, prevention strategies and the role of N‐hydroxylamines. Tetrahedron 2011; 67: 8595. [Google Scholar]
- 40. Tickler AK, Barrow CJ, Wade JD. Improved preparation of amyloid‐β peptides using DBU as Nα‐Fmoc deprotection reagent. J. Pept. Sci. 2001; 7: 488. [DOI] [PubMed] [Google Scholar]
- 41. Bodanszky M, Kwei JZ. Side reactions in peptide synthesis. Int. J. Pept. Protein Res. 1978; 12: 69. [PubMed] [Google Scholar]
- 42. Martinez J, Bodanszky M. Side reactions in peptide synthesis. Int. J. Pept. Protein Res. 1978; 12: 277. [PubMed] [Google Scholar]
- 43. Chandra K, Roy TK, Shalev DE, Loyter A, Gilon C, Gerber RB, Friedler A. A tandem in situ peptide cyclization through trifluoroacetic acid cleavage. Angew. Chem. Int. Ed. 2014; 53: 9450. [DOI] [PubMed] [Google Scholar]
- 44. Michels T, Doelling R, Haberkorn U, Mier W. Acid‐mediated prevention of aspartimide formation in solid phase peptide synthesis. Org. Lett. 2012; 14: 5218. [DOI] [PubMed] [Google Scholar]
- 45. Lauer JL, Fields CG, Fields GB. Sequence dependence of aspartimide formation during 9‐fluorenylmethoxycarbonyl solid‐phase peptide synthesis. Lett. Pept. Sci. 1995; 1: 197. [Google Scholar]
- 46. Mergler M, Dick F, Sax B, Staehelin C, Vorherr T. The aspartimide problem in Fmoc‐based SPPS. Part II. J. Pept. Sci. 2003; 9: 518. [DOI] [PubMed] [Google Scholar]
- 47. Quibell M, Owen D, Packman LC, Johnson T. Suppression of piperidine‐mediated side product formation for Asp(OBut) containing peptides by the use of N‐(2‐hydroxy‐4‐methoxybenzyl)(Hmb) backbone amide protection. J. Chem. Soc., Chem. Commun. 1994; 20: 2343. [Google Scholar]
- 48. Offer J, Quibell M, Johnson T. On‐resin solid‐phase synthesis of asparagine N‐linked glycopeptides: use of N‐(2‐acetoxy‐4‐methoxybenzyl)(AcHmb) aspartyl amide‐bond protection to prevent unwanted aspartimide formation. J. Chem. Soc., Perkin Trans. 1 1996; 2: 175. [Google Scholar]
- 49. Mergler M, Dick F. The aspartimide problem in Fmoc‐based SPPS. Part III. J. Pept. Sci. 2005; 11: 650. [DOI] [PubMed] [Google Scholar]
- 50. Karlström A, Undén A. A new protecting group for aspartic acid that minimizes piperidine‐catalyzed aspartimide formation in Fmoc solid phase peptide synthesis. Tetrahedron Lett. 1996; 37: 4243. [Google Scholar]
- 51. Mergler M, Dick F, Sax B, Weiler P, Vorherr T. The aspartimide problem in Fmoc‐based SPPS. Part I. J. Pept. Sci. 2003; 9: 36. [DOI] [PubMed] [Google Scholar]
- 52. Karlström A, Undén A. Design of protecting groups for the β‐carboxylic group of aspartic acid that minimize base‐catalyzed aspartimide formation. Int. J. Pept. Protein Res. 1996; 48: 305. [DOI] [PubMed] [Google Scholar]
- 53. Bodanszky M, Martinez J. Side reactions in peptide synthesis. 8. On the phenacyl group in the protection of the beta‐carboxyl function of aspartyl residues. J. Org. Chem. 1978; 43: 3071. [Google Scholar]
- 54. Dölling R, Beyermann M, Haenel J, Kernchen F, Krause E, Franke P, Brudel M, Bienert M. Piperidine‐mediated side product formation for Asp(OBut)‐containing peptides. J. Chem. Soc., Chem. Commun. 1994; 7: 853. [Google Scholar]
- 55. Ullmann V, Raedisch M, Boos I, Freund J, Poehner C, Schwarzinger S, Unverzagt C. Convergent solid‐phase synthesis of N‐glycopeptides facilitated by pseudoprolines at consensus‐sequence Ser/Thr residues. Angew. Chem. Int. Ed. 2012; 51: 11566. [DOI] [PubMed] [Google Scholar]
- 56. Wang P, Aussedat B, Vohra Y, Danishefsky SJ. An advance in the chemical synthesis of homogeneous N‐linked glycopolypeptides by convergent aspartylation. Angew. Chem. Int. Ed. 2012; 51: 11571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Behrendt R, Huber S, Martí R, White P. New t‐butyl based aspartate protecting groups preventing aspartimide formation in Fmoc SPPS. J. Pept. Sci. 2015; 21: 680. [DOI] [PubMed] [Google Scholar]
- 58. Subirós‐Funosas R, El‐Faham A, Albericio F. Use of Oxyma as pH modulatory agent to be used in the prevention of base‐driven side reactions and its effect on 2‐chlorotrityl chloride resin. Pept. Sci. 2012; 98: 89. [DOI] [PubMed] [Google Scholar]
- 59. Packman LC. N‐2‐Hydroxy‐4‐methoxybenzyl(Hmb) backbone protection strategy prevents double aspartimide formation in a ‘difficult’ peptide sequence. Tetrahedron Lett. 1995; 36: 7523. [Google Scholar]
- 60. Röder R, Henklein P, Weißhoff H, Mügge C, Pätzel M, Schubert U, Carpino LA, Henklein P. On the use of N‐dicyclopropylmethyl aspartyl‐glycine synthone for backbone amide protection. J. Pept. Sci. 2010; 16: 65. [DOI] [PubMed] [Google Scholar]
- 61. Conroy T, Jolliffe KA, Payne RJ. Synthesis of N‐linked glycopeptides via solid‐phase aspartylation. Org. Biomol. Chem. 2010; 8: 3723. [DOI] [PubMed] [Google Scholar]
- 62. El Oualid F, Merkx R, Ekkebus R, Hameed DS, Smit JJ, de Jong A, Hilkmann H, Sixma TK, Ovaa H. Chemical synthesis of ubiquitin, ubiquitin‐based probes, and diubiquitin. Angew. Chem. Int. Ed. 2010; 49: 10149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ramage R, Green J. NG‐2,2,5,7,8‐pentamethylchroman‐6‐sulfonyl‐l‐arginine: a new acid labile derivative for peptide synthesis. Tetrahedron Lett. 1987; 28: 2287. [Google Scholar]
- 64. Green J, Ogunjobi O, Ramage R, Stewart A, McCurdy S, Noble R. Application of the NG‐(2,2,5,7,8‐pentamethylchroman‐6‐sulphonyl) derivative of FMOC‐arginine to peptide synthesis. Tetrahedron Lett. 1988; 29: 4341. [Google Scholar]
- 65. Isidro A, Latassa D, Giraud M, Alvarez M, Albericio F. 1,2‐Dimethylindole‐3‐sulfonyl (MIS) as protecting group for the side chain of arginine. Org. Biomol. Chem. 2009; 7: 2565. [DOI] [PubMed] [Google Scholar]
- 66. Noda M, Kiffe M. New mild acid‐labile protecting groups for the guanidino function of Nα‐fluorenylmethoxycarbonyl‐l‐arginine in solid‐phase peptide synthesis: 10, 11‐dihydro‐5H‐dibenzo [a, d]. cyclohepten‐5‐yl, 2‐methoxy‐10, 11‐dihydoro‐5H‐dibenzo [a, d]. cyclohepten‐5‐yl and 5H‐dibenzo [a, d]. cyclohepten‐5‐yl groups. J. Pept. Res. 1997; 50: 329. [DOI] [PubMed] [Google Scholar]
- 67. Paul R, Anderson GW, Callahan FM. Some side reactions of nitro‐l‐arginine. J. Org. Chem. 1961; 26: 3347. [Google Scholar]
- 68. Fields GB, Noble RL. Solid phase peptide synthesis utilizing 9‐fluorenylmethoxycarbonyl amino acids. Int. J. Pept. Protein Res. 1990; 35: 161. [DOI] [PubMed] [Google Scholar]
- 69. Rink H, Sieber P, Raschdorf F. Conversion of N G urethane protected arginine to ornithine in peptide solid phase synthesis. Tetrahedron Lett. 1984; 25: 621. [Google Scholar]
- 70. Musiol HJ, Siedler F, Quarzago D, Moroder L. Redox‐active bis‐cysteinyl peptides. I. Synthesis of cyclic cystinyl peptides by conventional methods in solution and on solid supports. Biopolymers 1994; 34: 1553. [DOI] [PubMed] [Google Scholar]
- 71. Kaiser T, Nicholson G, Kohlbau H, Voelter W. Racemization studies of Fmoc‐Cys (Trt)‐OH during stepwise Fmoc‐solid phase peptide synthesis. Tetrahedron Lett. 1996; 37: 1187. [Google Scholar]
- 72. Palasek SA, Cox ZJ, Collins JM. Limiting racemization and aspartimide formation in microwave‐enhanced Fmoc solid phase peptide synthesis. J. Pept. Sci. 2007; 13: 143. [DOI] [PubMed] [Google Scholar]
- 73. Han Y, Albericio F, Barany G. Occurrence and minimization of cysteine racemization during stepwise solid‐phase peptide synthesis. J. Org. Chem. 1997; 62: 4307. [DOI] [PubMed] [Google Scholar]
- 74. Collins JM, Porter KA, Singh SK, Vanier GS. High‐efficiency solid phase peptide synthesis (HE‐SPPS). Org. Lett. 2014; 16: 940. [DOI] [PubMed] [Google Scholar]
- 75. Hibino H, Miki Y, Nishiuchi Y. Evaluation of acid‐labile S‐protecting groups to prevent Cys racemization in Fmoc solid‐phase peptide synthesis. J. Pept. Sci. 2014; 20: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mitchell MA, Runge TA, Mathews WR, Ichhpurani AK, Harn NK, Dobrowolski PJ, Eckenrode FM. Problems associated with use of the benzyloxymethyl protecting group for histidines. Formaldehyde adducts formed during cleavage by hydrogen fluoride 1. Int. J. Pept. Protein Res. 1990; 36: 350. [DOI] [PubMed] [Google Scholar]
- 77. Kumiko YK, Ishizu T, Isaka S, Tamura M, Rumi OT, Nishiuchi Y, Kimura T, in Peptide Science 2005: Proceedings of the 42nd Japanese Peptide Symposium (Ed.: Wakamiya T), Japanese Peptide Society, Osaka, 2006, pp. 163.
- 78. Hibino H, Nishiuchi Y. 4‐Methoxybenzyloxymethyl group as an Nπ‐protecting group for histidine to eliminate side‐chain‐induced racemization in the Fmoc strategy. Tetrahedron Lett. 2011; 52: 4947. [Google Scholar]
- 79. Ramos‐Tomillero I, Rodríguez H, Albericio F. Tetrahydropyranyl, a nonaromatic acid‐labile Cys protecting group for Fmoc peptide chemistry. Org. Lett. 2015, 17, 1680. [DOI] [PubMed] [Google Scholar]
- 80. Atherton E, Hardy P, Harris DE, Matthews BH, in Peptides 1990: Proceedings of the Twenty First European Peptide Symposium (Eds.: Giralt E, Andreu D), Escom, Leiden, 1991, pp. 243.
- 81. Windridge G, Jorgensen EC. 1‐Hydroxybenzotriazole as a racemization‐suppressing reagent for the incorporation of im‐benzyl‐l‐histidine into peptides. J. Am. Chem. Soc. 1971; 93: 6318. [DOI] [PubMed] [Google Scholar]
- 82. Jones JH, Ramage WI. An approach to the prevention of racemization in the synthesis of histidine‐containing peptides. J. Chem. Soc., Chem. Commun. 1978; 11: 472. [Google Scholar]
- 83. Jones JH, Ramage WI, Witty MJ. Mechanism of racemisation of histidine derivatives in peptide synthesis. Int. J. Pept. Protein Res. 1980; 15: 301. [DOI] [PubMed] [Google Scholar]
- 84. Brown T, Jones JH. Protection of histidine side‐chains with π‐benzyloxymethyl or π‐bromobenzyloxymethyl groups. J. Chem. Soc., Chem. Commun. 1981; 13: 648. [Google Scholar]
- 85. Brown T, Jones JH, Richards JD. Further studies on the protection of histidine side chains in peptide synthesis: the use of the π‐benzyloxymethyl group. J. Chem. Soc., Perkin Trans. 1 1982; 1553. [Google Scholar]
- 86. Colombo R, Colombo F, Jones JH. Acid‐labile histidine side‐chain protection: the N(π)‐tert‐butoxymethyl group. J. Chem. Soc., Chem. Commun. 1984; 5: 292. [Google Scholar]
- 87. Okada Y, Wang J, Yamamoto T, Mu Y, Yokoi T. Amino acids and peptides. Part 45. Development of a new Nπ‐protecting group of histidine, Nπ‐(1‐adamantyloxymethyl) histidine, and its evaluation for peptide synthesis. J. Chem. Soc., Perkin Trans. 1 1996; 2139. [Google Scholar]
- 88. Hibino H, Miki Y, Nishiuchi Y. Synthesis and application of Nα‐Fmoc‐Nπ‐4‐methoxybenzyloxymethylhistidine in solid phase peptide synthesis. J. Pept. Sci. 2012; 18: 763. [DOI] [PubMed] [Google Scholar]
- 89. Narita M, Ishikawa K, Chen JY, Kim Y. Prediction and improvement of protected peptide solubility in organic solvents. Int. J. Pept. Protein Res. 1984; 24: 580. [DOI] [PubMed] [Google Scholar]
- 90. Narita M, Fukunaga T, Wakabayashi A, Ishikawa K, Nakano H. Syntheses and properties of tertiary peptide bond‐containing polypeptides. Int. J. Pept. Protein Res. 1984; 23: 306. [PubMed] [Google Scholar]
- 91. Meyer DE, Chilkoti A. Purification of recombinant proteins by fusion with thermally‐responsive polypeptides. Nat. Biotechnol. 1999; 17: 1112. [DOI] [PubMed] [Google Scholar]
- 92. Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006; 75: 333. [DOI] [PubMed] [Google Scholar]
- 93. Nishiuchi Y, Inui T, Nishio H, Bódi J, Kimura T, Tsuji FI, Sakakibara S. Chemical synthesis of the precursor molecule of the Aequorea green fluorescent protein, subsequent folding, and development of fluorescence. Proc. Natl. Acad. Sci. 1998; 95: 13549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Sakakibara S. Chemical synthesis of proteins in solution. Pept. Sci. 1999; 51: 279. [DOI] [PubMed] [Google Scholar]
- 95. Hancock WS, Prescott DJ, Vagelos PR, Marshall GR. Solvation of the polymer matrix. Source of truncated and failure sequences in solid phase synthesis. J. Org. Chem. 1973; 38: 774. [Google Scholar]
- 96. Bedford J, Hyde C, Johnson T, Jun W, Owen D, Quibell M, Sheppard RC. Amino acid structure and ‘difficult sequences’ in solid phase peptide synthesis. Int. J. Pept. Protein Res. 1992; 40: 300. [DOI] [PubMed] [Google Scholar]
- 97. Van Woerkom W, Van Nispen J. Difficult couplings in stepwise solid phase peptide synthesis: predictable or just a guess? Int. J. Pept. Protein Res. 1991; 38: 103. [DOI] [PubMed] [Google Scholar]
- 98. Lahiri S, Brehs M, Olschewski D, Becker CF. Total chemical synthesis of an integral membrane enzyme: diacylglycerol kinase from Escherichia coli. Angew. Chem. Int. Ed. 2011; 50: 3988. [DOI] [PubMed] [Google Scholar]
- 99. Johnson EC, Malito E, Shen Y, Rich D, Tang W‐J, Kent SB. Modular total chemical synthesis of a human immunodeficiency virus type 1 protease. J. Am. Chem. Soc. 2007; 129: 11480. [DOI] [PubMed] [Google Scholar]
- 100. Hossain MA, Belgi A, Lin F, Zhang S, Shabanpoor F, Chan L, Belyea C, Truong H‐T, Blair AR, Andrikopoulos S, Tregear GW, Wade JD. Use of a temporary ‘solubilizing’ peptide tag for the Fmoc solid‐phase synthesis of human insulin glargine via use of regioselective disulfide bond formation. Bioconjugate Chem. 2009; 20: 1390. [DOI] [PubMed] [Google Scholar]
- 101. Wu C‐R, Stevens VC, Tregear GW, Wade JD. Continuous‐flow solid‐phase synthesis of a 74‐peptide fragment analogue of human β‐chorionic gonadotropin. J. Chem. Soc., Perkin Trans. 1 1989; 81. [Google Scholar]
- 102. Weygand F, Steglich W, Bjarnason J, Akhtar R, Khan NM. Leicht abspaltbare schutzgruppen für säureamidfunktionen 1. Mitteilung. Tetrahedron Lett. 1966; 7: 3483. [Google Scholar]
- 103. Narita M, Ishikawa K, Nakano H, Isokawa S. Tertiary peptide bond containing‐oligo (Leu) s. Int. J. Pept. Protein Res. 1984; 24: 14. [PubMed] [Google Scholar]
- 104. Hyde C, Johnson T, Owen D, Quibell M, Sheppard R. Some ‘difficult sequences’ made easy. Int. J. Pept. Protein Res. 1994; 43: 431. [PubMed] [Google Scholar]
- 105. Blaakmeer J, Tijsse‐Klasen T, Tesser GI. Enhancement of solubility by temporary dimethoxybenzyl‐substitution of peptide bonds. Towards the synthesis of defined oligomers of alanine and of lysyl‐glutamyl‐glycine. Int. J. Pept. Protein Res. 1991; 37: 556. [PubMed] [Google Scholar]
- 106. Johnson T, Quibell M, Owen D, Sheppard RC. A reversible protecting group for the amide bond in peptides. Use in the synthesis of difficult sequences. J. Chem. Soc., Chem. Commun. 1993; 4: 369. [Google Scholar]
- 107. Quibell M, Turnell WG, Johnson T. Reversible modification of the acid labile 2‐hydroxy‐4‐methoxybenzyl (Hmb) amide protecting group: a simple scheme yielding backbone substituted free peptides. Tetrahedron Lett. 1994; 35: 2237. [Google Scholar]
- 108. Quibell M, Turnell W, Johnson T. Preparation and purification of beta‐amyloid (1–43) via soluble, amide backbone protected intermediates. J. Org. Chem. 1994; 59: 1745. [Google Scholar]
- 109. Clippingdale A, Macris M, Wade J, Barrow C. Synthesis and secondary structural studies of penta (acetyl‐Hmb) Aβ (1–40). J. Pept. Res. 1999; 53: 665. [DOI] [PubMed] [Google Scholar]
- 110. Carpino LA, Nasr K, Abdel‐Maksoud AA, El‐Faham A, Ionescu D, Henklein P, Wenschuh H, Beyermann M, Krause E, Bienert M. Dicyclopropylmethyl peptide backbone protectant. Org. Lett. 2009; 11: 3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Isidro‐Llobet A, Just‐Baringo X, Alvarez M, Albericio F. EDOTn and MIM, new peptide backbone protecting groups. Pept. Sci. 2008; 90: 444. [DOI] [PubMed] [Google Scholar]
- 112. Ede NJ, Ang KH, James IW, Bray AM. Incorporation of 2‐hydroxy‐4‐methoxybenzyl protection during peptide synthesis via reductive alkylation on the solid phase. Tetrahedron Lett. 1996; 37: 9097. [Google Scholar]
- 113. Wöhr T, Wahl F, Nefzi A, Rohwedder B, Sato T, Sun X, Mutter M. Pseudo‐prolines as a solubilizing, structure‐disrupting protection technique in peptide synthesis. J. Am. Chem. Soc. 1996; 118: 9218. [Google Scholar]
- 114. Offer J, Johnson T, Quibell M. Application of reversible amide‐bond protection to suppress peptide segment epimerization. Tetrahedron Lett. 1997; 38: 9047. [Google Scholar]
- 115. Abdel‐Aal AM, Papageorgiou G, Quibell M, Offer J. Automated synthesis of backbone protected peptides. Chem. Commun. 2014; 50: 8316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Miranda LP, Meutermans WD, Smythe ML, Alewood PF. An activated O → N acyl transfer auxiliary: efficient amide‐backbone substitution of hindered ‘difficult’ peptides. J. Org. Chem. 2000; 65: 5460. [DOI] [PubMed] [Google Scholar]
- 117. Paradís‐Bas M, Tulla‐Puche J, Albericio F. 2‐Methoxy‐4‐methylsulfinylbenzyl: a backbone amide safety‐catch protecting group for the synthesis and purification of difficult peptide sequences. Chem. Eur. J. 2014; 20: 15031. [DOI] [PubMed] [Google Scholar]
- 118. Huang Y‐C, Guan C‐J, Tan X‐L, Chen C‐C, Guo Q‐X, Li Y‐M. Accelerated Fmoc solid‐phase synthesis of peptides with aggregation‐disrupting backbones. Org. Biomol. Chem. 2015; 13: 1500. [DOI] [PubMed] [Google Scholar]
- 119. Kemp D, Carey RI. Boc‐L‐Dmt‐OH as a fully N,S‐blocked cysteine derivative for peptide synthesis by prior thiol capture. Facile conversion of N‐terminal Boc‐l‐Dmt‐peptides to H‐Cys (Scm)‐peptides. J. Org. Chem. 1989; 54: 3640. [Google Scholar]
- 120. Barber M, Jones JH. An alternative synthesis of [7‐(thiazolidine‐4‐carboxylic acid)] oxytocin. Int. J. Pept. Protein Res. 1977; 9: 269. [DOI] [PubMed] [Google Scholar]
- 121. Goncalves V, Gautier B, Huguenot F, Leproux P, Garbay C, Vidal M, Inguimbert N. Total chemical synthesis of the D2 domain of human VEGF receptor 1. J. Pept. Sci. 2009; 15: 417. [DOI] [PubMed] [Google Scholar]
- 122. Mutter M, Chandravarkar A, Boyat C, Lopez J, Santos SD, Mandal B, Mimna R, Murat K, Patiny L, Saucède L, Tuchscherer G. Switch peptides in statu nascendi: induction of conformational transitions relevant to degenerative diseases. Angew. Chem. Int. Ed. 2004; 43: 4172. [DOI] [PubMed] [Google Scholar]
- 123. Carpino LA, Krause E, Sferdean CD, Schümann M, Fabian H, Bienert M, Beyermann M. Synthesis of ‘difficult’ peptide sequences: application of a depsipeptide technique to the Jung‐Redemann 10‐ and 26‐mers and the amyloid peptide Aβ(1–42). Tetrahedron Lett. 2004; 45: 7519. [Google Scholar]
- 124. Sohma Y, Sasaki M, Hayashi Y, Kimura T, Kiso Y. Novel and efficient synthesis of difficult sequence‐containing peptides through O–N intramolecular acyl migration reaction of O‐acyl isopeptides. Chem. Commun. 2004; 1: 124. [DOI] [PubMed] [Google Scholar]
- 125. Sohma Y, Kiso Y. Synthesis of O‐acyl isopeptides. The Chemical Record 2013; 13: 218. [DOI] [PubMed] [Google Scholar]
- 126. Coin I. The depsipeptide method for solid‐phase synthesis of difficult peptides. J. Pept. Sci. 2010; 16: 223. [DOI] [PubMed] [Google Scholar]
- 127. Miranda LP, Alewood PF. Challenges for protein chemical synthesis in the 21st century: bridging genomics and proteomics. Biopolymers 2000; 55: 217. [DOI] [PubMed] [Google Scholar]
- 128. Burlina F, Papageorgiou G, Morris C, White PD, Offer J. In situ thioester formation for protein ligation using α‐methylcysteine. Chem. Sci. 2014; 5: 766. [Google Scholar]
- 129. Zheng J‐S, Yu M, Qi Y‐K, Tang S, Shen F, Wang Z‐P, Xiao L, Zhang L, Tian C‐L, Liu L. Expedient total synthesis of small to medium‐sized membrane proteins via Fmoc chemistry. J. Am. Chem. Soc. 2014; 136: 3695. [DOI] [PubMed] [Google Scholar]
- 130. Asahina Y, Kamitori S, Takao T, Nishi N, Hojo H. Chemoenzymatic synthesis of the immunoglobulin domain of Tim‐3 carrying a complex‐type N‐glycan by using a one‐pot ligation. Angew. Chem. Int. Ed. 2013; 52: 9733. [DOI] [PubMed] [Google Scholar]
- 131. Prabakaran S, Lippens G, Steen H, Gunawardena J. Post‐translational modification: nature's escape from genetic imprisonment and the basis for dynamic information encoding. Wiley Interdiscip. Rev. Syst. Biol. Med. 2012; 4: 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Walsh CT, Garneau‐Tsodikova S, Gatto GJ. Protein posttranslational modifications: the chemistry of proteome diversifications. Angew. Chem. Int. Ed. 2005; 44: 7342. [DOI] [PubMed] [Google Scholar]
- 133. Lothrop AP, Torres MP, Fuchs SM. Deciphering post‐translational modification codes. FEBS Lett. 2013; 587: 1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Kouzarides T. Chromatin modifications and their function. Cell 2007; 128: 693. [DOI] [PubMed] [Google Scholar]
- 135. Perich JW, Ede NJ, Eagle S, Bray AM. Synthesis of phosphopeptides by the Multipinast method: evaluation of coupling methods for the incorporation of Fmoc‐Tyr (PO3Bzl, H)‐OH, Fmoc‐Ser (PO3Bzl, H)‐OH and Fmoc‐Thr (PO3Bzl, H)‐OH. Lett. Pept. Sci. 1999; 6: 91. [Google Scholar]
- 136. Wakamiya T, Saruta K, Yasuoka J‐i, Kusumoto S. An efficient procedure for solid‐phase synthesis of phosphopeptides by the Fmoc strategy. Chem. Lett. 1994; 6: 1099. [Google Scholar]
- 137. White P, Beythien J, in Innovations & perspectives in solid phase synthesis & combinatorial libraries, 4th International Symposium (Ed.: Epton R), Mayflower Scientific Ltd, Birmingham, 1996, pp. 557.
- 138. Kitas E, Perich J, Wade J, Johns R, Tregear G. Fmoc‐polyamide solid phase synthesis of an O‐phosphotyrosine‐containing tridecapeptide. Tetrahedron Lett. 1989; 30: 6229. [Google Scholar]
- 139. Perich JW, Reynolds EC. The facile one‐pot synthesis of Nα‐(9‐fluorenylmethoxycarbonyl)‐O‐(O′,O″‐dialkylphosphoro)‐l‐tyrosines using dialkyl N,N‐diethylphosphoramidites. Synlett 1991; 1991: 577. [Google Scholar]
- 140. Ottinger EA, Shekels LL, Bernlohr DA, Barany G. Synthesis of phosphotyrosine‐containing peptides and their use as substrates for protein tyrosine phosphatases. Biochemistry 1993; 32: 4354. [DOI] [PubMed] [Google Scholar]
- 141. Chao H‐G, Leiting B, Reiss PD, Burkhardt AL, Klimas CE, Bolen JB, Matsueda GR. Synthesis and application of Fmoc‐O‐[bis (dimethylamino)phosphono] tyrosine, a versatile protected phosphotyrosine equivalent. J. Org. Chem. 1995; 60: 7710. [Google Scholar]
- 142. Chao H‐G, Bernatowicz MS, Reiss PD, Matsueda GR. Synthesis and application of bis‐silylethyl‐derived phosphate‐protected Fmoc‐phosphotyrosine derivatives for peptide synthesis. J. Org. Chem. 1994; 59: 6687. [Google Scholar]
- 143. Chauhan SS, Varshney A, Verma B, Pennington MW. Efficient synthesis of protected l‐phosphonophenylalanine (Ppa) derivatives suitable for solid phase peptide synthesis. Tetrahedron Lett. 2007; 48: 4051. [DOI] [PubMed] [Google Scholar]
- 144. Marseigne I, Roques BP. Synthesis of new amino acids mimicking sulfated and phosphorylated tyrosine residues. J. Org. Chem. 1988; 53: 3621. [Google Scholar]
- 145. Gordeev MF, Patel DV, Barker PL, Gordon EM. N‐α‐Fmoc‐4‐phosphono (difluoromethyl)‐l‐phenylalanine: a new O‐phosphotyrosine isosteric building block suitable for direct incorporation into peptides. Tetrahedron Lett. 1994; 35: 7585. [Google Scholar]
- 146. Simpson LS, Widlanski TS. A comprehensive approach to the synthesis of sulfate esters. J. Am. Chem. Soc. 2006; 128: 1605. [DOI] [PubMed] [Google Scholar]
- 147. Simpson LS, Zhu JZ, Widlanski TS, Stone MJ. Regulation of chemokine recognition by site‐specific tyrosine sulfation of receptor peptides. Chem. Biol. 2009; 16: 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Ali AM, Taylor SD. Efficient solid‐phase synthesis of sulfotyrosine peptides using a sulfate protecting‐group strategy. Angew. Chem. Int. Ed. 2009; 48: 2024. [DOI] [PubMed] [Google Scholar]
- 149. Ali AM, Hill B, Taylor SD. Trichloroethyl group as a protecting group for sulfonates and its application to the synthesis of a disulfonate analog of the tyrosine sulfated PSGL‐143−50 peptide. J. Org. Chem. 2009; 74: 3583. [DOI] [PubMed] [Google Scholar]
- 150. White P, in Novabiochem innovations, 2006.
- 151. Rothbart SB, Krajewski K, Strahl BD, Fuchs SM. Peptide microarrays to interrogate the ‘histone code’. Methods Enzymol. 2012; 512: 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Meldal M, Bock K. Pentafluorophenyl esters for temporary carboxyl group protection in solid phase synthesis of N‐linked glycopeptides. Tetrahedron Lett. 1990; 31: 6987. [Google Scholar]
- 153. Meinjohanns E, Meldal M, Paulsen H, Dwek RA, Bock K. Novel sequential solid‐phase synthesis of N‐linked glycopeptides from natural sources. J. Chem. Soc., Perkin Trans. 1 1998; 549. [Google Scholar]
- 154. Paulsen H, Adermann K. Synthese von O‐Glycopeptid‐Sequenzen des N‐Terminus von Interleukin‐2. Liebigs Ann. Chem. 1989; 1989: 751. [Google Scholar]
- 155. Irazoqui FJ, Vides MA, Nores GA. Structural requirements of carbohydrates to bind Agaricus bisporus lectin. Glycobiology 1999; 9: 59. [DOI] [PubMed] [Google Scholar]
- 156. Liebe B, Kunz H. Solid‐phase synthesis of a tumor‐associated sialyl‐tn antigen glycopeptide with a partial sequence of the ‘tandem repeat’ of the MUC‐1 mucin. Angew. Chem. Int. Ed. 1997; 36: 618. [Google Scholar]
- 157. Komba S, Meldal M, Werdelin O, Jensen T, Bock K. Convenient synthesis of Thr and Ser carrying the tumor associated sialyl‐(2 → 3)‐T antigen as building blocks for solid‐phase glycopeptide synthesis. J. Chem. Soc., Perkin Trans. 1 1999; 415. [Google Scholar]
- 158. Zhang Y, Muthana SM, Farnsworth D, Ludek O, Adams K, Barchi JJ, Jr , Gildersleeve JC. Enhanced epimerization of glycosylated amino acids during solid‐phase peptide synthesis. J. Am. Chem. Soc. 2012; 134: 6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Arsequell G, Krippner L, Dwek RA, Wong SYC. Building blocks for solid‐phase glycopeptide synthesis: 2‐acetamido‐2‐deoxy‐ β‐d‐glycosides of FmocSerOH and FmocThrOH. J. Chem. Soc., Chem. Commun. 1994; 2383. [Google Scholar]
- 160. Cohen P. The regulation of protein function by multisite phosphorylation—a 25 year update. Trends Biochem. Sci. 2000; 25: 596. [DOI] [PubMed] [Google Scholar]
- 161. Besant PG, Attwood PV. Histidine phosphorylation in histones and in other mammalian proteins. Methods Enzymol. 2010; 471: 403. [DOI] [PubMed] [Google Scholar]
- 162. Cieśla J, Frączyk T, Rode W. Phosphorylation of basic amino acid residues in proteins: important but easily missed. Acta Biochim. Pol. 2011; 58: 137. [PubMed] [Google Scholar]
- 163. Perich JW, in Houben‐Weyl Methods of Organic Synthesis, Synthesis of Peptides and Peptidomimetics, Vol. E22b (Eds.: Goodman M, Felix A, Moroder L, Toniolo C.), Georg Thieme, Stuttgart, 2002, pp. 375. [Google Scholar]
- 164. Andrews D, Kitchin J, Seale P. Solid‐phase synthesis of a range of O‐phosphorylated peptides by post‐assembly phosphitylation and oxidation. Int. J. Pept. Protein Res. 1991; 38: 469. [DOI] [PubMed] [Google Scholar]
- 165. Petrillo DE, Mowrey DR, Allwein SP, Bakale RP. A general preparation of protected phosphoamino acids. Org. Lett. 2012; 14: 1206. [DOI] [PubMed] [Google Scholar]
- 166. Ottinger EA, Xu Q, Barany G. Intramolecular pyrophosphate formation during N: α‐9‐fluorenylmethyloxycarbonyl (Fmoc) solid‐phase synthesis of peptides containing adjacent phosphotyrosine residues. Pept. Res. 1995; 9: 223. [PubMed] [Google Scholar]
- 167. García‐Echeverría C. Potential pyrophosphate formation upon use of N α‐Fmoc‐Tyr (PO3H2)‐OH in solid‐phase peptide synthesis. Lett. Pept. Sci. 1995; 2: 93. [Google Scholar]
- 168. Kitas EA, Wade JD, Johns R, Perich JW, Tregear GW. Preparation and use of N α‐fluorenylmethoxycarbonyl‐O‐dibenzylphosphono‐l‐tyrosine in continuous flow solid phase peptide synthesis. J. Chem. Soc., Chem. Commun. 1991; 5: 338. [Google Scholar]
- 169. Kitas EA, Knorr R, Trzeciak A, Bannwarth W. Alternative strategies for the Fmoc solid‐phase synthesis of O 4‐phospho‐l‐tyrosine‐containing peptides. Helv. Chim. Acta 1991; 74: 1314. [Google Scholar]
- 170. Pascal R, in Peptides 2000, Proceedings of the 26th European Peptide Symposium (Eds.: Martinez J, Fehrentz A), Editions EDK, Paris, 2001, pp. 263.
- 171. Ishida A, Shigeri Y, Taniguchi T, Kameshita I. Protein phosphatases that regulate multifunctional Ca 2+/calmodulin‐dependent protein kinases: from biochemistry to pharmacology. Pharm. Ther. 2003; 100: 291. [DOI] [PubMed] [Google Scholar]
- 172. Lacombe J, Andriamanampisoa F, Pavia A. Solid‐phase synthesis of peptides containing phosphoserine using phosphate tert‐butyl protecting group. Int. J. Pept. Protein Res. 1990; 36: 275. [DOI] [PubMed] [Google Scholar]
- 173. Harris PW, Williams GM, Shepherd P, Brimble MA. The synthesis of phosphopeptides using microwave‐assisted solid phase peptide synthesis. Int. J. Pept. Res. Ther. 2008; 14: 387. [Google Scholar]
- 174. Vorherr T, Bannwarth W. Phospho‐serine and phospho‐threonine building blocks for the synthesis of phosphorylated peptides by the Fmoc solid phase strategy. Bioorg. Med. Chem. Lett. 1995; 5: 2661. [Google Scholar]
- 175. Attard TJ, O'Brien‐Simpson NM, Reynolds EC. Identification and suppression of β‐elimination byproducts arising from the use of Fmoc‐Ser (PO3Bzl, H)‐OH in peptide synthesis. Int. J. Pept. Res. Ther. 2009; 15: 69. [Google Scholar]
- 176. Burke TRJ, Smyth MS, Otaka A, Nomizu M, Roller PP, Wolf G, Case R, Shoelson SE. Nonhydrolyzable phosphotyrosyl mimetics for the preparation of phosphatase‐resistant SH2 domain inhibitors. Biochemistry 1994; 33: 6490. [DOI] [PubMed] [Google Scholar]
- 177. Engel R. Phosphonates as analogues of natural phosphates. Chem. Rev. 1977; 77: 349. [Google Scholar]
- 178. Berkowitz DB, Shen Q, Maeng J‐H. Synthesis of the (α,α‐difluoroalkyl) phosphonate analogue of phosphoserine. Tetrahedron Lett. 1994; 35: 6445. [Google Scholar]
- 179. Ruiz M, Ojea V, Shapiro G, Weber H‐P, Pombo‐Villar E. Asymmetric synthesis of a protected phosphonate isostere of phosphothreonine for solid‐phase peptide synthesis. Tetrahedron Lett. 1994; 35: 4551. [Google Scholar]
- 180. Berkowitz DB, Eggen M, Shen Q, Shoemaker RK. Ready access to fluorinated phosphonate mimics of secondary phosphates. Synthesis of the (α,α‐difluoroalkyl) phosphonate analogues of l‐phosphoserine, l‐phosphoallothreonine, and l‐phosphothreonine. J. Org. Chem. 1996; 61: 4666. [DOI] [PubMed] [Google Scholar]
- 181. Burke TR, Jr , Smyth MS, Nomizu M, Otaka A, Roller PR. Preparation of fluoro‐and hydroxy‐4‐(phosphonomethyl)‐d,l‐phenylalanine suitably protected for solid‐phase synthesis of peptides containing hydrolytically stable analogs of O‐phosphotyrosine. J. Org. Chem. 1993; 58: 1336. [Google Scholar]
- 182. Burke TR, Smyth MS, Otaka A, Roller PP. Synthesis of 4‐phosphono (difluoromethyl)‐d,l‐phenylalanine and N‐Boc and N‐Fmoc derivatives suitably protected for solid‐phase synthesis of nonhydrolyzable phosphotyrosyl peptide analogues. Tetrahedron Lett. 1993; 34: 4125. [Google Scholar]
- 183. Liu W‐Q, Olszowy C, Bischoff L, Garbay C. Enantioselective synthesis of (2S)‐2‐(4‐phosphonophenylmethyl)‐3‐aminopropanoic acid suitably protected for peptide synthesis. Tetrahedron Lett. 2002; 43: 1417. [Google Scholar]
- 184. Schenkels C, Erni B, Reymond J‐L. Phosphofurylalanine, a stable analog of phosphohistidine. Bioorg. Med. Chem. Lett. 1999; 9: 1443. [DOI] [PubMed] [Google Scholar]
- 185. Yang SH, Lee DJ, Brimble MA. Synthesis of an NDPK phosphocarrier domain peptide containing a novel triazolylalanine analogue of phosphohistidine using click chemistry. Org. Lett. 2011; 13: 5604. [DOI] [PubMed] [Google Scholar]
- 186. Kee J‐M, Villani B, Carpenter LR, Muir TW. Development of stable phosphohistidine analogues. J. Am. Chem. Soc. 2010; 132: 14327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Domchek SM, Auger KR, Chatterjee S, Burke TR, Jr , Shoelson SE. Inhibition of SH2 domain/phosphoprotein association by a nonhydrolyzable phosphonopeptide. Biochemistry 1992; 31: 9865. [DOI] [PubMed] [Google Scholar]
- 188. Burke TR, Jr . Design and synthesis of phosphonodifluoromethyl phenylalanine (F2Pmp): a useful phosphotyrosyl mimetic. Curr. Top. Med. Chem. 2006; 6: 1465. [DOI] [PubMed] [Google Scholar]
- 189. Burke TR, Jr . Development of Grb2 SH2 domain signaling antagonists: a potential new class of antiproliferative agents. Int. J. Pept. Res. Ther. 2006; 12: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Attwood P, Piggott M, Zu X, Besant P. Focus on phosphohistidine. Amino Acids 2007; 32: 145. [DOI] [PubMed] [Google Scholar]
- 191. McAllister TE, Webb ME. Triazole phosphohistidine analogues compatible with the Fmoc‐strategy. Org. Biomol. Chem. 2012; 10: 4043. [DOI] [PubMed] [Google Scholar]
- 192. McAllister TE, Horner KA, Webb ME. Evaluation of the interaction between phosphohistidine analogues and phosphotyrosine binding domains. ChemBioChem 2014; 15: 1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. McAllister TE, Nix MG, Webb ME. Fmoc‐chemistry of a stable phosphohistidine analogue. Chem. Commun. 2011; 47: 1297. [DOI] [PubMed] [Google Scholar]
- 194. Kee J‐M, Muir TW. Chasing phosphohistidine, an elusive sibling in the phosphoamino acid family. ACS Chem. Biol. 2011; 7: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Kee J‐M, Oslund RC, Perlman DH, Muir TW. A pan‐specific antibody for direct detection of protein histidine phosphorylation. Nature Chem. Biol. 2013; 9: 416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Bertran‐Vicente J, Schümann M, Schmieder P, Krause E, Hackenberger CPR. Direct access to site‐specifically phosphorylated‐lysine peptides from a solid‐support. Org. Biomol. Chem. 2015; 13: 6839. [DOI] [PubMed] [Google Scholar]
- 197. Yates LM, Marmelstein AM, Fiedler D. Synthetic pyrophosphorylation methods and their use in chemical Biology. Synlett 2014; 25: 2239. [Google Scholar]
- 198. Wu M, Chong LS, Capolicchio S, Jessen HJ, Resnick AC, Fiedler D. Elucidating diphosphoinositol polyphosphate function with nonhydrolyzable analogues. Angew. Chem. 2014; 126: 7320. [DOI] [PubMed] [Google Scholar]
- 199. Seibert C, Sakmar TP. Toward a framework for sulfoproteomics: synthesis and characterization of sulfotyrosine‐containing peptides. Pept. Sci. 2008; 90: 459. [DOI] [PubMed] [Google Scholar]
- 200. Sasaki N. Current status and future prospects for research on tyrosine sulfation. Curr. Pharm. Biotech. 2012; 13: 2632. [DOI] [PubMed] [Google Scholar]
- 201. Kehoe JW, Bertozzi CR. Tyrosine sulfation: a modulator of extracellular protein–protein interactions. Chem. Biol. 2000; 7: R57. [DOI] [PubMed] [Google Scholar]
- 202. Ludeman JP, Stone MJ. The structural role of receptor tyrosine sulfation in chemokine recognition. Br. J. Pharmacol. 2014; 171: 1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Moore KL. The biology and enzymology of protein tyrosine O‐sulfation. J. Biol. Chem. 2003; 278: 24243. [DOI] [PubMed] [Google Scholar]
- 204. Stone ML, Payne RJ. Homogenous sulfopeptides and sulfoproteins: synthetic approaches and applications to characterize the effects of tyrosine sulfation on biochemical function. Acc. Chem. Res. 2015; 48: 2251. [DOI] [PubMed] [Google Scholar]
- 205. Musiol HJ, Escherich A, Moroder L, in Houben‐Weyl Methods of Organic Synthesis, Synthesis of Peptides and Peptidomimetics, Vol. E22b (Eds.: Goodman M, Felix A, Moroder L, Toniolo C.), Georg Thieme, Stuttgart, 2002, pp. 424. [Google Scholar]
- 206. Young T, Kiessling LL. A strategy for the synthesis of sulfated peptides. Angew. Chem. 2002; 114: 3599. [DOI] [PubMed] [Google Scholar]
- 207. Bunschoten A, Kruijtzer JA, Ippel JH, de Haas CJ, van Strijp JA, Kemmink J, Liskamp RM. A general sequence independent solid phase method for the site specific synthesis of multiple sulfated‐tyrosine containing peptides. Chem. Commun. 2009; 21: 2999. [DOI] [PubMed] [Google Scholar]
- 208. Peters C, Wagner M, Völkert M, Waldmann H. Bridging the gap between cell biology and organic chemistry: chemical synthesis and biological application of lipidated peptides and proteins. Naturwissenschaften 2002; 89: 381. [DOI] [PubMed] [Google Scholar]
- 209. Novelli G, D'Apice MR. Protein farnesylation and disease. J. Inherit. Metab. Dis. 2012; 35: 917. [DOI] [PubMed] [Google Scholar]
- 210. Naider FR, Becker JM. Synthesis of prenylated peptides. Biopolymers 1997; 43: 3. [DOI] [PubMed] [Google Scholar]
- 211. Kragol G, Lumbierres M, Palomo JM, Waldmann H. Protein synthesis: solid‐phase synthesis of lipidated peptides. Angew. Chem. Int. Ed. 2004; 43: 5839. [DOI] [PubMed] [Google Scholar]
- 212. Pegoraro S, Moroder L, in Houben‐Weyl Methods of Organic Synthesis, Synthesis of Peptides and Peptidomimetics, Vol. E22b (Eds.: Goodman M, Felix A, Moroder L, Toniolo C.), Georg Thieme, Stuttgart, 2002, pp. 333. [Google Scholar]
- 213. O'Reilly N, Charbin A, Lopez‐Serra L, Uhlmann F. Facile synthesis of budding yeast a‐factor and its use to synchronize cells of α mating type. Yeast 2012; 29: 233. [DOI] [PubMed] [Google Scholar]
- 214. Kadereit D, Deck P, Heinemann I, Waldmann H. Acid‐labile protecting groups for the synthesis of lipidated peptides. Chem. Eur. J. 2001; 7: 1184. [DOI] [PubMed] [Google Scholar]
- 215. Millington CR, Quarrell R, Lowe G. Aryl hydrazides as linkers for solid phase synthesis which are cleavable under mild oxidative conditions. Tetrahedron Lett. 1998; 39: 7201. [Google Scholar]
- 216. Diaz‐Rodriguez V, Mullen DG, Ganusova E, Becker JM, Distefano MD. Synthesis of peptides containing C‐terminal methyl esters using trityl side‐chain anchoring: application to the synthesis of a‐factor and a‐factor analogs. Org. Lett. 2012; 14: 5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Paik WK, Paik DC, Kim S. Historical review: the field of protein methylation. Trends Biochem. Sci. 2007; 32: 146. [DOI] [PubMed] [Google Scholar]
- 218. Tian X, Fang J. Current perspectives on histone demethylases. Acta Biochim. Biophys. Sin. 2007; 39: 81. [DOI] [PubMed] [Google Scholar]
- 219. Clarke SG. Protein methylation at the surface and buried deep: thinking outside the histone box. Trends Biochem. Sci. 2013; 38: 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol. Cell 2009; 33: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005; 6: 838. [DOI] [PubMed] [Google Scholar]
- 222. Chirivi RG, Jenniskens GJ, Raats JM. Anti‐citrullinated protein antibodies as novel therapeutic drugs in rheumatoid arthritis. J. Clin. Cell Immunol. 2013; 4: 2. [Google Scholar]
- 223. Dalziel M, Crispin M, Scanlan CN, Zitzmann N, Dwek RA. Emerging principles for the therapeutic exploitation of glycosylation. Science 2014; 343: 1235681. [DOI] [PubMed] [Google Scholar]
- 224. Culyba EK, Price JL, Hanson SR, Dhar A, Wong C‐H, Gruebele M, Powers ET, Kelly JW. Protein native‐state stabilization by placing aromatic side chains in N‐glycosylated reverse turns. Science 2011; 331: 571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Piontek C, Varón Silva D, Heinlein C, Pöhner C, Mezzato S, Ring P, Martin A, Schmid FX, Unverzagt C. Semisynthesis of a homogeneous glycoprotein enzyme: ribonuclease C: part 2. Angew. Chem. Int. Ed. 2009; 48: 1941. [DOI] [PubMed] [Google Scholar]
- 226. Aussedat B, Vohra Y, Park PK, Fernández‐Tejada A, Alam SM, Dennison SM, Jaeger FH, Anasti K, Stewart S, Blinn JH. Chemical synthesis of highly congested gp120 V1V2 N‐glycopeptide antigens for potential HIV‐1‐directed vaccines. J. Am. Chem. Soc. 2013; 135: 13113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Jamsson AM, Hilaire PMS, Meldal M, in Houben‐Weyl Methods of Organic Synthesis, Synthesis of Peptides and Peptidomimetics, Vol. E22b (Eds.: Goodman M, Felix A, Moroder L, Toniolo C.), Georg Thieme, Stuttgart, 2002, pp. 235. [Google Scholar]
- 228. Unverzagt C, Kajihara Y. Chemical assembly of N‐glycoproteins: a refined toolbox to address a ubiquitous posttranslational modification. Chem. Soc. Rev. 2013; 42: 4408. [DOI] [PubMed] [Google Scholar]
- 229. Wang L‐X, Amin MN. Chemical and chemoenzymatic synthesis of glycoproteins for deciphering functions. Chem. Biol. 2014; 21: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Okamoto R, Mandal K, Ling M, Luster AD, Kajihara Y, Kent SB. Total chemical synthesis and biological activities of glycosylated and non‐glycosylated forms of the chemokines CCL1 and Ser‐CCL1. Angew. Chem. 2014; 126: 5288. [DOI] [PubMed] [Google Scholar]
- 231. Wilson RM, Stockdill JL, Wu X, Li X, Vadola PA, Park PK, Wang P, Danishefsky SJ. A fascinating journey into history: exploration of the world of isonitriles en route to complex amides. Angew. Chem. Int. Ed. 2012; 51: 2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Sharon N, Lis H, in Glycosciences: Status & Perspectives (Eds.: Gabius H‐J, Gabius S.), Chapman& Hall, Weinheim, 1997, pp. 133. [Google Scholar]
- 233. Sjölin P, Elofsson M, Kihlberg J. Removal of acyl protective groups from glycopeptides: base does not epimerize peptide stereocenters, and β‐elimination is slow. J. Org. Chem. 1996; 61: 560. [DOI] [PubMed] [Google Scholar]
- 234. Takano Y, Kojima N, Nakahara Y, Hojo H, Nakahara Y. Solid‐phase synthesis of core 2 O‐linked glycopeptide and its enzymatic sialylation. Tetrahedron 2003; 59: 8415. [Google Scholar]
- 235. Leppänen A, Mehta P, Ouyang Y‐B, Ju T, Helin J, Moore KL, van Die I, Canfield WM, McEver RP, Cummings RD. A novel glycosulfopeptide binds to P‐selectin and inhibits leukocyte adhesion to P‐selectin. J. Biol. Chem. 1999; 274: 24838. [DOI] [PubMed] [Google Scholar]
- 236. Galan MC, Benito‐Alifonso D, Watt GM. Carbohydrate chemistry in drug discovery. Org. Biomol. Chem. 2011; 9: 3598. [DOI] [PubMed] [Google Scholar]
- 237. Otvos L, Jr , Wroblewski K, Kollat E, Perczel A, Hollosi M, Fasman G, Ertl H, Thurin J. Coupling strategies in solid‐phase synthesis of glycopeptides. Pept. Res. 1988; 2: 362. [PubMed] [Google Scholar]
- 238. Otvos L, Urge L, Hollosi M, Wroblewski K, Graczyk G, Fasman GD, Thurin J. Automated solid‐phase synthesis of glycopeptides. Incorporation of unprotected mono‐and disaccharide units of N‐glycoprotein antennae into T cell epitopic peptides. Tetrahedron Lett. 1990; 31: 5889. [Google Scholar]
- 239. Cohen‐Anisfeld ST, Lansbury PT, Jr . A practical, convergent method for glycopeptide synthesis. J. Am. Chem. Soc. 1993; 115: 10531. [Google Scholar]
- 240. Vetter D, Tumelty D, Singh SK, Gallop MA. A versatile solid‐phase synthesis of N‐linked glycopeptides. Angew. Chem. Int. Ed. 1995; 34: 60. [Google Scholar]
- 241. Hojo H, Kwon Y, Kakuta Y, Tsuda S, Tanaka I, Hikichi K, Aimoto S. Development of a linker with an enhanced stability for the preparation of peptide thioesters and its application to the synthesis of a stable‐isotope‐labelled HU‐type DNA‐binding protein. Bull. Chem. Soc. Jpn. 1993; 66: 2700. [Google Scholar]
- 242. Hackeng TM, Griffin JH, Dawson PE. Protein synthesis by native chemical ligation: expanded scope by using straightforward methodology. Proc. Natl. Acad. Sci. 1999; 96: 10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Clippingdale AB, Barrow CJ, Wade JD. Peptide thioester preparation by Fmoc solid phase peptide synthesis for use in native chemical ligation. J. Pept. Sci. 2000; 6: 225. [DOI] [PubMed] [Google Scholar]
- 244. Ingenito R, Bianchi E, Fattori D, Pessi A. Solid phase synthesis of peptide C‐terminal thioesters by Fmoc/t‐Bu chemistry. J. Am. Chem. Soc. 1999; 121: 11369. [Google Scholar]
- 245. Hiedler P, Link A. N‐Acyl‐N‐alkyl‐sulfonamide anchors derived from Kenner's safety catch linker: powerful tools in bioorganic and medicinal chemistry. Biorg. Med. Chem. 2005; 13: 585. [DOI] [PubMed] [Google Scholar]
- 246. Mende F, Beisswenger M, Seitz O. Automated Fmoc‐based solid‐phase synthesis of peptide thioesters with self‐purification effect and application in the construction of immobilized SH3 domains. J. Am. Chem. Soc. 2010; 132: 11110. [DOI] [PubMed] [Google Scholar]
- 247. Macmillan D, Bertozzi CR. Modular assembly of glycoproteins: towards the synthesis of GlyCAM‐1 by using expressed protein ligation. Angew. Chem. 2004; 116: 1379. [DOI] [PubMed] [Google Scholar]
- 248. Flavell RR, Huse M, Goger M, Trester‐Zedlitz M, Kuriyan J, Muir TW. Efficient semisynthesis of a tetraphosphorylated analogue of the type I TGFβ receptor. Org. Lett. 2002; 4: 165. [DOI] [PubMed] [Google Scholar]
- 249. Burlina F, Morris C, Behrendt R, White P, Offer J. Simplifying native chemical ligation with an N‐acylsulfonamide linker. Chem. Commun. 2012; 48: 2579. [DOI] [PubMed] [Google Scholar]
- 250. Blanco‐Canosa JB, Dawson PE. An efficient Fmoc‐SPPS approach for the generation of thioester peptide precursors for use in native chemical ligation. Angew. Chem. Int. Ed. 2008; 47: 6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Pascal R, Chauvey D, Sola R. Carboxyl‐protecting groups convertible into activating groups. Carbamates of O‐aminoanilides are precursors of reactive N‐acyl ureas. Tetrahedron Lett. 1994; 35: 6291. [Google Scholar]
- 252. Pascal R, Sola R, Labéguère F, Jouin P. Synthesis of the novel amino acid 4‐amino‐3‐(aminomethyl) benzoic acid (AmAbz) and its protected derivatives as building blocks for pseudopeptide synthesis. Eur. J. Org. Chem. 2000; 2000: 3755. [Google Scholar]
- 253. Blanco‐Canosa JB, Nardone B, Albericio F, Dawson PE. Chemical protein synthesis using a second‐generation N‐acyl urea linker for the preparation of peptide‐thioester precursors. J. Am. Chem. Soc. 2015; 137: 7197. [DOI] [PubMed] [Google Scholar]
- 254. Meienhofer J, in The Peptides Analysis, Synthesis, Biology, Vol. 1 (Eds.: Meienhofer J, Gross E.), Academic Press: NY, 1979, pp. 197. [Google Scholar]
- 255. Zheng J‐S, Tang S, Huang Y‐C, Liu L. Development of new thioester equivalents for protein chemical synthesis. Acc. Chem. Res. 2013; 46: 2475. [DOI] [PubMed] [Google Scholar]
- 256. Zheng J‐S, Tang S, Qi Y‐K, Wang Z‐P, Liu L. Chemical synthesis of proteins using peptide hydrazides as thioester surrogates. Nat. Protoc. 2013; 8: 2483. [DOI] [PubMed] [Google Scholar]
- 257. Reif A, Siebenhaar S, Tröster A, Schmälzlein M, Lechner C, Velisetty P, Gottwald K, Pöhner C, Boos I, Schubert V. Semisynthesis of biologically active glycoforms of the human cytokine interleukin 6. Angew. Chem. Int. Ed. 2014; 53: 12125. [DOI] [PubMed] [Google Scholar]
- 258. Siman P, Karthikeyan SV, Nikolov M, Fischle W, Brik A. Convergent chemical synthesis of histone H2B protein for the site‐specific ubiquitination at Lys34. Angew. Chem. Int. Ed. 2013; 52: 8059. [DOI] [PubMed] [Google Scholar]
- 259. Adams AL, Cowper B, Morgan RE, Premdjee B, Caddick S, Macmillan D. Cysteine promoted C‐terminal hydrazinolysis of native peptides and proteins. Angew. Chem. Int. Ed. 2013; 52: 13062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Kang J, Richardson JP, Macmillan D. 3‐Mercaptopropionic acid‐mediated synthesis of peptide and protein thioesters. Chem. Commun. 2009; 4: 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Offer J. Native chemical ligation with Nα acyl transfer auxiliaries. Pept. Sci. 2010; 94: 530. [DOI] [PubMed] [Google Scholar]
- 262. Kawakami T, Sumida M, Vorherr T, Aimoto S. Peptide thioester preparation based on an N–S acyl shift reaction mediated by a thiol ligation auxiliary. Tetrahedron Lett. 2005; 46: 8805. [Google Scholar]
- 263. Szabó J, Vinkler E, Varga I. Chemical properties of 4H‐benzo [e]‐1,3‐thiazine derivatives. I. Hydrolysis of 2‐phenyl‐6,7‐dimethoxy‐4H‐benzo [e]‐1,3‐thiazine. Acta Chim. Acad. Sci. Hung. 1968; 58: 179. [Google Scholar]
- 264. Nakamura K, Kanao T, Uesugi T, Hara T, Sato T, Kawakami T, Aimoto S. Synthesis of peptide thioesters via an N–S acyl shift reaction under mild acidic conditions on an N‐4, 5‐dimethoxy‐2‐mercaptobenzyl auxiliary group. J. Pept. Sci. 2009; 15: 731. [DOI] [PubMed] [Google Scholar]
- 265. Hojo H, Onuma Y, Akimoto Y, Nakahara Y, Nakahara Y. N‐Alkyl cysteine‐assisted thioesterification of peptides. Tetrahedron Lett. 2007; 48: 25. [Google Scholar]
- 266. Kang J, Macmillan D. Peptide and protein thioester synthesis via N → S acyl transfer. Org. Biomol. Chem. 2010; 8: 1993. [DOI] [PubMed] [Google Scholar]
- 267. Botti P, Villain M, Manganiello S, Gaertner H. Native chemical ligation through in situ O to S acyl shift. Org. Lett. 2004; 6: 4861. [DOI] [PubMed] [Google Scholar]
- 268. Johnson EC, Kent SB. Insights into the mechanism and catalysis of the native chemical ligation reaction. J. Am. Chem. Soc. 2006; 128: 6640. [DOI] [PubMed] [Google Scholar]
- 269. Ollivier N, Dheur J, Mhidia R, Blanpain A, Melnyk O. Bis (2‐sulfanylethyl) amino native peptide ligation. Org. Lett. 2010; 12: 5238. [DOI] [PubMed] [Google Scholar]
- 270. Boll E, Drobecq H, Ollivier N, Blanpain A, Raibaut L, Desmet R, Vicogne J, Melnyk O. One‐pot chemical synthesis of small ubiquitin‐like modifier protein–peptide conjugates using bis (2‐sulfanylethyl) amido peptide latent thioester surrogates. Nat. Protoc. 2015; 10: 269. [DOI] [PubMed] [Google Scholar]
- 271. Tsuda S, Shigenaga A, Bando K, Otaka A. N → S acyl‐transfer‐mediated synthesis of peptide thioesters using anilide derivatives. Org. Lett. 2009; 11: 823. [DOI] [PubMed] [Google Scholar]
- 272. Sato K, Shigenaga A, Tsuji K, Tsuda S, Sumikawa Y, Sakamoto K, Otaka A. N‐Sulfanylethylanilide peptide as a crypto‐thioester peptide. ChemBioChem 1840; 2011: 12. [DOI] [PubMed] [Google Scholar]
- 273. Sato K, Shigenaga A, Kitakaze K, Sakamoto K, Tsuji D, Itoh K, Otaka A. Chemical synthesis of biologically active monoglycosylated GM2‐activator protein analogue using N‐sulfanylethylanilide peptide. Angew. Chem. Int. Ed. 2013; 125: 8009. [DOI] [PubMed] [Google Scholar]
- 274. Cowper B, Shariff L, Chen W, Gibson SM, Di W‐L, Macmillan D. Expanding the scope of N → S acyl transfer in native peptide sequences. Org. Biomol. Chem. 2015; 13: 7469. [DOI] [PubMed] [Google Scholar]
- 275. Terrier VP, Adihou H, Arnould M, Delmas AF, Aucagne V. A straightforward method for automated Fmoc‐based synthesis of bio‐inspired peptide crypto‐thioesters. Chem. Sci 2015. DOI: 10.1039/C5SC02630J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Ruff Y, Garavini V, Giuseppone N. Reversible native chemical ligation: a facile access to dynamic covalent peptides. J. Am. Chem. Soc. 2014; 136: 6333. [DOI] [PubMed] [Google Scholar]
- 277. Yu HM, Chen ST, Wang KT. Enhanced coupling efficiency in solid‐phase peptide synthesis by microwave irradiation. J. Org. Chem. 1992; 57: 4781. [Google Scholar]
- 278. Pedersen SL, Tofteng AP, Malik L, Jensen KJ. Microwave heating in solid‐phase peptide synthesis. Chem. Soc. Rev. 2012; 41: 1826. [DOI] [PubMed] [Google Scholar]
- 279. Echalier C, Al‐Halifa S, Kreiter A, Enjalbal C, Sanchez P, Ronga L, Puget K, Verdié P, Amblard M, Martinez J. Heating and microwave assisted SPPS of C‐terminal acid peptides on trityl resin: the truth behind the yield. Amino Acids 2013; 45: 1395. [DOI] [PubMed] [Google Scholar]
- 280. Yang X, Lin H, Lu W, Wang D. Compatibility study of Merrifield linker in Fmoc strategy peptide synthesis. Prot. Pept. Lett. 2013; 20: 140. [DOI] [PubMed] [Google Scholar]
- 281. Loffredo C, Assuncao NA, Gerhardt J, Miranda M. Microwave‐assisted solid‐phase peptide synthesis at 60 °C: alternative conditions with low enantiomerization. J. Pept. Sci. 2009; 15: 808. [DOI] [PubMed] [Google Scholar]
- 282. Bacsa B, Horváti K, Bõsze S, Andreae F, Kappe CO. Solid‐phase synthesis of difficult peptide sequences at elevated temperatures: a critical comparison of microwave and conventional heating technologies. J. Org. Chem. 2008; 73: 7532. [DOI] [PubMed] [Google Scholar]
- 283. Bacsa B, Bősze S, Kappe CO. Direct solid‐phase synthesis of the β‐amyloid (1−42) peptide using controlled microwave heating. J. Org. Chem. 2010; 75: 2103. [DOI] [PubMed] [Google Scholar]
- 284. Roodbeen R, Pedersen SL, Hosseini M, Jensen KJ. Microwave heating in the solid‐phase synthesis of N‐methylated peptides: when is room temperature better? Eur. J. Org. Chem. 2012; 2012: 7106. [Google Scholar]
- 285. Nissen F, Kraft TE, Ruppert T, Eisenhut M, Haberkorn U, Mier W. Hot or not—the influence of elevated temperature and microwave irradiation on the solid phase synthesis of an affibody. Tetrahedron Lett. 2010; 51: 6216. [Google Scholar]
- 286. Tofteng AP, Jensen KJ, Schäffer L, Hoeg‐Jensen T. Total synthesis of desB30 insulin analogues by biomimetic folding of single‐chain precursors. ChemBioChem 2008; 9: 2989. [DOI] [PubMed] [Google Scholar]
- 287. Marek P, Woys AM, Sutton K, Zanni MT, Raleigh DP. Efficient microwave‐assisted synthesis of human islet amyloid polypeptide designed to facilitate the specific incorporation of labeled amino acids. Org. Lett. 2010; 12: 4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Harris PWR, Kowalczyk R, Hay DL, Brimble MA. A single pseudoproline and microwave solid phase peptide synthesis facilitates an efficient synthesis of human amylin 1–37. Int. J. Pept. Res. Ther. 2013; 19: 147. [Google Scholar]