

Abstract
Adiponectin (Ad) is an insulin-sensitizing adipocytokine with anti-inflammatory and vasoprotective properties. Cleavage of native full-length Ad (fAd) by elastases from activated monocytes generates globular Ad (gAd). Increased gAd levels are observed in the proximity of atherosclerotic lesions, but the physiological meaning of this proteolytic Ad fragment in the cardiovascular system is controversial. We compared molecular and biological properties of fAd and gAd in human aortic endothelial cells (HAEC). In control HAEC, both fAd and gAd acutely stimulated nitric oxide (NO) production by AMPK-dependent pathways. With respect to fAd, gAd more efficiently increased activation of NF-κB signaling pathways, resulting in cyclooxygenase-2 (COX-2) overexpression and COX-2-dependent prostacyclin 2 (PGI2) release. In contrast with fAd, gAd also increased p38 MAPK phosphorylation and VCAM-1 expression, ultimately enhancing adhesion of monocytes to endothelial cells. In HAEC lacking AdipoR1 (by siRNA), both activation of NF-κB as well as COX-2 overexpression by gAd were abrogated. Conversely, gAd-mediated p38MAPK activation and VCAM-1 expression were unaffected, and monocyte adhesion was greatly enhanced. In HAEC lacking COX-2 (by siRNA), reduced levels of PGI2 further increased gAd-dependent monocyte adhesion. Our findings suggest that biological activities of fAd and gAd in endothelium do not completely overlap, with gAd possessing both AdipoR1-dependent ability to stimulate COX-2 expression and AdipoR1-independent effects related to expression of VCAM-1 and adhesion of monocytes to endothelium.
Keywords: vascular cell adhesion molecule-1, adiponectin receptor 1, nuclear factor-κB, atherosclerosis, cyclooxygenase-2, endothelial function
over the last two decades, results from basic and clinical research have suggested that the insulin-sensitizing adiponectin (Ad) is more than an innocent bystander and a convenient marker for cardiovascular integrity. In obesity and insulin-resistant states, circulating levels of Ad correlate inversely with increased risk of ischemic heart diseases and atherosclerosis (18, 20).
The anti-inflammatory and antiatherogenic potential of Ad reported in experimental and human studies (23, 33, 41) is driven by multiple signaling mechanisms. At the endothelial level, Ad stimulates nitric oxide (NO) production following activation of phosphatidylinositol 3-kinase (PI3K) and AMP-activated protein kinase (AMPK)-dependent pathways (5, 13). Inhibition of TNFα-mediated NF-κB activation accounts for Ad-dependent decrease of leukocyte adhesion in TNFα-inflamed microvascular networks (34). Cyclooxygenase-2 (COX-2) signaling mechanisms are required for Ad protection from cardiac ischemia-reperfusion injury (17, 40) as well as for revascularization of ischemic muscle (31).
Nevertheless, the many potential benefits of Ad have currently been questioned by a number of studies indicating that increased Ad levels may predict increased cardiovascular mortality in high-risk populations such as patients with chronic heart failure (4, 21), coronary disease (37), or chronic kidney disease (27). In addition, Ad activates NF-κB in adipose tissue (25), osteoblasts (16), prostate cancer cells (43), and human hepatocytes (52).
Native full-length Ad (fAd) circulates in plasma as trimer, hexamer, and multimeric complexes that are likely to differ in signaling effects and biological activity (12, 36, 39, 46, 49). Cleavage of fAd by proteases secreted from activated monocytes and/or neutrophils generates globular Ad (gAd) fragment (50), which accumulates in vascular walls when the endothelial barrier is injured (32).
The common assumption that fAd and gAd share overlapping properties has been challenged by the observation that gAd activates proinflammatory transduction mechanisms in monocytes (14) as well as in vascular endothelial cells (11). Since pathophysiological relevance of these effects is still under debate (8), an extensive reassessment of fAd and gAd profile is necessary to understand whether these two molecules may differ in signaling and biological activities in the vascular compartment. This is particularly important since the replenishment of Ad might provide a novel treatment modality for insulin resistance and type 2 diabetic patients, and gAd would be easier to manufacture and administer than fAd (15).
In this study, we analyzed molecular and cellular properties of fAd and gAd in human aortic endothelial cells (HAEC). We found that gAd is more effective than fAd to enhance the expression of COX-2 and increase the levels of prostacyclin (PGI2). These effects were mediated by NF-κB signaling pathways and dependent on adiponectin receptor 1 (AdipoR1). In contrast with fAd, only gAd activated p38 MAPK pathways and augmented synthesis and release of adhesion molecules, including E-selectin, monocyte chemoattractant protein-1 (MCP-1), and vascular cell adhesion molecule-1 (VCAM-1), with no significant difference between control HAEC and HAEC lacking AdipoR1. Increased expression of adhesion molecules by gAd correlates with enhanced adhesion of monocytes to endothelial cells. Of note, abrogation of AdipoR1 or inhibition of COX-2 expression with subsequent reduction of PGI2 levels further increases both gAd-mediated VCAM-1 expression and gAd-dependent monocyte adhesion to endothelial cells.
MATERIALS AND METHODS
Reagents.
Pharmacological inhibitors (E)-3-(4-methylphenylsulphonyl)-2-propenenenitrile (BAY 11-7082), 4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)1H-imidazole (SB-203580), 2′-amino-3′-methoxyflavone (PD-98059), and N-(2-cyclohexyloxy-4-nitrophenyl) methanesulfonamide (NS-398) were purchased from Alexis Biochemical (San Diego, CA). Tumor necrosis factor-α (TNFα) was from R & D Systems Europe (Lille, France). Recombinant human full-length adiponectin (cat. no. ALX-522-063) expressed in human embryonic kidney was from Alexis Biochemical. Recombinant human globular adiponectin (cat. no. 450-21) expressed in E. coli was from PeproTech EC (London, UK). Purity of recombinant proteins was ≥98% (by SDS-PAGE and HPLC analyses), and endotoxin levels were <0.2 ng/μg for both gAd and fAd.
Cell cultures.
HAEC (Lonza, Basel, Switzerland) in primary culture were grown in EGM-2 (Lonza) and used between passages 3 and 4. At confluence, HAEC were stimulated with fAd (10 μg/ml), gAd (10 μg/ml), and TNFα (10 ng/ml) for various time intervals in the absence and in the presence of the NF-κB inhibitor BAY 11-7082 (20 μM), p38 MAPK inhibitor SB-203580 (10 μM), MEK inhibitor PD-98059 (20 μM), or COX-2 inhibitor NS-398 (20 μM). Potential endotoxin contamination in recombinant adiponectin-mediated effects was excluded by incubation of HAEC with the LPS inhibitor polimixin B (PMB; 10 μg/ml, cat no. P1004; Sigma-Aldrich) (6).
Human monocyte U937 (ATCC, Rockville, MD) was grown in RPMI medium 1640 (ATCC) containing 10% FBS and 1% penicillin-streptomycin (Euroclone, Milan, Italy).
Western blot analysis.
Activation of p42/44 MAPK, p38 MAPK, and NF-κB pathways was assessed using prototypical proinflammatory cytokine TNFα as positive control. Phosphorylation at Thr202/Tyr204 and at Thr180/Tyr182 was considered an indicator of activation for p42/44 and p38 MAPK, respectively. Phosphorylation of IκBα at Ser32/36, as well as phosphorylation of p65 subunit at Ser536, was assumed as an indicator for NF-κB activation. Cell lysates were prepared using 300 μl of lysis buffer (100 mM NaCl, 40 mM HEPES, pH 7.5, 1% Triton X-100, 1 mM Na3VO4, 4 mM Na4P2O7, 10 mM EDTA, 1 mM PMSF, 10 mM NaF, 2 μg/ml aprotinin, and 2 μg/ml leupeptin). Equal amounts of protein (25 μg) were separated by 10% SDS-PAGE and subjected to immunoblotting with the following primary antibodies (dilution 1:1,000): α-COX-2, α-p65, α-ph-p65, α-VCAM-1 (Santa Cruz Biotecnology, Santa Cruz, CA); α-IκBα, α-ph-IkBα, α-ERK1/2, α-ph-ERK1/2, α-p38 MAPK, and α-ph-p38 MAPK, (Cell Signaling Technology). The β-actin antibody was from Sigma. Incubation with horseradish peroxidase-linked anti-mouse, anti-rabbit, and anti-goat secondary antibodies (Santa Cruz Biotecnology) (1: 3,000) was performed for 1 h at room temperature. Immunoblotting results were visualized by Molecular Imager ChemiDoc XRS System (Bio-Rad Laboratories). Images were captured with QuantityOne Software (Bio-Rad Laboratories) and blots quantified by scanning densitometry (Image J; National Institutes of Health, Bethesda, MD).
Short interfering RNA transfection.
Nontargeting (scrambled) and human AdipoR1 short interfering RNA (siRNA; forward: 5′-ACCAUGCUCAGACCAAAUAUGUACT-3′; reverse: 3′-ACUGGUACGAGUCUGGUUUAUACAUGA-5′; Integrated DNA Technologies) or human COX-2 siRNA (forward: 5′-GGCUAAUACUGAUAGGAG-3′; reverse: 3′-AUAGUCUCUCCUAUCAGU-5′; Integrated DNA Technologies) were prepared in 20 μM stocks. HAECs were transfected with Lipofectamine (Invitrogen) according to the manufacturer's protocol. After 48 h, HAEC were stimulated with fAd and gAd for 8 h and subjected to monocyte adhesion assay, as described below. In analogous experiments, HAEC were stimulated with fAd and gAd and lysates analyzed for COX-2 and VCAM-1 expression.
RNA extraction and RT-PCR.
For RT-PCR, 1 μg of total RNA (Fast Pure RNA kit; Takara) was reversely transcribed into cDNA using oligo(dT) primer (Takara) and PrimeScript Reverse Transcriptase (Takara). Equal amounts of each reverse-transcribed cDNA were amplified (Takara Taq), with β-actin as internal control. The following primers (all obtained from Eurofins MWG Operon, Ebersberg, Germany) were used: human (h)COX-2: foward 5′-CAAGACAGATCATAAGCGAG, reverse 5′-CAGTTTGAAGTGATAGCCAC; hVCAM-1: forward 5′-GAATCTACAGCACCTTTCTG, reverse 5′-TTCAATCTCCAGCCGGTCAA; β-actin: forward 5′-ACGAGGCCCAGAGCAAGAGA, reverse 5′-AAGGTAGTTTCGTGGATGCC. The amplified cDNA fragments were resolved on a 2% (wt/vol) agarose gel and detected using Molecular Imager ChemiDoc XRS System (Bio-Rad Laboratories). Images were captured with QuantityOne Software (Bio-Rad Laboratories).
Measurement of PGI2, thromboxane B2, VCAM-1, E-selectin, and MCP-1 proteins.
Conditioned media from HAEC under basal conditions or stimulated with fAd, gAd, and TNFα without or with pretreatment with NS-398 were collected. Protein concentrations of 6-keto-prostaglandin F1α (6-keto-PGF-1α) and thromboxane B2 (TXB2) were determined by EIA kit (Cayman Chemical, Ann Arbor, MI) according to the manufacturer's protocols.
Protein concentrations of VCAM-1, E-selectin, and MCP-1 were determined in conditioned media or whole cell lysates by ELISA kit (Boster Biological Technology) according to the manufacturer's protocol.
Adhesion assay.
HAEC were left untreated or stimulated with TNFα, fAd, or gAd in the presence or absence of p38 MAPK inhibitor SB-203580, COX-2 inhibitor NS-398, Ad-neutralizing antibody (Alexis Biochemical), or VCAM-1-neutralizing antibody (R & D Systems). In some experiments, HAEC stimulated with TNFα were cotreated with fAd or gAd. In other experiments, HAEC transfected with nontargeting siRNA, human AdipoR1 siRNA, or human COX-2 siRNA were left untreated or stimulated with TNFα, fAd, or gAd.
Human monocytes U937 were labeled with CellTrace Calcein Green AM (Molecular Probes, Invitrogen) and then incubated for 1 h with HAEC. Nonadherent cells were removed and monolayers fixed in 1% paraformaldehyde (PFA). Interaction was calculated by the ratio between the number of fluorescent monocytes over the number of HAEC per field. VCAM-1 cell distribution was analyzed in HAEC unstimulated or treated with gAd and TNFα and incubated with α-VCAM-1. Confocal scans were taken in both the xy and xz planes for all the fields examined under basal and stimulated conditions.
Statistical analysis.
Two-way ANOVA for repeated measurements followed by post hoc Student's t-test for paired or unpaired data was used as appropriate. Values of P < 0.05 were considered to indicate statistical significance. Results are expressed as means ± SE of at least 3 independent experiments for each condition.
RESULTS
gAd (but not fAd) activates p38 MAPK and NF-κB signaling pathways in endothelial cells.
Molecular weight (MW) is ∼30 kDa for fAd and 20 kDa for the truncated form of gAd, respectively (7, 35). To assess molecular weight and oligomerization status of fAd and gAd under our conditions, 50 μl of conditioned media (CM) collected from HAEC stimulated with gAd or fAd was loaded on a 12% SDS-nonreducing PAGE, and membranes were subsequently probed with α-Ad antibody. As expected, and consistent with fAd's ability to form trimers, a 90-kDa MW band was found in the CM from fAd-treated HAEC (Fig. 1A). Conversely, a 19-kDa MW band was detected in the CM from HAEC treated with gAd, suggesting that this fragment maintains a monomeric conformation in our experimental conditions. As reported previously (5, 13), stimulation with both fAd or gAd acutely increased phosphorylation levels of Akt and AMPK and stimulated NO production in HAEC (Fig. 1, B and C). Thus, gAd and fAd share similar ability to activate PI3K- and AMPK-dependent signaling related to production of NO in HAEC. Compared with basal levels, phosphorylation of p42/44 MAPK, p38 MAPK, p65, and IκBα proteins was increased significantly in HAEC treated with TNFα (Fig. 2, A and C). Acute treatment with either fAd or gAd also increased phosphorylation of p42/44 MAPK but did not significantly change phosphorylated forms of p38 MAPK or NF-κB components (Fig. 2A).
Fig. 1.
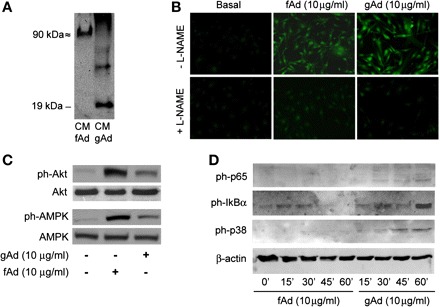
Overlapping and distinct effects of globular (gAd) and full-length adiponectin (fAd) in human aortic endothelial cells (HAEC). A: electrophoresis mobility and oligomerization status of gAd and fAd. Fifty microliters of conditioned media (CM) collected from HAEC treated with fAd or gAd (10 μg/ml, 1 h) was loaded on a 12% nonreducing SDS-PAGE, and membranes were probed with anti-adiponectin antibody. A 90-kDa molecular mass band was found in the CM of fAd-treated HAEC cells. Conversely, a 19-kDa molecular mass band was detected in HAEC treated with gAd. B: HAEC were serum starved (3 h) and loaded with the nitric oxide (NO)-specific fluorescent dye 4,5-diaminofluorescein diacetate (3 μM; Cayman Chemical, Ann Arbor, MI) in the absence or presence of NO synthase inhibitor NG-nitro-l-arginine methyl ester (l-NAME; 100 μM). HAEC were then stimulated with fAd or gAd (10 mg/ml, 10 min), fixed in 2% paraformaldehyde (PFA) for 5 min at 4°C, and then viewed using an epifluorescent microscope. Emission of green light (510 nm) from cells excited at 480 nm is indicative of NO production. Representative images are shown for experiments that were repeated independently 3 times. C: lysates from HAEC stimulated with fAd or gAd (10 mg/ml, 10 min) were subjected to immunoblotting for phosphorylated (ph) and total forms of Akt and AMP-activated protein kinase (AMPK) proteins. Both fAd and gAd increased phosphorylation levels of Akt and AMPK. D: time course experiments (0, 15, 30, 45, and 60 min) indicate that increased phosphorylation of p65, IκBα, and p38 MAPK in response to treatment with gAd peaks after 1-h stimulation. Treatment with fAd did not significantly enhance phosphorylation levels for NF-κB or p38 MAPK at any of the time points considered.
Fig. 2.
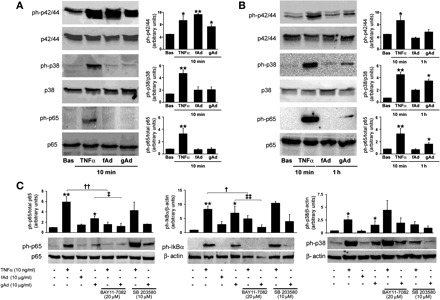
Differential activation of proinflammatory signaling by gAd and fAd in HAEC. HAEC cultured as described in materials and metthods were treated with TNFα (10 ng/ml), fAd (10 μg/ml), or gAd (10 μg/ml) for the indicated time points. HAEC lysates were subjected to immunoblotting with antibodies for total and phosphorylated forms of p42/44 MAPK, p38 MAPK, p65, and IκBα. A: representative immunoblots for results obtained in HAEC stimulated with TNFα (10 ng/ml), fAd (10 μg/ml), or gAd (10 μg/ml) for 10 min. Each bar represents the mean ± SE of densitometric analysis for phosphorylated proteins normalized to their respective total forms. B: representative immunoblots for results obtained in HAEC stimulated with TNFα (10 ng/ml) for 10 min and fAd (10 μg/ml) or gAd (10 μg/ml) for 1 h. Each bar represents the mean ± SE of densitometric analysis for phosphorylated proteins normalized to their respective total forms. C: experiments described in B were repeated in the absence or presence of NF-κB inhibitor BAY 11-7082 (20 μM, 1-h preincubation) or p38 MAPK inhibitor SB-203580 (10 μM, 1-h preincubation). Representative immunoblots from at least 3 independent experiments are shown for each condition. Each bar represents the mean ± SE of densitometric analysis for phosphorylated proteins normalized to their respective total forms. *P < 0.05, **P < 0.01 vs. basal (Bas) conditions; †P < 0.05, ††P < 0.01 vs. TNFα; ‡P < 0.05, ‡‡P < 0.01 vs. gAd.
Time course experiments (0, 10, 30, 45, and 60 min) indicate that TNFα-dependent activation of p42/44, p38 MAPK, and NF-κB signaling pathways peaks during the first 10 min and gradually returns to basal levels within 1 h (data not shown). In response to fAd, phosphorylation of IκBα and p65 was hardly noticeable; phosphorylation of p38 MAPK protein was undetectable (Fig. 1D). Conversely, when considered at indicated intervals, a progressive increase in phosphorylation levels of NF-κB and p38 MAPK proteins was observed in HAEC treated with gAd, with peaking effects at 60 min of stimulation (Fig. 1D). Thus, although both fAd and gAd are able to acutely activate p42/44, Akt, and AMPK signaling pathways, only gAd significantly phosphorylates p38 MAPK and NF-κB components in HAEC. However, compared with TNFα stimulation, a longer time is required for gAd to maximally activate these proinflammatory cascades. Therefore, rather than similar stimulation time, maximal activation of indicated signaling pathways was considered to compare TNFα-, fAd-, and gAd-mediated effects in HAEC (Fig. 2, B and C). Quantitative analysis of phosphorylation levels also reveals a different order of magnitude for TNFα- and gAd-mediated phosphorylation of p38 MAPK, IκBα, and p65 subunits (Fig. 2, B and C). No significant quantitative or qualitative differences in gAd concentrations were observed at time 0 or after 1 h incubation in CM from HAEC treated with gAd (data not shown). Furthermore, no significant increases in TNFα levels were measured in HAEC stimulated for 1 h with gAd (data not shown). Importantly, no significant change in activation of NF-κB or p38 MAPK signaling pathways in response to gAd was observed in HAEC pretreated with the LPS inhibitor PMB (10 μg/ml; data not shown) (6). These observations help to rule out the possibility that activation of signaling pathways observed in HAEC stimulated with gAd may not be dependent on biological properties of gAd itself.
NF-κB and p38 MAPK pathways are independently activated by gAd.
To clarify whether NF-κB and p38 MAPK signaling are independently activated by both TNFα and gAd in our conditions, experiments were repeated in the presence of the IKK inhibitor BAY 11-7082 or the p38 MAPK inhibitor SB-203580. Pretreatment with BAY 11-7082 significantly reduced phosphorylation of IκBα and p65 subunits (Fig. 2C) and inhibited nuclear translocation of p65 in gAd-treated HAEC (data not shown). Phosphorylation of p38 MAPK by TNFα or gAd was not changed under inhibition of NF-κB signaling (Fig. 2C). Phosphorylation levels of NF-κB were not changed under pretreatment with SB-203580 in HAEC stimulated with TNFα or gAd (Fig. 2C). Treatment with SB-203580 was not expected to reduce phosphorylated levels of p38 MAPK in response to both TNFα and gAd treatment, because SB-203580 inhibits p38 catalytic activity without affecting phosphorylation of p38 MAPK by upstream kinases (24). Thus, in our experimental conditions, phosphorylation of NF-κB and p38 MAPK seems to be independently triggered by gAd or TNFα. However, these findings do not exclude a downstream reciprocal modulation of NF-κB- and p38 MAPK-mediated signaling.
gAd induces overexpression of COX-2 and VCAM-1 by NF-κB and p38 MAPK pathways.
Consistent with other studies demonstrating Ad's ability to increase COX-2 expression in cardiomyocytes (40) and endothelial cells (31), mRNA levels of COX-2 were increased significantly in HAEC treated with TNFα, fAd, or gAd (Fig. 3A). Accordingly, levels of 6-keto-PGF-1α, the major prostanoid product of COX-2 in endothelial cells, were higher in response to gAd than to fAd (Fig. 3B). As shown in Fig. 3C, inhibition of NF-κB or p38 MAPK reduced COX-2 expression in response to TNFα or gAd, whereas no significant effect was observed when p42/44 MAPK was inhibited by pretreatment with PD-98059. Thus, in HAEC cells, gAd-mediated activation of NF-κB and p38 MAPK is associated with increased expression and function of COX-2. With respect to gAd, fAd was less effective in promoting COX-2 expression (Fig. 5C), although a slight increase in COX-2 protein was observed in HAEC treated with 30 μg/ml fAd for 8 h (Fig. 5C) or in HAEC treated with 10 μg/ml fAd for 24 h (data not shown). Similarly, activation of NF-κB signaling by 30 μg/ml fAd was still lower than activation promoted by 10 μg/ml gAd (data not shown).
Fig. 3.
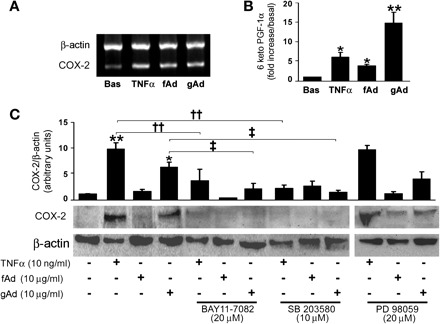
gAd induces overexpression of cyclooxygenase-2 (COX-2) by p38 MAPK and NF-κB pathways. A: mRNA levels of COX-2 detected in HAEC treated for 8 h with TNFα (10 ng/ml), fAd, or gAd (10 μg/ml). B: protein concentrations of 6-keto-prostaglandin F1α (6-keto-PGF-1α) in CM from HAEC treated as in A. C: levels of COX-2 protein expression in lysates of HAEC treated as in A in the absence or presence of NF-κB inhibitor BAY 11-7082 (20 μM, 1-h preincubation), p38 MAPK inhibitor SB-203580 (10 μM, 1-h preincubation), or p42/44 MAPK inhibitor PD-98059 (20 μM, 1-h preincubation). Representative immunoblots from at least 3 independent experiments are shown. Bar graphs indicate the mean ± SE of densitometric analysis for COX-2 protein normalized to β-actin expression. *P < 0.05, **P < 0.01 vs. basal; ††P < 0.01 vs. TNFα; ‡P < 0.05 vs. gAd.
Fig. 5.
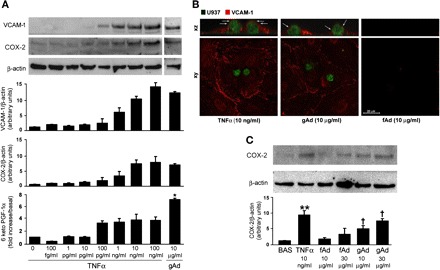
gAd- and TNFα-enhanced expression of COX-2 and VCAM-1 in HAEC. A: HAEC were left untreated or stimulated with TNFα (100 fg/ml to 100 ng/ml) or gAd (10 μg/ml) for 8 h. Lysates were subjected to immunoblotting for COX-2 or VCAM-1, and conditioned medium was analyzed for prostacyclin content. Representative immunoblots are shown for experiments that were independently repeated 3 times. B: confocal fluorescence images in the xy and xz planes showing immunolocalization of VCAM-1 protein (red fluorescence) and U937 human monocytes (green fluorescence) in mock transfected HAEC subjected to adhesion assay. Arrows in the xz scans indicate VCAM-1 protein surrounding monocytes at the level of plasma membrane. A similar pattern is shown for membrane-located VCAM-1 in HAEC stimulated with either 10 ng/ml TNFα or 10 μg/ml gAd. C: gAd- and fAd-dependent COX-2 expression. Representative blot shows protein levels of COX-2 in HAEC treated for 8 h with 10 and 30 μg/ml gAd and fAd. Bar graphs indicate the mean ± SE of densitometric analysis for VCAM-1 and COX-2 protein normalized to β-actin expression. *P < 0.05, **P < 0.01 vs. basal; †P < 0.05 vs. fAd.
A significant increase in mRNA and protein levels of VCAM-1 was measured in lysates (Fig. 4, A and C) and conditioned medium (Fig. 4B) from HAEC treated with either TNFα or gAd, but not with fAd. Likewise, significantly increased levels of E-selectin and MCP-1 were found in HAEC stimulated with TNFα or gAd (but not with fAd; Table 1). As for COX-2, no significant effect on VCAM-1 expression was observed when p42/44 MAPK pathway was inhibited (Fig. 4C). Conversely, pharmacological inhibition of NF-κB or p38 MAPK markedly decreased VCAM-1 expression in response to TNFα or gAd. These findings suggest that, despite a concomitant and independent phosphorylation of p65 and p38 MAPK subunits in response to gAd, a reciprocal modulation of NF-κB and p38 MAPK signaling cascades may occur downstream of early signaling events.
Fig. 4.
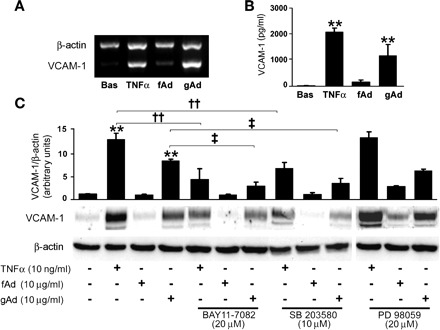
gAd induces overexpression of vascular cell adhesion molecule-1 (VCAM-1) by p38 MAPK and NF-κB pathways. A: mRNA levels of VCAM-1 detected in HAEC treated for 8 h with TNFα (10 ng/ml), fAd, or gAd (10 μg/ml). B: soluble VCAM-1 protein in CM from HAEC treated as in A. C: levels of VCAM-1 protein expression in lysates of HAEC treated as in A in the absence or presence of NF-κB inhibitor BAY 11-7082 (20 μM, 1-h preincubation), p38 MAPK inhibitor SB-203580 (10 μM, 1-h preincubation), or p42/44 MAPK inhibitor PD-98059 (20 μM, 1-h preincubation). Representative immunoblots from at least 3 independent experiments are shown. Bar graphs indicate the mean ± SE of densitometric analysis for VCAM-1 protein normalized to β-actin expression. **P < 0.01 vs. basal; ‡P < 0.05; ‡‡P < 0.01 vs. gAd.
Table 1.
VCAM-1, E-selectin, and MCP-1 concentration in HAEC lysates
| VCAM-1 | E-selectin | MCP-1 | |
|---|---|---|---|
| Basal | 4.6 ± 0.2 | 0.135 ± 0.02 | 0.06 ± 0.02 |
| TNFα (10 ng/ml) | 278.5 ± 49.6** | 85.5 ± 10.5** | 2.9 ± 0.10* |
| fAd (10 μg/ml) | 1.8 ± 0.3 | 0.2 ± 0.05 | 0.1 ± 0.002 |
| gAd (10 μg/ml) | 72.1 ± 11.2*§ | 20.2 ± 2.4*§ | 1.0 ± 0.07* |
Values are means ± SE of 4 independent experiments performed in duplicate and in ng/ml. VCAM-1, vascular cell adhesion molecule-1; MCP-1, monocyte chemoattractant protein-1; HAEC, human aortic endothelial cells; fAd, full-length adiponectin; gAd, globular adiponectin. Endothelial markers of atherogenesis were measured in cell lysates of HAEC under basal and stimulated conditions (8 h).
P < 0.05,
P < 0.01 vs. basal value;
P < 0.05 vs. TNFα
Interestingly, both VCAM-1 and COX-2 protein levels were comparable in HAEC treated with 10 ng/ml TNFα or 10 μg/ml gAd (Fig. 5A). Immunofluorescent analysis suggests that 10 ng/ml TNFα and 10 μg/ml gAd result in a similar pattern for membrane-located VCAM-1 in HAEC (Fig. 5B). Moreover, complete inclusion of monocytes surrounded by VCAM-1 protein was found at the apical membranes of HAEC stimulated with either TNFα or gAd, but not fAd (Fig. 5B). Nevertheless, PGI2 levels were significantly higher in HAEC stimulated with gAd than in HAEC treated with TNFα (Fig. 5A). These data suggest that, at doses able to induce a comparable VCAM-1 expression, gAd is significantly more effective than TNFα in enhancing COX-2 activity.
Increased VCAM-1 expression correlates with enhanced monocyte adhesion to endothelial cells.
To evaluate the pathophysiological significance of COX-2 and VCAM-1 overexpression in response to gAd, a monocyte/endothelial cell adhesion assay was carried out. In control HAEC, the number of adhering monocytes was increased significantly under TNFα and gAd treatment with respect to unstimulated conditions (Fig. 6, top). Consistent with involvement of p38 MAPK signaling in gAd-mediated VCAM-1 expression, pretreatment with SB-203580 substantially decreased gAd- and TNFα-mediated monocyte adhesion (Fig. 6, top middle). Conversely, pharmacological inhibition of COX-2 activity by NS-398 enhanced gAd- and TNFα-mediated monocyte adhesion (Fig. 6, middle). These effects were likely related to gAd ability to increase VCAM-1 expression, because pretreatment with VCAM-1-neutralizing antibody significantly reduced monocyte adherence in response to both TNFα and gAd (Fig. 6, bottom middle). As expected, pretreatment with adiponectin-neutralizing antibody abrogated gAd-mediated (but not to TNFα-mediated) monocyte adhesion (Fig. 6, bottom). Taken together, these results suggest that gAd ability to elicit monocyte adhesion depends on biological properties of gAd itself to increase VCAM-1 expression. Interestingly, cotreatment with either fAd or gAd greatly reduced monocyte adhesion in response to TNFα (data not shown). These results suggest that, despite its ability to increase monocyte adhesion, overall gAd-mediated effects may help to limit proadhesive effects of TNFα related to VCAM-1 expression.
Fig. 6.
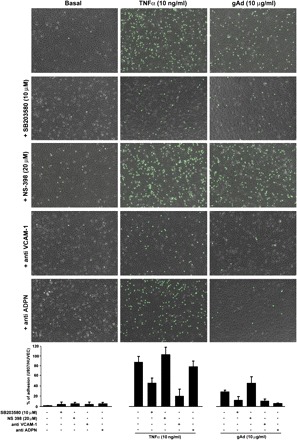
Increased VCAM-1 expression correlates with enhanced monocyte adhesion to endothelial cells. A: HAEC cultured as described in materials and methods were left untreated or stimulated for 8 h with TNFα (10 ng/ml) or gAd (10 μg/ml) in the absence (top) or presence of p38 inhibitor SB-203580, COX-2 inhibitor NS-398, VCAM-1-neutralizing antibody, or Ad-neutralizing antibody. HAEC were then incubated for 1 h with calcein green-labeled U937 human monocytes and fixed in 1% PFA. Overlapping of bright-field images with green fluorescent images shows U937 monocytes over HAEC for each tested condition. Bar graphs indicate the mean ± SE of U937/HAEC ratio for experiments independently repeated at least 3 times. ADPN, adiponectin.
Abrogation of AdipoR1 or COX-2 enhances VCAM-1-mediated monocyte adhesion in response to gAd.
Our RT-PCR analysis of AdipoR1/AdipoR2 relative expression demonstrates that the signal intensity of AdipoR1 is ∼4.5-fold greater than that of AdipoR2 in HAEC (data not shown). Since gAd differs from fAd in the ability to activate cellular signaling related to expression of COX-2 and VCAM-1, we then asked whether AdipoR1, the predominant adiponectin receptor in endothelium, would be involved in gAd effects. Interestingly, a further increase in monocyte adhesion was observed in HAEC transfected with AdipoR1 siRNA and stimulated with gAd (but not with TNFα; Fig. 7, A and B).
Fig. 7.
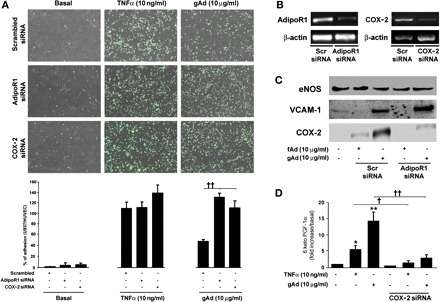
Abrogation of adiponectin receptor 1 (AdipoR1) or COX-2 enhances VCAM-1-mediated monocyte adhesion in response to gAd. A: HAEC cultured as described in materials and methods were transfected with scrambled siRNA, AdipoR1, or COX-2 siRNA. Forty-eight hours after transfection, HAEC were left untreated or stimulated for 8 h with TNFα (10 ng/ml) or gAd (10 μg/ml), incubated for 1 h with calcein green-labeled U937 human monocytes, and then fixed in 1% PFA. Overlapping of bright-field images with green fluorescent images shows U937 monocytes over HAEC for each tested condition. Bar graphs indicate the mean ± SE of U937/HAEC ratio for experiments independently repeated at least 3 times. B: mRNA levels of AdipoR1 and COX-2 detected in mock-transfected or specific siRNA-transfected HAEC under basal conditions and after stimulation with TNFα (10 ng/ml) or gAd (10 μg/ml). C: HAEC treated as described in A were stimulated with fAd (10 μg/ml) and gAd (10 μg/ml) for 8 h and lysates subjected to immunoblotting for endothelial nitric oxide synthase (eNOS; control), VCAM-1, or COX-2 expression. Representative immunoblots are shown for experiments that were independently repeated 3 times. D: protein concentrations of 6-keto-PGF-1α in the CM from mock-transfected and siCOX-2-transfected HAEC under basal conditions and after stimulation for 8 h with TNFα or gAd. Bar graphs indicate the mean ± SE of at least 3 independent experiments. *P < 0.05, **P < 0.01 vs. basal; †P < 0.05, ††P < 0.01 vs. mock transfected cells.
Consistent with increased monocyte adhesion, gAd-mediated VCAM-1 expression was not reduced but rather increased in HAEC lacking the AdipoR1 receptor (Fig. 7C). In parallel, gAd-mediated p38 MAPK phosphorylation was still comparable in HAEC transfected with scrambled or AdipoR1 siRNA (data not shown). Thus, these findings suggest that gAd's ability to increase the expression of VCAM-1 and activate p38 MAPK may be unrelated to AdipoR1 activation. Conversely, abrogation of AdipoR1 was associated with reduced gAd-mediated phosphorylation of both p65 and IκBα (data not shown) and with a substantial decrease of gAd-dependent COX-2 expression (Fig. 7C).
Finally, to further evaluate the role of COX-2-increased expression in response to gAd, adhesion experiments were repeated in HAEC transfected with COX-2 siRNA. Under these conditions, PGI2 levels were reduced, and the number of adhering monocytes was increased significantly in cells stimulated with either gAd or TNFα (Fig. 5, A–C). This is consistent with decreased PGI2 levels, increased levels of VCAM-1 protein, and increased monocyte adhesion observed in control HAEC pretreated with COX-2 inhibitor NS-398 and stimulated with either gAd or TNFα. Taken together, our findings suggest that the gAd's ability to increase COX-2 expression is dependent on AdipoR1 and that COX-2 activity is important to limit the grade of monocyte adhesion promoted by gAd itself via VCAM-1 upregulation.
DISCUSSION
Differences in the function for the various isoforms of Ad are currently under investigation (see Ref. 9 for review). The COOH-terminal fragment of Ad, known as globular Ad (gAd), is a proteolytic cleavage product of full-length Ad whose tissue distribution and physiological meaning in the cardiovascular district are still under debate.
This study provides novel evidence that, in contrast with the shared ability of fAd and gAd to acutely induce NO production and activate p42/44 MAPK and AMPK signaling, gAd is significantly more effective than fAd in activating NF-κB and p38 MAPK signaling in human endothelial cells. Of note, activation of these pathways results in increased expression of COX-2 and VCAM-1 proteins. Importantly, gAd-mediated activation of NF-κB signaling and subsequent upregulation of COX-2 are specifically mediated by activation of AdipoR1 receptor in HAEC cells. Conversely, gAd-mediated activation of p38 MAPK and expression of VCAM-1 do not require AdipoR1 activation. Increased VCAM-1 by gAd is related to increased monocyte adhesion to endothelial cells. In addition, we found that gAd-mediated upregulation of COX-2 with subsequently increased levels of PGI2 is important to counteract gAd-mediated VCAM-1-dependent endothelial binding of monocytes.
Consistent with findings in human monocytes (14), vascular endothelial cells (11), and cardiac fibroblasts (10), we show that gAd elicits activation of NF-κB and p38 MAPK in HAEC. These effects were not related to potential endotoxin contamination of recombinant adiponectin because pretreatment with LPS inhibitor polimixin B did not affect gAd-induced activation of NF-κB and p38 MAPK signaling pathways. Moreover, gAd-mediated monocyte adhesion was completely abrogated in HAEC pretreated with Ad-neutralizing antibody. In addition, degradation or unspecific enzymatic cleavage of gAd may be excluded, since equal amounts of gAd were resolved on SDS-PAGE from conditioned media of HAEC incubated with gAd for 10 min or 1 h. Similarly, activation of NF-κB and p38 MAPK could not be ascribed to gAd-induced de novo synthesis of TNFα, because no significant increases in TNFα concentration were detected from HAEC stimulated for 1 h with gAd compared with basal levels. Therefore, it is reasonable to infer that activation of signaling pathways observed in HAEC stimulated with gAd is dependent on biological properties of gAd itself.
In our experiments, gAd-mediated activation of NF-κB is dependent on the AdipoR1 and delayed with respect to activation of the p42/44 MAPK or AMPK/endothelial nitric oxide synthase (eNOS) pathways. Both AMPK activity and NO release are important for controlling NF-κB transcription activity (3), because activation of NF-κB is significantly enhanced by inhibitors of AMPK or inhibitors of NO production (12, 51). The gAd-mediated AMPK activation and increased NO production are maximal after 10 min and then rapidly decline. Conversely, activation of NF-κB in response to gAd increases progressively during the first hour. Thus, one possibility to explain the belated activation of NF-κB signaling in response to gAd may involve the earlier and transient activation of AMPK/eNOS signaling. Alternatively, gAd-dependent activation of NF-κB may be related to the mechanism by which gAd binds to the AdipoR1 receptor and to the events that occur after binding. In this regard, AdipoR1 receptor interaction may facilitate the transactivation of NF-kB, since antibodies against the extracellular domain of AdipoR1 strongly activate NF-κB (10). Interestingly, Ad may influence dimerization of the AdipoR1 by segments of the collagen-like domain (22), which is cleaved off in the gAd fragment.
As mentioned in results, acute p38 MAPK phosphorylation in response to gAd does not seem to depend on NF-κB signaling in HAEC, and pharmacological inhibition of p38 MAPK blunts VCAM-1 expression as well as monocyte adhesion mediated by gAd. This observation suggests that p38 MAPK signaling is required for gAd-mediated VCAM-1 overexpression in HAEC cells. Interestingly, both gAd-mediated p38 MAPK activation and VCAM-1 upregulation do not appear to depend on activation of AdipoR1 receptor in our experiments. Previous reports have shown that, in addition to AdipoR1, other signaling molecules, including calreticulin or its adaptor protein CD91, may mediate certain Ad effects in endothelial cells (31). Other studies on Ad's ability to modulate endothelial progenitor cell migration have failed to associate PI3K/Cdc42/Rac1 signaling with AdipoR1/R2 binding (29). Further experiments are needed to explore which signaling molecules are upstream gAd-mediated p38 MAPK and VCAM-1 activation in HAEC. The possibility that gAd effects might depend, at least partially, on AdipoR2 activation cannot be completely ruled out from our experiments. Nevertheless, the preferential binding of gAd to AdipoR1, the low expression of AdipoR2 in endothelial cells, and the very same proinflammatory nature of these biological effects suggest that the involvement of AdipoR2 in gAd-dependent VCAM-1 expression may be, if any, of limited importance. At present, our preliminary results seem to rule out the potential role of TNFα receptors in these effects. Altogether, our data strongly suggest that the unique ability of gAd to increase VCAM-1-mediated proadhesive effects is independent on AdipoR1 activation.
Previous reports indicate that gAd accumulates in the arterial wall preferentially in places of endothelial injury (namely, the same places where VCAM-1 is expressed) (32). We found that gAd-induced VCAM-1 was associated with increased monocyte adhesion in HAEC. Interestingly, striking similarities were observed between TNFα- and gAd-dependent VCAM-1's ability to surround and encompass adhering monocytes. These findings are reminiscent of the first step of monocyte diapedesis in vivo and underline the potential proadhesive effects of gAd in endothelium. Although our results are in agreement with investigations describing the ability of gAd to increase mRNA levels of VCAM-1, intracellular adhesion molecule-1, E-selectin, and MCP-1 in endothelial cells (19, 45), alternative explanations are possible; for example, by acting as a partial agonist, gAd may limit VCAM-1 expression levels elicited by TNFα and therefore protect the vascular wall from subsequent atherosclerotic lesions (48). This hypothesis is supported by the observation that TNFα-mediated monocyte adhesion is reduced significantly by concomitant treatment with gAd in our experiments. On the other hand, the enhanced monocyte adhesion promoted by gAd may help to explain the positive association between VCAM-1 and Ad levels in patients with type 1 diabetes (38) or with manifest cardiovascular diseases (48). In this regard, the observation that biological properties of gAd are quite different from fAd may be important for the characterization of risk/benefit profile of recombinant adiponectin in view of its possible use as replacement therapy.
We found that both fAd and gAd increased endothelial COX-2 expression, with gAd-mediated upregulation of COX-2 dependent on AdipoR1 and NF-κB activation. Interestingly, gAd was significantly more effective than TNFα in enhancing COX-2 activity at doses that were able to induce comparable VCAM-1 expression. The stronger effect of gAd in overexpressing COX-2 with respect to fAd may be partially explained by the different oligomerization status of the globular fragment and depend on the higher binding affinity to AdipoR1 for gAd with respect to fAd (42, 53). Consistent with this, a more sustained COX-2 expression was observed in HAEC treated with fAd at higher concentrations or longer exposure time. In endothelial cells, upregulation of COX-2 and subsequently increased levels of PGI2 oppose impaired NO production at the onset of diabetic condition (28). In addition to PGI2, other mediators, including NO, may contribute to counteract monocyte adhesion to endothelial cells. Indeed, both gAd and fAd are able to acutely increase NO production in HAEC, whereas TNFα is known to impair physiological expression and activation of eNOS (47). In addition, both gAd (1) and TNFα (2) may increase inducible nitric oxide synthase expression and activity. Nevertheless, our findings strongly suggest that COX-2-dependent prostacyclin levels are central to counteract monocyte adhesion in HAEC. In this respect, we are the first to demonstrate the physiological relevance of COX-2 by gAd in endothelium.
Consistent with the important role of COX-2 activity in the Ad-mediated protection from ischemia-reperfusion injury (17, 31, 40), gAd-mediated monocyte adhesion was greatly enhanced under pharmacological inhibition of COX-2 or in HAEC lacking endogenous COX-2. Concomitantly, abrogation of AdipoR1 with subsequent inhibition of gAd-dependent COX-2 activity was accompanied by enhanced VCAM-1 expression.
In summary, the results obtained in this study suggest that biological effects of fAd and gAd in endothelium may impact differently on endothelial activation. Our data strongly imply that the globular form of adiponectin [whose levels are reportedly increased in the proximity of vascular lesions (50)] possesses both receptor-mediated and receptor-unrelated effects, with the former replicating the protective vascular effects of full-length adiponectin and the latter holding proatherogenic potential. Differences among various Ad oligomers as well as differences in their ability to activate cognate receptors may help to explain some controversies between studies underscoring the antiatherogenic potential of Ad (26, 54) and studies failing to detect a direct association between high levels of Ad and protection from atherosclerotic lesions (30, 44). Whether gAd-mediated effects might help to reduce the proatherogenic ability of other inflammatory cytokines remains to be clearly established.
GRANTS
This work was supported in part by the Juvenile Diabetes Research Foundation (CDA 2-2006-32; to M. Montagnani).
DISCLOSURES
The authors declare no conflict of interest.
REFERENCES
- 1. Akifusa S, Kamio N, Shimazaki Y, Yamaguchi N, Yamashita Y. Regulation of globular adiponectin-induced apoptosis by reactive oxygen/nitrogen species in RAW264 macrophages. Free Radic Biol Med 45: 1326–1339, 2008. [DOI] [PubMed] [Google Scholar]
- 2. Bogdan C. Nitric oxide and the immune response. Nat Immunol 2: 907–916, 2001. [DOI] [PubMed] [Google Scholar]
- 3. Cacicedo JM, Yagihashi N, Keaney JF, Jr, Ruderman NB, Ido Y. AMPK inhibits fatty acid-induced increases in NF-kappaB transactivation in cultured human umbilical vein endothelial cells. Biochem Biophys Res Commun 324: 1204–1209, 2004. [DOI] [PubMed] [Google Scholar]
- 4. Cavusoglu E, Ruwende C, Chopra V, Yanamadala S, Eng C, Clark LT, Pinsky DJ, Marmur JD. Adiponectin is an independent predictor of all-cause mortality, cardiac mortality, and myocardial infarction in patients presenting with chest pain. Eur Heart J 27: 2300–2309, 2006. [DOI] [PubMed] [Google Scholar]
- 5. Chen H, Montagnani M, Funahashi T, Shimomura I, Quon MJ. Adiponectin stimulates production of nitric oxide in vascular endothelial cells. J Biol Chem 278: 45021–45026, 2003. [DOI] [PubMed] [Google Scholar]
- 6. Fan D, Li L, Wang C, Cui XB, Zhou Y, Wu LL. Adiponectin induces interleukin-6 production and its underlying mechanism in adult rat cardiac fibroblasts. J Cell Physiol 226: 1793–1802, 2011. [DOI] [PubMed] [Google Scholar]
- 7. Fruebis J, Tsao TS, Javorschi S, Ebbets-Reed D, Erickson MR, Yen FT, Bihain BE, Lodish HF. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc Natl Acad Sci USA 98: 2005–2010, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goldstein BJ, Scalia R, Ma XL, Mahadev K, Wu X, Ouedraogo R. Comment on: Hattori et al. (2007) Globular adiponectin activates nuclear factor-kappaB and activating protein-1 and enhances angiotensin II-induced proliferation in cardiac fibroblasts: Diabetes 56:804–808. Diabetes 56: e7–e8; author reply e9–e10, 2007. [DOI] [PubMed] [Google Scholar]
- 9. Goldstein BJ, Scalia RG, Ma XL. Protective vascular and myocardial effects of adiponectin. Nat Clin Pract Cardiovasc Med 6: 27–35, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hattori Y, Hattori S, Akimoto K, Nishikimi T, Suzuki K, Matsuoka H, Kasai K. Globular adiponectin activates nuclear factor-kappaB and activating protein-1 and enhances angiotensin II-induced proliferation in cardiac fibroblasts. Diabetes 56: 804–808, 2007. [DOI] [PubMed] [Google Scholar]
- 11. Hattori Y, Hattori S, Kasai K. Globular adiponectin activates nuclear factor-kappaB in vascular endothelial cells, which in turn induces expression of proinflammatory and adhesion molecule genes. Diabetes Care 29: 139–141, 2006. [DOI] [PubMed] [Google Scholar]
- 12. Hattori Y, Nakano Y, Hattori S, Tomizawa A, Inukai K, Kasai K. High molecular weight adiponectin activates AMPK and suppresses cytokine-induced NF-kappaB activation in vascular endothelial cells. FEBS Lett 582: 1719–1724, 2008. [DOI] [PubMed] [Google Scholar]
- 13. Hattori Y, Suzuki M, Hattori S, Kasai K. Globular adiponectin upregulates nitric oxide production in vascular endothelial cells. Diabetologia 46: 1543–1549, 2003. [DOI] [PubMed] [Google Scholar]
- 14. Haugen F, Drevon CA. Activation of nuclear factor-kappaB by high molecular weight and globular adiponectin. Endocrinology 148: 5478–5486, 2007. [DOI] [PubMed] [Google Scholar]
- 15. Heiker JT, Klöting N, Blüher M, Beck-Sickinger AG. Access to gram scale amounts of functional globular adiponectin from E. coli inclusion bodies by alkaline-shock solubilization. Biochem Biophys Res Commun 398: 32–37, 2010. [DOI] [PubMed] [Google Scholar]
- 16. Huang CY, Lee CY, Chen MY, Tsai HC, Hsu HC, Tang CH. Adiponectin increases BMP-2 expression in osteoblasts via AdipoR receptor signaling pathway. J Cell Physiol 224: 475–483, 2010. [DOI] [PubMed] [Google Scholar]
- 17. Ikeda Y, Ohashi K, Shibata R, Pimentel DR, Kihara S, Ouchi N, Walsh K. Cyclooxygenase-2 induction by adiponectin is regulated by a sphingosine kinase-1 dependent mechanism in cardiac myocytes. FEBS Lett 582: 1147–1150, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev 26: 439–451, 2005. [DOI] [PubMed] [Google Scholar]
- 19. Kase H, Hattori Y, Jojima T, Okayasu T, Tomizawa A, Suzuki K, Banba N, Monden T, Satoh H, Akimoto K, Kasai K. Globular adiponectin induces adhesion molecule expression through the sphingosine kinase pathway in vascular endothelial cells. Life Sci 81: 939–943, 2007. [DOI] [PubMed] [Google Scholar]
- 20. Kawano T, Saito T, Yasu T, Nakamura T, Namai K, Tamemoto H, Kawakami M, Saito M, Ishikawa SE. Close association of hypoadiponectinemia with arteriosclerosis obliterans and ischemic heart disease. Metabolism 54: 653–656, 2005. [DOI] [PubMed] [Google Scholar]
- 21. Kistorp C, Faber J, Galatius S, Gustafsson F, Frystyk J, Flyvbjerg A, Hildebrandt P. Plasma adiponectin, body mass index, and mortality in patients with chronic heart failure. Circulation 112: 1756–1762, 2005. [DOI] [PubMed] [Google Scholar]
- 22. Kosel D, Heiker JT, Juhl C, Wottawah CM, Blüher M, Mörl K, Beck-Sickinger AG. Dimerization of adiponectin receptor 1 is inhibited by adiponectin. J Cell Sci 123: 1320–1328, 2010. [DOI] [PubMed] [Google Scholar]
- 23. Kubota N, Terauchi Y, Yamauchi T, Kubota T, Moroi M, Matsui J, Eto K, Yamashita T, Kamon J, Satoh H, Yano W, Froguel P, Nagai R, Kimura S, Kadowaki T, Noda T. Disruption of adiponectin causes insulin resistance and neointimal formation. J Biol Chem 277: 25863–25866, 2002. [DOI] [PubMed] [Google Scholar]
- 24. Kumar S, Jiang MS, Adams JL, Lee JC. Pyridinylimidazole compound SB 203580 inhibits the activity but not the activation of p38 mitogen-activated protein kinase. Biochem Biophys Res Commun 263: 825–831, 1999. [DOI] [PubMed] [Google Scholar]
- 25. Lappas M, Permezel M, Rice GE. Leptin and adiponectin stimulate the release of proinflammatory cytokines and prostaglandins from human placenta and maternal adipose tissue via nuclear factor-kappaB, peroxisomal proliferator-activated receptor-gamma and extracellularly regulated kinase 1/2. Endocrinology 146: 3334–3342, 2005. [DOI] [PubMed] [Google Scholar]
- 26. Luo N, Liu J, Chung BH, Yang Q, Klein RL, Garvey WT, Fu Y. Macrophage adiponectin expression improves insulin sensitivity and protects against inflammation and atherosclerosis. Diabetes 59: 791–799, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Menon V, Li L, Wang X, Greene T, Balakrishnan V, Madero M, Pereira AA, Beck GJ, Kusek JW, Collins AJ, Levey AS, Sarnak MJ. Adiponectin and mortality in patients with chronic kidney disease. J Am Soc Nephrol 17: 2599–2606, 2006. [DOI] [PubMed] [Google Scholar]
- 28. Nacci C, Tarquinio M, De Benedictis L, Mauro A, Zigrino A, Carratù MR, Quon MJ, Montagnani M. Endothelial dysfunction in mice with streptozotocin-induced type 1 diabetes is opposed by compensatory overexpression of cyclooxygenase-2 in the vasculature. Endocrinology 150: 849–861, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nakamura N, Naruse K, Matsuki T, Hamada Y, Nakashima E, Kamiya H, Matsubara T, Enomoto A, Takahashi M, Oiso Y, Nakamura J. Adiponectin promotes migration activities of endothelial progenitor cells via Cdc42/Rac1. FEBS Lett 583: 2457–2463, 2009. [DOI] [PubMed] [Google Scholar]
- 30. Nawrocki AR, Hofmann SM, Teupser D, Basford JE, Durand JL, Jelicks LA, Woo CW, Kuriakose G, Factor SM, Tanowitz HB, Hui DY, Tabas I, Scherer PE. Lack of association between adiponectin levels and atherosclerosis in mice. Arterioscler Thromb Vasc Biol 30: 1159–1165, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ohashi K, Ouchi N, Sato K, Higuchi A, Ishikawa TO, Herschman HR, Kihara S, Walsh K. Adiponectin promotes revascularization of ischemic muscle through a cyclooxygenase 2-dependent mechanism. Mol Cell Biol 29: 3487–3499, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Okamoto Y, Arita Y, Nishida M, Muraguchi M, Ouchi N, Takahashi M, Igura T, Inui Y, Kihara S, Nakamura T, Yamashita S, Miyagawa J, Funahashi T, Matsuzawa Y. An adipocyte-derived plasma protein, adiponectin, adheres to injured vascular walls. Horm Metab Res 32: 47–50, 2000. [DOI] [PubMed] [Google Scholar]
- 33. Okamoto Y, Kihara S, Ouchi N, Nishida M, Arita Y, Kumada M, Ohashi K, Sakai N, Shimomura I, Kobayashi H, Terasaka N, Inaba T, Funahashi T, Matsuzawa Y. Adiponectin reduces atherosclerosis in apolipoprotein E-deficient mice. Circulation 106: 2767–2770, 2002. [DOI] [PubMed] [Google Scholar]
- 34. Ouedraogo R, Gong Y, Berzins B, Wu X, Mahadev K, Hough K, Chan L, Goldstein BJ, Scalia R. Adiponectin deficiency increases leukocyte-endothelium interactions via upregulation of endothelial cell adhesion molecules in vivo. J Clin Invest 117: 1718–1726, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pajvani UB, Du X, Combs TP, Berg AH, Rajala MW, Schulthess T, Engel J, Brownlee M, Scherer PE. Structure-function studies of the adipocyte-secreted hormone Acrp30/adiponectin. Implications for metabolic regulation and bioactivity. J Biol Chem 2002. [DOI] [PubMed] [Google Scholar]
- 36. Pajvani UB, Hawkins M, Combs TP, Rajala MW, Doebber T, Berger JP, Wagner JA, Wu M, Knopps A, Xiang AH, Utzschneider KM, Kahn SE, Olefsky JM, Buchanan TA, Scherer PE. Complex distribution, not absolute amount of adiponectin, correlates with thiazolidinedione-mediated improvement in insulin sensitivity. J Biol Chem 279: 12152–12162, 2004. [DOI] [PubMed] [Google Scholar]
- 37. Pilz S, Mangge H, Wellnitz B, Seelhorst U, Winkelmann BR, Tiran B, Boehm BO, Marz W. Adiponectin and mortality in patients undergoing coronary angiography. J Clin Endocrinol Metab 91: 4277–4286, 2006. [DOI] [PubMed] [Google Scholar]
- 38. Schalkwijk CG, Chaturvedi N, Schram MT, Fuller JH, Stehouwer CD. Adiponectin is inversely associated with renal function in type 1 diabetic patients. J Clin Endocrinol Metab 91: 129–135, 2006. [DOI] [PubMed] [Google Scholar]
- 39. Schraw T, Wang ZV, Halberg N, Hawkins M, Scherer PE. Plasma adiponectin complexes have distinct biochemical characteristics. Endocrinology 149: 2270–2282, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shibata R, Sato K, Pimentel DR, Takemura Y, Kihara S, Ohashi K, Funahashi T, Ouchi N, Walsh K. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat Med 11: 1096–1103, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Takemura Y, Osuga Y, Koga K, Tajima T, Hirota Y, Hirata T, Morimoto C, Harada M, Yano T, Taketani Y. Selective increase in high molecular weight adiponectin concentration in serum of women with preeclampsia. J Reprod Immunol 73: 60–65, 2007. [DOI] [PubMed] [Google Scholar]
- 42. Takeuchi T, Adachi Y, Ohtsuki Y, Furihata M. Adiponectin receptors, with special focus on the role of the third receptor, T-cadherin, in vascular disease. Med Mol Morphol 40: 115–120, 2007. [DOI] [PubMed] [Google Scholar]
- 43. Tang CH, Lu ME. Adiponectin increases motility of human prostate cancer cells via adipoR, p38, AMPK, and NF-kappaB pathways. Prostate 69: 1781–1789, 2009. [DOI] [PubMed] [Google Scholar]
- 44. Thorp E, Kuriakose G, Shah YM, Gonzalez FJ, Tabas I. Pioglitazone increases macrophage apoptosis and plaque necrosis in advanced atherosclerotic lesions of nondiabetic low-density lipoprotein receptor-null mice. Circulation 116: 2182–2190, 2007. [DOI] [PubMed] [Google Scholar]
- 45. Tomizawa A, Hattori Y, Kasai K. Induction of gene expression in response to globular adiponectin in vascular endothelial cells. Life Sci 85: 457–461, 2009. [DOI] [PubMed] [Google Scholar]
- 46. Tsao TS, Tomas E, Murrey HE, Hug C, Lee DH, Ruderman NB, Heuser JE, Lodish HF. Role of disulfide bonds in Acrp30/adiponectin structure and signaling specificity. Different oligomers activate different signal transduction pathways. J Biol Chem 278: 50810–50817, 2003. [DOI] [PubMed] [Google Scholar]
- 47. Valerio A, Cardile A, Cozzi V, Bracale R, Tedesco L, Pisconti A, Palomba L, Cantoni O, Clementi E, Moncada S, Carruba MO, Nisoli E. TNF-alpha downregulates eNOS expression and mitochondrial biogenesis in fat and muscle of obese rodents. J Clin Invest 116: 2791–2798, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vaverkova H, Karasek D, Novotny D, Jackuliakova D, Halenka M, Lukes J, Frohlich J. Positive association of adiponectin with soluble vascular cell adhesion molecule sVCAM-1 levels in patients with vascular disease or dyslipidemia. Atherosclerosis 197: 725–731, 2008. [DOI] [PubMed] [Google Scholar]
- 49. Waki H, Yamauchi T, Kamon J, Ito Y, Uchida S, Kita S, Hara K, Hada Y, Vasseur F, Froguel P, Kimura S, Nagai R, Kadowaki T. Impaired multimerization of human adiponectin mutants associated with diabetes. Molecular structure and multimer formation of adiponectin. J Biol Chem 278: 40352–40363, 2003. [DOI] [PubMed] [Google Scholar]
- 50. Waki H, Yamauchi T, Kamon J, Kita S, Ito Y, Hada Y, Uchida S, Tsuchida A, Takekawa S, Kadowaki T. Generation of globular fragment of adiponectin by leukocyte elastase secreted by monocytic cell line THP-1. Endocrinology 146: 790–796, 2005. [DOI] [PubMed] [Google Scholar]
- 51. Wang C, Li L, Zhang ZG, Fan D, Zhu Y, Wu LL. Globular adiponectin inhibits angiotensin II-induced nuclear factor kappaB activation through AMP-activated protein kinase in cardiac hypertrophy. J Cell Physiol 222: 149–155, 2009. [DOI] [PubMed] [Google Scholar]
- 52. Wanninger J, Neumeier M, Weigert J, Bauer S, Weiss TS, Schaffler A, Krempl C, Bleyl C, Aslanidis C, Scholmerich J, Buechler C. Adiponectin-stimulated CXCL8 release in primary human hepatocytes is regulated by ERK1/ERK2, p38 MAPK, NF-κB, and STAT3 signaling pathways. Am J Physiol Gastrointest Liver Physiol 297: G611–G618, 2009. [DOI] [PubMed] [Google Scholar]
- 53. Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, Murakami K, Ohteki T, Uchida S, Takekawa S, Waki H, Tsuno NH, Shibata Y, Terauchi Y, Froguel P, Tobe K, Koyasu S, Taira K, Kitamura T, Shimizu T, Nagai R, Kadowaki T. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 423: 762–769, 2003. [DOI] [PubMed] [Google Scholar]
- 54. Zhang P, Wang Y, Fan Y, Tang Z, Wang N. Overexpression of adiponectin receptors potentiates the antiinflammatory action of subeffective dose of globular adiponectin in vascular endothelial cells. Arterioscler Thromb Vasc Biol 29: 67–74, 2009. [DOI] [PubMed] [Google Scholar]