

Abstract
The formation of functional amyloid must be carefully regulated to prevent the accumulation of potentially toxic products. Premelanosome protein (PMEL) forms non-toxic functional amyloid fibrils that assemble into sheets upon which melanins ultimately are deposited within the melanosomes of pigment cells. PMEL is synthesized in the endoplasmic reticulum but forms amyloid only within post-Golgi melanosome precursors; thus, PMEL must traverse the secretory pathway in a non-amyloid form. Here, we identified two pre-amyloid PMEL intermediates that likely regulate the timing of fibril formation. Analyses by non-reducing SDS-PAGE, size exclusion chromatography, and sedimentation velocity revealed two native high Mr disulfide-bonded species that contain Golgi-modified forms of PMEL. These species correspond to disulfide bond-containing dimeric and monomeric PMEL isoforms that contain no other proteins as judged by two-dimensional PAGE of metabolically labeled/immunoprecipitated PMEL and by mass spectrometry of affinity-purified complexes. Metabolic pulse-chase analyses, small molecule inhibitor treatments, and evaluation of site-directed mutants suggest that the PMEL dimer forms around the time of endoplasmic reticulum exit and is resolved by disulfide bond rearrangement into a monomeric form within the late Golgi or a post-Golgi compartment. Mutagenesis of individual cysteine residues within the non-amyloid cysteine-rich Kringle-like domain stabilizes the disulfide-bonded dimer and impairs fibril formation as determined by electron microscopy. Our data show that the Kringle-like domain facilitates the resolution of disulfide-bonded PMEL dimers and promotes PMEL functional amyloid formation, thereby suggesting that PMEL dimers must be resolved to monomers to generate functional amyloid fibrils.
Keywords: cysteine-mediated cross-linking, disulfide, endosome, fibril, melanogenesis, membrane trafficking, oxidation-reduction (redox), protein aggregation, Pmel17, melanosome
Introduction
Amyloid is a “cross-β-sheet” polymeric protein conformation in which β-strands are stacked perpendicular to the long axis of the amyloid fibril (1). The amyloid fold is traditionally associated with pathological protein misfolding in neurodegenerative diseases such as Alzheimer and Parkinson diseases (2). However, the amyloid fold has also been exploited for diverse physiological processes in a wide range of organisms (3) including biofilm formation in bacteria (4), learning and memory in Drosophila (5), and programmed necrosis in mammals (6). The molecular mechanisms that limit the toxicity of such “functional amyloids” or their folding intermediates during synthesis are incompletely understood but likely regulate when and where these proteins assemble into amyloid fibrils. For example, curli biofilm formation in Escherichia coli is regulated by the accessory proteins CsgB, CsgC, and CsgF. CsgB is a minor curli subunit required to nucleate amyloid fibril formation in vivo (7). CsgC is a chaperone protein that prevents premature amyloid formation in the bacterial periplasm (8), and CsgF is an extracellular protein important for the attachment of curli fibers to the cell surface (9). However, much less is known about functional amyloid regulation in mammals, where the formation of large, self-assembling, insoluble protein structures likely carries increased risk because of the multicellular nature and longer life span of higher organisms.
The best characterized mammalian functional amyloid protein is premelanosome protein (PMEL2; also referred to as Pmel17, Silver, ME20, or gp100). Within immature melanosomes of pigment cells in the skin and eye, PMEL polymerizes into amyloid fibrils (10) that associate laterally into sheets upon which melanins are deposited as they are synthesized during melanosome maturation (reviewed in Ref. 11). The amyloid sheets are critical determinants of the ellipsoid melanosome shape (12, 13), which is required for proper melanosome motility into the apical processes of retinal pigment epithelial cells (14). The amyloid sheets have also been proposed to accelerate melanin polymerization (10, 15, 16), and organisms that lack PMEL or carry mutations in the PMEL gene are characterized by various degrees of hypopigmentation (13, 17–21). Because PMEL is synthesized in the endoplasmic reticulum (ER) as a type I transmembrane protein (22–24) but only initiates amyloid fibril formation within the lumen of endosomal membrane compartments (25–27), PMEL must navigate the secretory pathway from the ER to endosomes in a non-amyloid form. It is thus an excellent model system by which to dissect the regulation of amyloid formation within the endomembrane system.
Our current understanding of the processes involved in PMEL biosynthesis and amyloidogenesis is as follows. Following synthesis and addition of four core N-linked glycans within the ER (generating a “P1” precursor form), PMEL is exported to the Golgi where it is modified extensively by O-glycosylation (28, 29) and maturation of the N-linked glycans to the complex type (26). This generates a full-length “P2” form that is subsequently cleaved by a proprotein convertase into two disulfide-linked fragments, Mα and Mβ (30), either in the trans-Golgi network (31) or within endosomes (32) (see Fig. 1A). A subsequent proteolytic cleavage by BACE2 within endosomes (33) or by an ADAM (a disintegrin and a metalloproteinase) protease at the plasma membrane (34) cleaves Mβ into two fragments; the C-terminal fragment (CTF) contains the transmembrane domain and is eventually degraded in a γ-secretase-dependent manner in lysosomes (35), whereas the other fragment (Mβ′) is liberated from the membrane covalently bound to Mα (33, 36). No longer anchored to the membrane, most of the Mα polymerizes into amyloid fibrils within endosomes (30, 37) and concomitantly or subsequently undergoes additional proteolytic processing events to form smaller fragments; some of these fragments, called Mα′, are derived from the central RPT domain and are detected within preparations of the parallel arrays of functional amyloid fibrils characteristic of early stage melanosomes (27, 29, 38). Proprotein convertase and BACE2 cleavage are necessary for functional amyloid fibril formation, as interference with either of these two proteolytic processing events results in the formation of disordered PMEL aggregates instead of ordered fibrils (30, 33). However, these cleavage events are not sufficient to induce fibril formation because a small fraction of cleaved PMEL is secreted from cells in culture (26, 36, 39) in a non-amyloid form.3 Therefore, additional regulatory mechanisms must govern PMEL fibril formation.
FIGURE 1.

Golgi-matured PMEL exists predominantly in high Mr disulfide-bonded species. A, schematic diagram of the mature PMEL primary structure showing the sites of antibody recognition for αPMEL-N, αPMEL-I, HMB45, and αPMEL-C (note that αPMEL-I recognizes only the immature form of PMEL lacking O-glycans), cleavage sites for a proprotein convertase (PC) and BACE2 (red arrows), and resulting cleavage fragments Mα, Mβ, Mβ′, and CTF. Also noted are the domain structure, N-linked glycosylation sites (green triangles; double triangles indicate sites that are modified to the complex type in the Golgi), O-linked glycosylation sites (blue clovers), and cysteine residues (black circles). The N-terminal region (NTR), a region with homology to a repeated domain in polycystin 1 (PKD), a highly glycosylated RPT domain, and the KLD are indicated in light gray, and the transmembrane domain (TM) is indicated in blue. B–E, MNT-1 melanoma cells (MNT, lanes 1 and 3) or transiently transfected HeLa cells (lanes 2 and 4) expressing wild-type PMEL were lysed in buffer containing 1% (w/v) Triton X-100, and the soluble material was fractionated by SDS-PAGE under non-reducing (NR) or reducing (R) conditions. Proteins transferred to membranes were then probed with the PMEL antibodies HMB45 (B), αPMEL-I (C), αPMEL-N (D), or αPMEL-C (E). The migration of molecular weight standards is indicated to the right of each blot. Bands corresponding to p250, p160, p160*, Mα+Mβ′, Mα, immature PMEL conformers (cfmrs), and P1 are indicated by arrows. Note that Mα and P1 comigrate in MNT-1 cells and that free Mβ was run off the end of these gels. *, an alternatively spliced PMEL product (PMEL-ss). The lane numbers are indicated at the bottom.
Regulated disulfide bond rearrangements have the potential to affect both protein conformation and protein function. For example, interaction of the β-barrel protein LptD with the LptE subunit of the LPS translocon in the E. coli outer membrane triggers a disulfide bond rearrangement within LptD that activates the translocon (40). PMEL has 13 cysteine residues (Fig. 1A, black circles), of which 11 are exposed to the lumen of the endomembrane system and thus might participate in disulfide bonds. Indeed, the existence of intermolecular disulfide bonds can be inferred from data showing that P2 and nonfibrillar Mα and Mβ fragments are incorporated into higher order oligomers when samples are analyzed by non-reducing SDS-PAGE (26). Here we have shown that disulfide bonds maintain PMEL in a tethered dimeric form and that resolution of the dimer requires the integrity of a cysteine-rich Kringle-like domain (KLD) that is outside of the amyloidogenic region. Mutagenesis of this region blocks dimer resolution and impairs amyloidogenesis. Our data support a model in which the generation and destruction of a disulfide-stabilized dimer contributes an additional layer of regulation to the timing of PMEL amyloid formation.
Experimental Procedures
Antibodies and Reagents
Unless otherwise specified, chemicals were obtained from Sigma-Aldrich and tissue culture reagents from Life Technologies. Protease inhibitors were purchased from Roche Diagnostics, gene amplification primers from Integrated DNA Technologies (Coralville, IA), GoTaq DNA polymerase from Promega Corp. (Madison, WI), and restriction enzymes and T4 DNA ligase from New England Biolabs (Ipswich, MA). HMB45, a mouse monoclonal antibody to PMEL, was purchased from Enzo Life Sciences (catalogue No. ENZ-C34930; Farmingdale, NY). Rabbit polyclonal antibodies PMEL-I, PMEL-N, and PMEL-C were affinity-purified as described previously (26, 29, 41). The mouse monoclonal antibody NKI-beteb was obtained from Lab Vision (Fremont, CA), and H4A3 to LAMP1 (lysosome-associated membrane protein 1) was from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City). Secondary antibodies conjugated to Alexa Fluor 488 or Alexa Fluor 594 for immunofluorescence microscopy or to alkaline phosphatase for immunoblotting were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA). Secondary antibodies conjugated to IRDye 800CW or IRDye 680LT for immunoblotting were obtained from LI-COR Biotechnology (Lincoln, NE).
Cell Culture and Transfection Conditions
The highly pigmented MNT-1 human melanoma cell line was cultured as described previously (25). HeLa cells were cultured in DMEM supplemented with 10% FBS, transiently transfected using FuGENE 6 (Roche), FuGENE HD (Promega), or Xfect transfection reagent (Clontech Laboratories, Mountain View, CA), and analyzed 24–72 h post-transfection.
DNA Constructs and Cloning
Wild-type hPMEL (long form) and the cleavage site mutant ΔCS in the pCI vector (Promega) have been described previously (26, 30). The cysteine mutants were generated by site-directed mutagenesis using two-step gene amplification as described (42), and plasmid inserts were verified by DNA sequencing. The sequences of primers used for gene amplification and for the production of all mutants are available upon request.
Immunoblotting
Cells were harvested by centrifugation following release from plates with PBS, 5 mm EDTA, then treated with PBS, 50 mm N-ethylmaleimide (NEM), and then lysed (∼107 cells/ml) in lysis buffer (1% (w/v) Triton X-100, 150 mm sodium chloride, 0.02% (w/v) sodium azide, 10 mm HEPES pH 7.4, supplemented with protease inhibitors and 50 mm NEM). After centrifugation at 19,000 × g for 20 min, 1 volume of 6× SDS sample buffer (0.4 M Tris, pH 6.8, 12% (w/v) SDS, 34% glycerol, 0.02% (w/v) bromphenol blue) with or without 11% β-mercaptoethanol was added to 5 volumes of detergent-soluble lysate. Samples were then heated at 60 °C for 20 min, fractionated by SDS-PAGE on Tris-glycine gels containing 7, 10, or 12% acrylamide, and transferred electrophoretically to PVDF membranes. Membranes were blocked using blocking buffer (TBS, pH 7.4, and 0.2% Tween 20 with 5% (w/v) nonfat dry milk) or Odyssey blocking buffer (LI-COR Biotechnology), incubated with primary antibodies diluted in blocking buffer, and washed with TBST (TBS, pH 7.4, 0.2% Tween 20). In some experiments, membranes were subsequently incubated with secondary antibodies conjugated to alkaline phosphatase diluted in blocking buffer, washed with TBST, detected using enhanced chemifluorescence (GE Healthcare Life Sciences), and analyzed using a Molecular Dynamics Storm 860 Molecular Imager PhosphorImager (GE Healthcare Life Sciences). In other experiments, membranes were probed using secondary antibodies conjugated to IRDye-800CW or IRDye-680LT diluted in blocking buffer, washed with TBST, and analyzed using an Odyssey infrared imaging system (LI-COR Biotechnology). Quantification of bands was performed using the ImageJ gel analyzer tool (National Institutes of Health) or Image Studio Lite (LI-COR).
Sucrose Gradient Sedimentation
Cells treated with PBS, 50 mm NEM were lysed in fractionation buffer (150 mm sodium chloride, 0.02% (w/v) sodium azide, 100 mm Tris, pH 7.4, supplemented with protease inhibitor tablets, 50 mm NEM, and 20 mm iodoacetamide) containing 250 mm n-octylglucoside. The lysate was clarified by centrifugation at 19,000 × g for 15 min, layered on top of 5–20% sucrose gradients in 150 mm sodium chloride, 0.02% (w/v) sodium azide, 25 mm n-octylglucoside, 100 mm Tris, pH 8, and fractionated by sedimentation velocity as described (43). Briefly, gradients were subjected to ultracentrifugation in a Beckman L8–70 M Ultracentrifuge (Beckman Coulter Inc.) for 38 h at 30,000 rpm in an SW-41 rotor. Fractions were collected from bottom to top using a peristaltic pump, and aliquots of each fraction were analyzed by non-reducing SDS-PAGE and immunoblotting. Standard proteins (ovalbumin, rabbit IgG, bovine liver catalase, and/or bovine thyroid thyroglobulin) were fractionated on parallel gradients; these fractions were analyzed by reducing SDS-PAGE followed by staining with Coomassie Brilliant Blue R-250.
Size Exclusion Chromatography
Cells were lysed in fractionation buffer containing either 250 mm n-octylglucoside, 40 mm dodecyl-β-d-maltoside, or 4% (w/v) Triton X-100. After clarification at 19,000 × g for 15 min, detergent-soluble lysates were analyzed by size exclusion chromatography on an ÄKTAFPLC system (GE Healthcare Life Sciences) using a Superose 6 10/300 GL column (GE Healthcare Life Sciences) in running buffer (150 mm sodium chloride, 0.02% (w/v) sodium azide, 10 mm HEPES, pH 7.4) containing a lower concentration of the same detergent used for lysis, either 25 mm n-octylglucoside, 1 mm dodecyl-β-d-maltoside, or 0.1% (w/v) Triton X-100. The column was eluted at a flow rate of 0.4–0.45 ml/min with a fraction volume of 300 or 600 μl. Aliquots of each fraction were analyzed by non-reducing SDS-PAGE and immunoblotting. The elution volumes of thyroglobulin (669 kDa), α2-macroglobulin (720 kDa), rabbit IgG (150 kDa), ovalbumin (45 kDa), and/or cytochrome C (12 kDa) were also evaluated in separate runs with fractions analyzed by SDS-PAGE followed by staining with Coomassie Brilliant Blue R-250.
Molecular Weight Calculations
Molecular weights were calculated using the formula mass = (6πNas20,wη20,w)/(1-υρ20,w), where N is Avogadro's number, a is the Stokes radius as measured by size exclusion chromatography, s20,w is the sedimentation coefficient at 20 °C in water, η20,w is the viscosity of water at 20 °C, υ is the partial specific volume (see below), and ρ20,w is the density of water at 20 °C (44). We assumed that p250 and p160 associate with one detergent micelle each and that Mα+Mβ′ and Mα do not associate with detergent at all. Octylglucoside micelle size was estimated to be 25 kDa (45). The partial specific volume of each species was estimated by summing the partial specific volumes of each amino acid, the N-linked glycan of the high mannose or complex type, 20 kDa of O-linked glycans for each RPT domain, and the octylglucoside detergent micelle for those species in which it was warranted (43).
Metabolic Labeling and Immunoprecipitation
Metabolic labeling with [35S]methionine/cysteine and immunoprecipitation analyses were conducted essentially as described (26). Briefly, cells were harvested by centrifugation following digestion with trypsin-EDTA, washed, and cultured in suspension for 30 min in methionine/cysteine-free DMEM containing 5% dialyzed FBS. Cells were then pelleted and labeled metabolically in suspension in the same medium containing 35S-Met/Cys Express mix (PerkinElmer Life Sciences) for 10–15 min (pulse-chase analyses) or 2 h (steady-state labeling for two-dimensional PAGE). For pulse-chase analyses, cells were pelleted again and chased for the indicated periods of time in standard growth medium containing excess methionine/cysteine as described (26). Cells were then lysed in lysis buffer and clarified by centrifugation at 19,000 × g for 20 min. After first preclearing the lysates over unconjugated protein A beads (Life Technologies) and then over protein A beads conjugated to normal rabbit serum, normal mouse serum, or a mouse isotype control antibody, the samples were subjected to immunoprecipitation using the specified antibodies. The immunoprecipitated material was fractionated by SDS-PAGE, and the resulting gels were dried and analyzed on a PhosphorImager as described above.
Two-dimensional Non-reducing/Reducing SDS-PAGE
Triton X-100 lysates from cells metabolically labeled for 2 h were subjected to immunoprecipitation using the PMEL antibodies αPMEL-N, NKI-beteb, or αPMEL-C after preclearing first with unconjugated protein A beads and then with protein A beads conjugated to either normal rabbit serum or normal mouse serum as described above. The immunoprecipitated material was then fractionated by SDS-PAGE in a 7% polyacrylamide tube gel under non-reducing conditions. The tube gel was reduced by incubation in 0.5% (w/v) DTT, 0.5% (w/v) SDS, 125 mm Tris, pH 6.8, for 2 h at room temperature and then mounted and sealed (with 1% agarose) on top of a standard SDS-polyacrylamide gel. After fractionation by SDS-PAGE, the gels were dried and analyzed on a PhosphorImager as detailed above.
Immunoprecipitation/Immunoblotting
HeLa cells expressing wild-type hPMEL or the C301S variant were released from plates using PBS, 5 mm EDTA and harvested by centrifugation. The cells were treated with PBS, 50 mm NEM on ice, pelleted, frozen, and subsequently lysed (∼3 × 106 cells/ml) in lysis buffer. Following clarification at 19,000 × g, the lysates were precleared over unconjugated protein A beads prior to immunoprecipitation using NKI-beteb, αPMEL-C, normal rabbit serum, or a mouse isotype control antibody prebound to protein A beads. The beads were washed four times with wash buffer (0.1% (w/v) Triton X-100, 150 mm sodium chloride, 0.02% (w/v) sodium azide, 10 mm HEPES, pH 7.4) followed by one wash with PBS. The immunoprecipitated material was then eluted by adding 2× SDS sample buffer containing 3.6% β-mercaptoethanol and heating the samples for 20 min at 60 °C. After fractionation by SDS-PAGE and transfer to PVDF membranes, immunoblots were probed with αPMEL-C or HMB45 and analyzed using the Odyssey imaging system detailed above.
Proteomic Analysis of p250 and p160
Affinity Purification
Pellets of 2.6–7.8 × 108 NEM-treated MNT-1 cells were either homogenized with a Dounce homogenizer and enriched for the total membrane fraction as described previously (30) or lysed directly in lysis buffer and clarified by centrifugation at 19,000 × g for 15 min. The detergent-soluble fraction was then loaded onto three columns in tandem containing Sepharose 4B (Sigma), rabbit IgG-conjugated agarose (Sigma), and αPMEL-C-conjugated Sepharose (generated by combining 3 ml of 1 mg/ml αPMEL-C with 3 ml of packed CNBr-activated Sepharose 4B from GE Healthcare Life Sciences according to manufacturer's instructions). The αPMEL-C column was subsequently washed with 600 ml of wash buffer and eluted using 0.63 μg/ml PMEL-C peptide (CPIGENSPLLSGQQV, Peptide 2.0 Inc., Chantilly, VA). Eluate was collected in 0.5–1-ml fractions, and aliquots were analyzed by immunoblotting for PMEL content. Peak fractions were precipitated using a 5-fold volume of either methanol or acetone at −20 °C. The concentrated eluate was then fractionated on a 3–8% Tris acetate protein gel (Life Technologies) and stained with Coomassie Brilliant Blue G-250.
In-gel Digestion
Each sample was excised from the gel and cut into 1-mm cubes, destained in a solution containing 50% methanol, 1.25% acetic acid, reduced with 5 mm DTT (Thermo Fisher Scientific), and alkylated with 20 mm iodoacetamide (Sigma). Gel pieces were then washed with 20 mm ammonium bicarbonate (Sigma) and dehydrated with acetonitrile (Thermo Fisher Scientific). Trypsin (Promega; 5 ng/ml in 20 mm ammonium bicarbonate) was added to the gel pieces, and proteolysis was allowed to proceed overnight at 37 °C. Peptides were extracted with 0.3% trifluoroacetic acid (J. T. Baker, Avantor Performance Materials, Center Valley, PA) followed by 50% acetonitrile. The extracts were combined, and the volume was reduced by vacuum centrifugation.
Mass Spectrometry Analysis
Tryptic digests were analyzed by LC-MS/MS on a hybrid LTQ Orbitrap Elite mass spectrometer (Thermo Fisher Scientific) coupled with a nanoLC Ultra (Eksigent Technologies Inc., Dublin, CA). Peptides were separated by reverse phase-HPLC on a nanocapillary column (75 μm × 15 cm Reprosil-pur 3 μm, 120 Å (Dr. Maisch, Ammerbuch, Germany)) in a Nanoflex chip system (Eksigent Technologies). Mobile phase A consisted of 1% methanol (Thermo Fisher Scientific) and 0.1% formic acid (Thermo Fisher Scientific) and mobile phase B of 1% methanol, 0.1% formic acid, and 80% acetonitrile. Peptides were eluted into the mass spectrometer at 300 nl/min with each reversed phase-HPLC run comprising a 90-min gradient from 10 to 25% B in 65 min and then 25 to 40% B in 25 min. The mass spectrometer was set to repetitively scan m/z from 300 to 1800 (r = 240,000 for LTQ-Orbitrap Elite) followed by data-dependent MS/MS scans on the 20 most abundant ions with a minimum signal of 1500, dynamic exclusion with a repeat count of 1, repeat duration of 30 s, exclusion size of 500, and duration of 60 s, isolation width of 2.0, normalized collision energy of 33, and waveform injection and dynamic exclusion enabled. The Fourier transform mass spectroscopy full-scan automatic gain control target value was 1E6, and the MSn automatic gain control was 1E4. Fourier transform mass spectroscopy full-scan maximum fill time was 500 ms, and ion trap MSn fill time was 50 ms; microscans were set at 1. The FT preview mode, charge state screening, and monoisotopic precursor selection were all enabled with rejection of unassigned and 1+ charge states.
Protein Identification
MaxQuant/Andromeda version 1.5.2.8was used to identify proteins by searching against a human complete proteome sequence database downloaded from UniProt on July 14, 2014. The MaxQuant search parameters are detailed in the supplemental Table S1 (see parameters and summary tabs), and the identified protein groups and their estimated MS1 summed spectral intensities for each gel slice are listed in the proteinGroups tab of Supplemental Table S1.
Immunofluorescence Microscopy
Cells grown on coverslips were fixed with 2% formaldehyde in PBS, permeabilized and labeled with primary antibodies and fluorochrome-conjugated secondary antibodies in PBS, 0.2% (w/v) saponin, 0.1% (w/v) BSA as described previously (26), and analyzed on either a DM IRBE microscope (Leica Microsystems, Wetzlar, Germany) equipped with a ×100 Plan Apochromat objective lens (1.4 N.A., Leica), a Retiga Exi Fast 1394 digital camera (QImaging, Surrey, Canada), and OpenLab software (Improvision, Lexington, MA) or a DMI 6000B microscope (Leica Microsystems) equipped with the same ×100 objective, an Orca Flash 4.0 V2 camera (Hamamatsu, Bridgewater, NJ), and Leica Application Suite X software (Leica Microsystems). Images captured in sequential z-planes separated by 0.2 μm were deconvolved and manipulated using the Volume Deconvolution module in OpenLab or Gold's three-dimensional deconvolution algorithm with three iterations in the Leica Application Suite. Insets were magnified and further adjustments in brightness and contrast were performed using Adobe Photoshop (Adobe Systems, Mountain View, CA).
Electron Microscopy
For electron microscopy, HeLa cells transfected with pCI-PMEL wild type, C301S, or C566S with or without pEGFP-C1 (Clontech Laboratories) in a 9:1 ratio were grown in 10-cm dishes and either fixed in situ, high-pressure frozen, or sorted for GFP expression prior to fixation in suspension. For fixation in situ, cells were incubated in Karnovsky's fixative (4% paraformaldehyde, 4 mm calcium chloride, 72 mm sodium cacodylate, pH 7.4) containing 0.5% glutaraldehyde for 1–2 h. This solution was then removed and replaced with Karnovsky's fixative containing 2% glutaraldehyde, and the cells were fixed overnight at room temperature. After the cells were scraped using a rubber policeman, the overnight fixative was replaced with Karnovsky's fixative containing 0.5% glutaraldehyde and the cells stored at 4 °C. For high-pressure freezing, cells were released from plates using PBS, 5 mm EDTA, pelleted by centrifugation, and frozen under high pressure in an HPM 010 machine (ABRAFluid, Widnow, Switzerland) at the University of Pennsylvania Electron Microscopy Resource Laboratory. Sample dehydration by freeze substitution was performed in a Leica AFSII (Leica Microsystems) over a period of 72 h at −90 °C in glass-distilled acetone supplemented with 0.1% uranyl acetate and 2% OsO4. Cells were then embedded in EPON resin at room temperature followed by polymerization for 48 h at 60 °C. HeLa cells sorted prior to fixation were co-transfected with pEGFP, released from plates using PBS and 5 mm EDTA, and sorted for GFP expression on a Moflo Astrios EQ (Beckman Coulter Inc.) by the Flow Cytometry Core Laboratory (Children's Hospital of Philadelphia). Similar to the cells sorted in situ, these sorted cells were fixed for 1–2 h in Karnovsky's fixative containing 0.5% glutaraldehyde, overnight in Karnovsky's fixative containing 2% glutaraldehyde, and stored in Karnovsky's fixative with 0.5% glutaraldehyde at 4 °C. Both the cells fixed in situ and the cells sorted prior to fixation were subsequently dehydrated and embedded in EMbed-812 (Electron Microscopy Sciences, Fort Washington, PA) at the Electron Microscopy Resource Laboratory. Ultrathin sections were cut on either a Reichert-Jung Ultracut E or a Reichert Ultracut S microtome (now Leica Microsystems), placed on either uncoated copper mesh grids or Formvar-coated copper slot grids, and stained with 50% ethanol, 1% uranyl acetate, 1% lead citrate as described previously (46). The specimens were then examined with a JEOL 1010 electron microscope fitted with a Hamamatsu C4742-95 digital camera (Hamamatsu, Bridgewater, NJ) and AMT Advantage image capture software (Advanced Microscopy Techniques, Woburn, MA). For quantification of fibril-containing organelles, a combined total of at least 30 cells from the three independent experiments were analyzed for each transfected cell type. The number of multivesicular bodies that lacked fibrils and the number of multivesicular bodies that contained fibrils (or fibrillar compartments lacking other internal structure) were quantified, and the percentage of fibril-containing multivesicular bodies was calculated for each cell. These data were analyzed and plotted using Prism 6 (GraphPad Prism 6, La Jolla, CA), and statistical analyses were performed using a two-tailed unpaired t test.
Results
p250 and p160 Are Disulfide-bonded Species Composed of Golgi-modified PMEL
The Golgi-matured P2 form of PMEL and the products of proprotein convertase cleavage, Mα and Mβ (Fig. 1A), migrate with Mr of ∼120,000, 100,000, and 28,000, respectively, when analyzed by reducing SDS-PAGE (26, 38). However, when analyzed by non-reducing SDS-PAGE, most detergent-soluble forms of Golgi-matured PMEL migrate with a higher Mr (26), indicating that the vast majority of pre-amyloid P2, Mα, and Mβ isoforms exist as components of disulfide-bonded species. To further investigate the nature of these species, immunoblots were performed using antibodies that can distinguish between immature and mature PMEL isoforms. MNT-1 human melanoma cells (which endogenously express PMEL) or transfected HeLa cells transiently expressing full-length PMEL (isoform 1) were treated with NEM prior to and during lysis in TritonX-100 to prevent the formation of artifactual disulfide bonds. Detergent-soluble lysates were then fractionated by SDS-PAGE on low percentage polyacrylamide gels under non-reducing or reducing conditions and analyzed by immunoblotting using the HMB45, αPMEL-I, αPMEL-N, or αPMEL-C antibody. The HMB45 monoclonal antibody recognizes the O-glycosylated RPT region of mature PMEL isoforms present only in post-Golgi compartments (29, 47). The αPMEL-I antibody recognizes the same domain in its non-glycosylated form as is characteristic of immature PMEL isoforms located in the ER (29). The αPMEL-N and αPMEL-C antibodies recognize the N terminus and C terminus of PMEL, respectively, regardless of maturation status (26, 41).
Under non-reducing conditions, HMB45 detected two major species that migrate with Mr of ∼160,000 (p160) and ∼250,000 (p250). Also detected were larger PMEL species retained at the boundary between the stacking and separating gel and minor amounts of p160*, Mα+Mβ′, and free Mα (Fig. 1B, lanes 1 and 2). The p160* species is a slightly faster migrating version of p160 resulting from an alternatively spliced PMEL variant expressed in melanocytic cells but not transfected HeLa cells (41). The Mα+Mβ′ species is composed of Mα covalently bound to Mβ′ based on the following criteria: (i) it is detected by the HMB45 and αPMEL-N antibodies but not the αPMEL-C antibody, indicating that it lacks the PMEL C terminus; (ii) it comigrates with the major form of secreted PMEL (see Fig. 5A), which consists predominantly of disulfide-bonded Mα+Mβ′ complexes (36); and (iii) it is depleted following inhibition of BACE2 (see Fig. 6A), the enzyme that cleaves Mβ into CTF and Mβ′ fragments. As only a single band corresponding to Mα was observed under reducing conditions (Fig. 1B, lanes 3 and 4; note that Mα migrates with a higher Mr when derived from MNT-1 cells than when derived from HeLa cells, due to differences in terminal glycosylation (26)), disulfide bonds must be necessary to maintain the integrity of p250 and p160 as well as that of p160* and Mα+Mβ′. Differences in the relative abundance of these species in lysates of MNT-1 cells versus PMEL-expressing HeLa cells likely reflect cell type-specific differences in the rates at which these species are formed and consumed by interconversion, secretion, or incorporation into insoluble amyloid fibrils.
FIGURE 5.

p250 and p160 are transient PMEL intermediates present only in cell lysates. A and B, MNT-1 cells were metabolically labeled with [35S]methionine/cysteine for 15 min and chased for the times indicated (min). PMEL in detergent cell lysates or culture media, as indicated, was then immunoprecipitated using NKI-beteb and fractionated by SDS-PAGE under non-reducing (A) or reducing (B) conditions. C, transiently transfected HeLa cells expressing wild-type PMEL (long form) were metabolically pulse labeled with [35S]methionine/cysteine for 10 min and chased for the indicated amounts of time. Cell lysates were then immunoprecipitated using αPMEL-C and fractionated by SDS-PAGE under non-reducing (NR, lanes 1–5) or reducing (R, lanes 6–10) conditions. The migration of molecular weight standards is shown to the right of each gel. Bands corresponding to p250, p160, Mα+Mβ′, P1, and Mα on the non-reducing gels and P2, P1, Mα, and Mβ on the reducing gels are indicated by arrows. Note that P1 and Mα largely comigrate in MNT-1 cells. *, product of alternatively spliced PMEL mRNA (PMEL-ss). Lane numbers are indicated across the bottom of each gel.
FIGURE 6.

Potential precursor-product relationships among p250, p160, and Mα monomers. A and B, MNT-1 cells were untreated (lane 1) or treated for 3 h with 10 μm monensin (lane 2), 100 μm β-secretase inhibitor IV (lane 3), or 1 mg/ml E-64 (lanes 4 and 5). Cells treated with E-64 were then either immediately harvested (lane 4) or additionally treated with 10 μg/ml cycloheximide (CHX) in the presence of E-64 for 2 h (lane 5). After lysis in Triton X-100, detergent-soluble cell lysates were analyzed by non-reducing (NR) SDS-PAGE followed by immunoblotting with HMB45 (A) or αPMEL-C (B). C, MNT-1 cells that were untreated (lane 1) or treated for 2 days with 1 mg/ml E-64 (lane 2) were lysed in Triton X-100, and detergent-insoluble cell lysates were analyzed by reducing (R) SDS-PAGE followed by immunoblotting with HMB45. The migration of molecular weight standards is indicated to the left of each blot, and bands corresponding to p250, p160, Mα+Mβ′, Mα, P1, and Mα′ fragments and higher Mr multimers (mtmrs) are indicated with arrows.
The p250 and p160 bands were not labeled by αPMEL-I under non-reducing conditions; instead, this antibody detected a band that corresponds to monomeric P1 as well as a series of high Mr bands ranging from ∼220,000 to the top of the separating gel (Fig. 1C, lanes 1 and 2). Because the high Mr bands collapse into P1 under reducing conditions (Fig. 1C, lanes 3 and 4; note that the slightly faster migrating band (*) corresponds to the P1 form of the alternatively spliced PMEL variant, PMEL-ss (41)), these species most likely represent partially folded disulfide-bonded conformers of PMEL in the ER (Fig. 1C, cfmrs). The αPMEL-N and αPMEL-C antibodies recognized p250 and p160 as well as the immature PMEL conformers detected by αPMEL-I, indicating that all of these high Mr species contain either full-length PMEL or Mα disulfide-bonded to Mβ (Fig. 1, D and E). Therefore, only Golgi-matured forms of full-length or proprotein convertase-cleaved PMEL are present in p250 and p160. Furthermore, the presence of p250 and p160 both in MNT-1 cells that endogenously express PMEL and in non-pigment HeLa cells expressing a PMEL transgene suggests that no additional pigment cell-specific proteins are required for the formation of mature disulfide-bonded PMEL species.
p250 and p160 Represent Distinct Physiological Complexes
To test whether the disulfide-bonded p250 and p160 species observed by non-reducing SDS-PAGE correspond to physiological PMEL-containing complexes, we assessed the biophysical properties of p250 and p160 under non-denaturing conditions. First, we examined the PMEL species by sedimentation velocity analysis. Lysates of MNT-1 cells were prepared using n-octylglucoside, a non-ionic detergent with a small micelle size and a relatively high partial specific volume (45). After fractionation through sucrose gradients containing n-octylglucoside, mature PMEL species were identified by immunoblot analysis of gradient fractions separated by non-reducing SDS-PAGE (Fig. 2A). Sedimentation coefficients were then calculated by comparing the peak position of mature PMEL species, detected using the HMB45 antibody, with the peak position of globular protein standards (Fig. 2, B and C). Distinct sedimentation coefficients were calculated for p250, p160, Mα+Mβ′, and “free” Mα. Consistent with the relative order of their migration by non-reducing SDS-PAGE, p250 had the highest calculated s value followed by p160, Mα+Mβ′, and lastly free Mα. This suggests that p250 and p160 are not artifacts of non-reducing SDS-PAGE but distinct native disulfide-bonded PMEL complexes. The sedimentation coefficients of all four PMEL species were substantially smaller than expected given the magnitude of their migration by non-reducing SDS-PAGE (Table 1), suggesting that these species might have extended glycoprotein structures.
FIGURE 2.
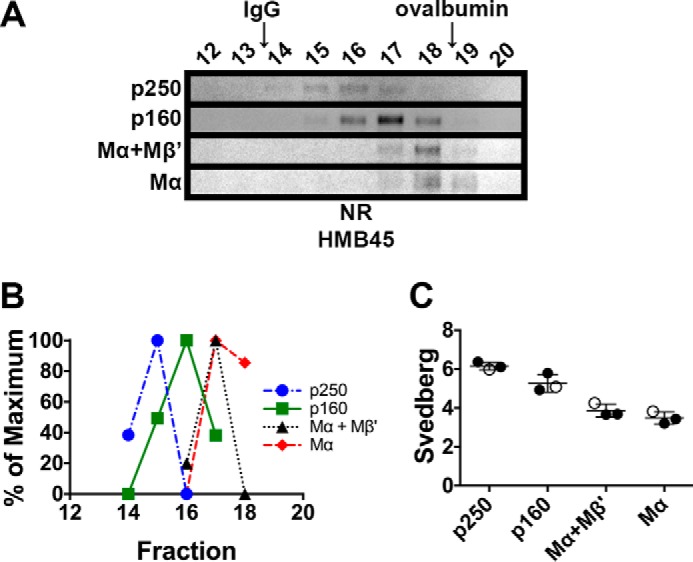
p250, p160, and Mα monomers have distinct sedimentation properties. MNT-1 cell lysates prepared using 250 mm n-octylglucoside were subject to sedimentation velocity analyses in 5–20% sucrose gradients. A, aliquots of the gradient fractions were analyzed by non-reducing (NR) SDS-PAGE and immunoblotted using HMB45. Shown are regions of the immunoblots corresponding to p250, p160, Mα+Mβ′, and Mα, as indicated to the left. Fraction numbers are indicated above each lane (bottom of the gradient is to the left, and top of the gradient is to the right), and the migration of the globular protein standards, IgG and ovalbumin, are indicated with arrows at the top. Note that immunoblot contrast was optimally adjusted for each individual species. B, HMB45 immunoreactivity was quantified to determine the peak elution fraction for p250, p160, Mα+Mβ′, and Mα. The lowest value quantified for each species was set to 0%, and the highest value was set to 100%. C, Svedberg values were calculated for each species by comparing peak fractions with those of protein standards with known s values in three independent experiments. Circles, values calculated from each experiment; open circles, values calculated from the experiment shown in A and B; horizontal lines, mean value; error bars, S.D. Mean ± S.D. are also shown in Table 1.
TABLE 1.
Summary of calculated sedimentation coefficients and Stokes radii
| Species | Predicted s20,wa | s20,wa | Stokes radius |
Calculated mass (protein)b | ||
|---|---|---|---|---|---|---|
| n-Octylglucoside | Dodecyl-β-d-maltoside | Triton X-100 | ||||
| S | S | nm | kDa | |||
| p250 | 11.4 | 6.15 ± 0.20 | 10.38 ± 0.34 | 9.82 ± 0.16 | 10.09 ± 0.07 | 241 ± 12 |
| p160 | 7.4 | 5.27 ± 0.46 | 8.76 ± 0.19 | 8.41 ± 0.08 | 8.84 ± 0.08 | 176 ± 18 |
| Mα+Mβ′ | 5.9 | 3.86 ± 0.33 | 8.63 ± 0.12 | 7.96 ± 0.12 | 8.12 ± 0.06 | 130 ± 11 |
| Mα | 5.4 | 3.49 ± 0.31 | 8.03 ± 0.29 | 7.54 ± 0.09 | 7.68 ± 0.08 | 107 ± 10 |
a Predicted sedimentation values based on comparison with the globular standard most similar in molecular weight using the formula S1/S2 = (Mr1/Mr2)2/3. Actual sedimentation values are based on the distance traveled relative to globular standards. Standards and values were: ovalbumin (3.6 S, Mr 42,881), rabbit IgG (7.1 S, Mr 150,000), bovine liver catalase (11.3 S, Mr 247,000), and/or bovine thyroglobulin (19.3 S, Mr 667,000).
b Calculated based on s20,w and Stokes radius in octylglucoside and assuming association of p250 and p160 with a single detergent micelle as described under “Experimental Procedures.“
To further characterize the PMEL species under native conditions, MNT-1 cell lysates were fractionated by size exclusion chromatography in n-octylglucoside. Aliquots of each fraction were separated by non-reducing SDS-PAGE, and mature PMEL species were detected by immunoblotting with HMB45. As observed in the sedimentation analyses, the four PMEL species, p250, p160, Mα+Mβ′, and Mα, migrated with distinct elution volumes by size exclusion chromatography (Fig. 3, A and B). However, when the peak elution volume of each species was compared with that of globular protein standards, the mature PMEL species had peak elution volumes much smaller, and therefore Stokes radii much larger, than expected for globular proteins of similar molecular weight (Fig. 3C and Table 1). For example, the Stokes radius of p250 (10.4 nm) was calculated to be larger than that of α2-macroglobulin (8.8 nm), a 720-kDa tetrameric protein (49). To test whether the exceptionally large Stokes radii reflected association with multiple detergent micelles, cell lysates were prepared and analyzed by size exclusion chromatography using detergents with different micelle sizes: n-octylglucoside (average micelle size of 8 kDa (50)), dodecyl-β-d-maltoside (average micelle size of 50–76 kDa (50)) and Triton X-100 (average micelle size of 81 kDa (51)). If any of the PMEL species had been associated with multiple detergent micelles, those species would have eluted with distinct volumes in each of the three detergents. However, the elution volume of each PMEL species was roughly similar in all three detergents (Fig. 3 and Table 1), suggesting that detergent micelles do not contribute substantially to the large Stokes radii of the mature PMEL species. Therefore, the unusually high Stokes radii and low sedimentation coefficients of p250, p160, Mα, and Mα+Mβ′ most likely reflect extended glycoprotein conformations resulting from the heavily glycosylated RPT domain of mature PMEL. Remarkably, the estimated molecular weight calculated using the sedimentation and size exclusion chromatography properties of each of the four PMEL species closely approximated the molecular weight of each species as determined by SDS-PAGE (Table 1).
FIGURE 3.

p250, p160, and Mα monomers have distinct Stokes radii that are minimally altered by solubilization in detergents with different micelle sizes. MNT-1 cells were lysed using n-octylglucoside (A–C), dodecyl-β-d-maltoside (D–F), or Triton X-100 (G–I), and the detergent extracts were fractionated by size exclusion chromatography in the corresponding detergent. A, D, and G, eluted fractions were analyzed by SDS-PAGE under non-reducing (NR) conditions and immunoblotted using the HMB45 antibody. Shown are regions of the immunoblots corresponding to p250, p160, Mα+Mβ′, and Mα as indicated to the left. Fraction numbers are indicated above each lane, and the migration of α2-macroglobulin is indicated with an arrow. Note that immunoblot contrast was optimally adjusted for each individual species in each experiment. B, E, and H, HMB45 immunoreactivity was quantified to determine the elution volume of p250, p160, Mα+Mβ′, and Mα. The lowest value quantified for each species was set to 0% and the highest value was set to 100%. C, F, and I, the Stokes radius of each species was then calculated by comparing the peak elution fraction of that species with those of globular protein standards in three independent experiments. Circles, values calculated from each experiment; open circles, values calculated from the experiments shown in A, D, and G; horizontal lines, mean value; error bars, S.D. Mean ± S.D. are also shown in Table 1.
PMEL Is the Sole Component of Both p250 and p160
To determine whether p250 and p160 contain additional polypeptides besides PMEL, we analyzed the contents of PMEL immunoprecipitates by two-dimensional non-reducing/reducing SDS-PAGE. We used transfected HeLa cells transiently expressing wild-type PMEL in this experiment because Mα is more clearly identified in HeLa cell lysates analyzed by reducing SDS-PAGE. This is because, unlike melanocytic cells, transfected HeLa cells typically do not express the alternatively spliced form of PMEL (PMEL-ss) that comigrates with Mα by reducing SDS-PAGE (see Fig. 1, B–D). In addition, Mα comigrates with P1 in MNT-1 cells but not in transfected HeLa cells as a result of differences in terminal glycosylation. After extensively labeling biosynthesized proteins with [35S]methionine/cysteine, PMEL species in detergent cell lysates were immunoprecipitated using αPMEL-N, αPMEL-C, or NKI-beteb (52), a monoclonal antibody to the PKD (a region with homology to a repeated domain in polycystin 1 (29)) and then fractionated by two-dimensional PAGE and analyzed by phosphorimaging. Using this technique, the higher Mr bands observed by standard SDS-PAGE (Fig. 1) were resolved into their individual components. Similar results were obtained using all three PMEL antibodies (Fig. 4). Spots corresponding to the core-glycosylated P1 and the Golgi-modified P2 forms of PMEL were identified close to the diagonal and thus represent the monomeric forms of these PMEL species. The p160 band typically observed by one-dimensional non-reducing SDS-PAGE resolved into two species by two-dimensional PAGE; p160a contained the Mα and Mβ products of proprotein convertase cleavage, whereas p160b contained uncleaved, full-length, Golgi-modified P2. The p250 band also resolved into both the cleaved and uncleaved forms of Golgi-modified PMEL. A number of other bands present in the non-reducing dimension resolved into P1 under reducing conditions and thus most likely represent disulfide-bonded ER folding intermediates. Importantly, no other radiolabeled polypeptide was reproducibly detected within the p250 or p160 bands. Furthermore, three independent attempts to identify additional components by tandem mass spectrometry analysis of affinity-purified PMEL from MNT-1 cells yielded no reasonable candidates other than chaperones and protein disulfide isomerases, which were present in vastly substoichiometric amounts (Table 2 and supplemental Table S1) and were associated only with immature forms of PMEL.4 Other identified peptides either derived from common contaminants such as keratins, derived from proteins with molecular weights that were inconsistent with that of p250 and p160, or were very poorly recovered relative to PMEL-derived peptides. We therefore concluded that p250 and p160 are composed exclusively of PMEL or PMEL fragments.
FIGURE 4.

p250 and p160 comprise Golgi-matured P2 and Mα/Mβ fragments of PMEL. Transiently transfected HeLa cells expressing wild-type PMEL were metabolically labeled for 2 h with [35S]methionine/cysteine, immunoprecipitated using αPMEL-N (A), NKI-beteb (B), or αPMEL-C (C) and analyzed by two-dimensional PAGE under non-reducing conditions in the first dimension (left to right; −BME, β-mercaptoethanol) and under reducing conditions in the second dimension (top to bottom; +BME). The migrations of p250, p160a, p160b, P2, and P1 in the first dimension are indicated by arrows at the top; the migration of molecular weight standards in the second dimension is indicated to the right of each blot; and the positions of P2, P1, Mα, and Mβ in the second dimension are indicated on the left.
TABLE 2.
Analysis of p250 and p160 by LC-MS/MS
Affinity-purified PMEL was fractioned on a non-reducing SDS-polyacrylamide gel, relevant bands excised and digested using trypsin or AspN, and the samples analyzed by mass spectrometry. This table is a brief summary of the top 50 “hits” obtained. Summed intensity refers to the sum of the extracted ion currents from three independent experiments analyzing higher Mr disulfide-bonded PMEL species. The proteins listed are of human origin unless otherwise noted. More detailed results, with each band in each of the three experiments analyzed, are provided in supplemental Table S1.
| Summed intensity | Protein | Mol. mass |
|---|---|---|
| kDa | ||
| 10,459,000,000 | PMEL | 120 |
| 9,310,000,000 | Trypsin (Sus scrofa) | 24 |
| 3,643,900,000 | AspN (Pseudomonas fragi) | 16 |
| 2,444,500,000 | Clathrin heavy chain 1 | 188 |
| 1,113,200,000 | Mucin-19 | 357 |
| 1,060,400,000 | Keratin, type I cytoskeletal 13 | 50 |
| 1,052,500,000 | UDP-glucose:glycoprotein glucosyltransferase 1 | 175 |
| 908,870,000 | Keratin, type II cytoskeletal 1 | 66 |
| 720,070,000 | Trypsin | 15 |
| 477,000,000 | Keratin, type I cytoskeletal 10 | 59 |
| 431,880,000 | Keratin, type II cytoskeletal 6A | 60 |
| 352,060,000 | Keratin, type II cytoskeletal 4 | 57 |
| 351,310,000 | Keratin, type II cytoskeletal 2 epidermal | 65 |
| 350,080,000 | Methionine synthase | 135 |
| 292,260,000 | Solute carrier family 12, member 7 | 119 |
| 264,970,000 | Filamin-A | 280 |
| 248,250,000 | Protein disulfide-isomerase A3 | 57 |
| 194,490,000 | Myosin-9 | 227 |
| 161,510,000 | Keratin, type I cytoskeletal 9 | 62 |
| 152,110,000 | Endoplasmic reticulum resident protein 44 | 47 |
| 125,550,000 | κ-Casein (Bos taurus) | 21 |
| 120,610,000 | Ubiquitin | 10 |
| 117,270,000 | Unconventional myosin-Ic | 118 |
| 113,980,000 | Unconventional myosin-Id | 116 |
| 112,810,000 | Fatty acid synthase | 273 |
| 105,730,000 | Keratin, type I cytoskeletal 19 | 44 |
| 100,700,000 | Vinculin | 117 |
| 100,210,000 | Protein disulfide isomerase A4 | 73 |
| 97,113,000 | Transferrin receptor protein 1 | 85 |
| 86,599,000 | 4F2 cell-surface antigen heavy chain | 65 |
| 70,376,000 | 78-kDa glucose-regulated protein (BiP) | 72 |
| 67,557,000 | Protein disulfide isomerase A6 | 48 |
| 65,533,000 | Myosin-10 | 231 |
| 64,584,000 | Ras GTPase-activating-like protein IQGAP1 | 189 |
| 64,038,000 | Protein S100-A9 | 13 |
| 61,131,000 | Protein disulfide isomerase | 57 |
| 60,047,000 | α-Synuclein | 14 |
| 54,174,000 | Exportin-1 | 123 |
| 54,029,000 | Small proline-rich protein 3 | 17 |
| 44,466,000 | Keratin, type II cytoskeletal 5 | 62 |
| 43,245,000 | Actin | 42 |
| 42,124,000 | Protein S100-A8 | 11 |
| 40,829,000 | Heat shock protein β-1 | 23 |
| 39,343,000 | Tetraspanin-10 | 36 |
| 36,125,000 | Solute carrier family 12, member 4 | 119 |
| 34,172,000 | Annexin A1 | 39 |
| 32,707,000 | Transcription elongation factor SPT5 | 121 |
| 32,563,000 | Transmembrane protein 65 | 25 |
| 31,993,000 | Filamin-B | 276 |
| 31,932,000 | Talin-1 | 270 |
As detailed above, the molecular weight of p250 and p160 calculated from the combined size exclusion chromatography and velocity sedimentation data are ∼241,000 and ∼176,000, respectively, closely correlating with their migration by SDS-PAGE. Given these data and the absence of any additional protein components, p250 most likely represents a PMEL dimer, and p160b and p160a most likely represent extended disulfide-dependent conformations of a single PMEL monomer and a single Mα/Mβ complex, respectively.
p250 and p160 Are Intermediates of PMEL Fibril Formation
The reactivity of p250 and p160 with HMB45 but not with αPMEL-I suggests that these two species are primarily present in post-ER compartments. To define the kinetics of their formation, we performed a metabolic pulse-chase and immunoprecipitation analysis. MNT-1 cells were metabolically labeled for 15 min with [35S]methionine/cysteine and then chased in medium containing excess unlabeled amino acids for 0–120 min. PMEL isoforms in detergent-soluble cell lysates and the culture medium from each time point were immunoprecipitated using NKI-beteb and analyzed by non-reducing or reducing SDS-PAGE. At the pulse, the primary PMEL species detected under non-reducing conditions was monomeric P1, which migrated with a Mr of ∼110,000 (Fig. 5A, lane 1). The p250 band first appeared after 5 min of chase and peaked at 30 min (Fig. 5A, lanes 2–4). This resembles the kinetics of P2 formation/accumulation, as observed under reducing conditions (Fig. 5B, lanes 1–6), suggesting that dimer formation occurs just prior to ER exit or in the early Golgi. The p160 band was first observed after 15 min of chase and peaked at 30–60 min (Fig. 5A, lanes 3–5). This resembles the kinetics of proprotein convertase cleavage, as indicated by the appearance of the Mβ cleavage product observed under reducing conditions (Fig. 5B, lanes 1–6). Essentially similar results were obtained upon pulse-chase analysis of PMEL expressed in transiently transfected HeLa cells (Fig. 5C). Thus, p160 is likely generated in the trans-Golgi network or in a post-Golgi compartment. Importantly, at no point did monomeric forms of P2 or Mα appear in cell lysates analyzed under non-reducing conditions. This indicates that the vast majority of post-ER/prefibrillar PMEL must be present in disulfide bond-dependent intermediates.
As observed previously (24, 26, 36, 52), a small fraction of PMEL was secreted into the medium of MNT-1 cells starting at 30 min of chase and peaking at 120 min (Fig. 5, A and B, lanes 7–12). Consistent with those reports (36), PMEL secreted in this manner comprised primarily Mα+Mβ′ with lesser amounts of free Mα.
To verify band identity and to probe for potential precursor-product relationships, MNT-1 cells were treated with small molecule inhibitors of trafficking and processing steps. Detergent-soluble lysates were then analyzed by non-reducing SDS-PAGE and immunoblotted with HMB45 or αPMEL-C. Treatment of MNT-1 cells with monensin, an inhibitor of intra-Golgi transport (53), led to the accumulation of p250 and higher Mr oligomers as well as the concomitant depletion of p160 (Fig. 6, A and B, lane 2). This supports the conclusion that p250 forms during ER exit or in the early Golgi and that p160 forms in the trans-Golgi network or later. It also suggests that p250 might be the precursor of p160. Treatment with β-secretase inhibitor IV at concentrations that inhibit BACE2 cleavage (54) resulted in the substantial accumulation of p160 and the minor accumulation of p250 and higher Mr oligomers (Fig. 6, A and B, lane 3). This result indicates that the resolution of p160, and perhaps a fraction of p250, requires BACE2 cleavage, which is known to cleave Mβ to Mβ′ and the CTF (33). Because BACE2 inhibition has been shown to impede PMEL fibril formation (33), this also suggests that p160 cannot directly assemble into functional amyloid fibrils. Lastly, treatment with the cysteine protease inhibitor E-64 resulted in the accumulation of free Mα and Mα+Mβ′ (as detected by HMB45 but not by αPMEL-C) but had no effect on p250 or p160 (Fig. 6, A and B, lane 4). Prolonged treatment with E-64 also resulted in the accumulation of Mα and the depletion of proteolytic fragments of Mα containing the RPT domain (Mα′) in the detergent-insoluble fraction of MNT-1 cells (Fig. 6C), suggesting that cysteine protease inhibitors, like ADAM protease inhibitors (55), interfere with the processing of Mα and Mα+Mβ′ into smaller fragments.
Importantly, treatment of these cells for an additional 2 h in the presence of both E-64 and the protein synthesis inhibitor cycloheximide to allow for further processing in the absence of new PMEL biosynthesis led to the depletion of p160 but not of Mα or Mα+Mβ′ (Fig. 6, A and B, lane 5). This suggests that p160 is a true intermediate in PMEL processing that precedes the formation of free Mα and Mα+Mβ′. Finally, none of the inhibitor treatments (except for cycloheximide) affected the level of P1 detected by αPMEL-C (Fig. 6B), which is consistent with the notion that immature PMEL monomers are located predominantly in pre-Golgi compartments (i.e. the ER).
Cysteine 301 Participates in a Cross-subunit Disulfide Bond in p250 and a Disulfide Bond between Mα and Mβ Derived from the Same PMEL Precursor in p160
To determine which cysteine residues participate in the disulfide bonds that stabilize p250 and p160, we evaluated the effect of single cysteine mutations on the disulfide-bonded PMEL species. The 11 luminal cysteine residues within PMEL (Fig. 7A) were each individually mutagenized to serine and the single point mutants expressed in HeLa cells by transient transfection. Like wild-type PMEL, all mutants were able to exit the ER and localize to a subset of late endosomes and lysosomes labeled with LAMP1 by immunofluorescence microscopy (Fig. 8). The mutants were also all appropriately cleaved in post-Golgi compartments by a proprotein convertase, as each one produced Mβ fragments when analyzed by reducing SDS-PAGE followed by immunoblotting with αPMEL-C (Fig. 7B). These data indicate that no single disulfide bond is necessary for PMEL to pass ER quality control checkpoints or to be appropriately modified by Golgi enzymes and proprotein convertase. To assess p250 and p160 formation, lysates were then fractionated by non-reducing SDS-PAGE and immunoblotted with HMB45 (Fig. 7, C–E). Of the four cysteine residues within the region of PMEL that corresponds to Mα, individual mutations in three of them had little to no effect on p250 and p160. Mutagenesis of Cys-60 resulted in disulfide-bonded PMEL species that were undistinguishable from those produced by wild-type PMEL (Fig. 7C, lane 2), and mutagenesis of Cys-130 or Cys-138 produced only slight increases in the Mr of p250 and p160 (Fig. 7C, lanes 3 and 4). This suggests that although Cys-130 and Cys-138 likely participate in an intramolecular disulfide bond, neither Cys-60, Cys-130, nor Cys-138 is critical for maintaining the integrity of p250 or p160. By contrast, mutagenesis of Cys-301 ablated the formation of both p250 and p160, causing most mature PMEL to migrate as free Mα by non-reducing SDS-PAGE (Fig. 7C, lane 5). Because Cys-301 was the only cysteine residue within the Mα region of PMEL required to stabilize both disulfide-bonded PMEL species, Cys-301 must participate in a disulfide bond with a cysteine residue in the Mβ region of PMEL to generate p250 and p160.
FIGURE 7.

p250 has a Cys-301-dependent intermolecular disulfide bond, and p160 has a Cys-301-dependent intramolecular disulfide bond. A, schematic from Fig. 1A showing luminal cysteine residues labeled with their amino acid numbers. For definitions of abbreviations, see the legend for Fig. 1. B–E, lysates from transiently transfected HeLa cells expressing wild-type (WT) PMEL, the indicated cysteine-to-serine mutants of PMEL, or the ΔCS variant was analyzed by reducing (R) SDS-PAGE and immunoblotted with αPMEL-C (B) or by non-reducing (NR) SDS-PAGE and immunoblotted with HMB45 (C–E). Single point mutants are analyzed in B, C, and E; double and triple mutants are analyzed in D together with ΔCS. The positions of molecular weight standards are indicated to the right of each blot, and relevant PMEL bands are indicated with arrows.
FIGURE 8.

PMEL cysteine mutants traffic appropriately to late endosomal compartments when expressed in HeLa cells. HeLa cells transiently transfected with wild-type PMEL (a–c), PMEL C60S (d–f), PMEL C130S (g–i), C138S (j–l), C301S (m–o), C475S (p–r), C516S (s–u), C525S (v–x), C533S (y, z, and aa), C541S (ab–ad), C550S (ae–ag), or C566S (ah–aj) were fixed, labeled with antibodies to PMEL (NKI-beteb, red: a, d, g, j, m, p, s, v, y, ab, ae, and ah) and LAMP1 (H4A3, green: b, e, h, k, n, q, t, w, z, ac, af, and ai), and analyzed by deconvolution immunofluorescence microscopy. Shown are representative images of each label separately and merged together (c, f, i, l, o, r, u, x, aa, ad, ag, and aj). Arrows indicate examples of overlap between PMEL and LAMP1. Insets show ×4 magnification of the boxed regions. Scale bar represents 10 μm.
To generate p250, a disulfide bond must connect the two PMEL monomers within the dimer. Because p250 contains both Mα and Mβ products of proprotein convertase cleavage (Fig. 4), a disulfide bond must also form a bridge between the Mα and Mβ fragments. If Cys-301 is the only cysteine residue in the Mα region of PMEL that actively functions to fulfill both of these requirements, then Cys-301 of one PMEL molecule must form an intermolecular disulfide bond with the Mβ region of the other PMEL molecule within the p250 dimer. Alternatively, the other cysteine residues in the Mα region of PMEL could participate in a disulfide bond with Mβ in a redundant manner such that mutagenesis of any one is insufficient to disrupt disulfide bond formation. We therefore tested whether combined mutagenesis of the other Mα cysteine residues would impact p250 or p160 formation. Relative to single point mutations in Cys-130 or Cys-138, simultaneous mutation of two (Cys-130 and Cys-138) or all three (Cys-60, Cys-130, and Cys-138) of these cysteine residues had no additional impact on the migration or the formation of p250 (Fig. 7D, lanes 2 and 3). Therefore, Cys-301 must participate in cross-subunit disulfide bonds that both (a) link the two PMEL monomers present within the p250 dimer and (b) link Mα to Mβ within p250.
To generate p160a, a disulfide bond must connect Cys-301 of Mα to a cysteine residue of Mβ. However, this disulfide bond could form between Mα and Mβ fragments derived from the same PMEL molecule or from different PMEL molecules. To distinguish between these possibilities, we analyzed a mutagenized form of PMEL lacking the proprotein convertase cleavage site (ΔCS (30)) by non-reducing SDS-PAGE. As predicted by the migration of uncleaved P2 in p160b by two-dimensional SDS-PAGE (Fig. 4), the ΔCS variant generated both p250 and p160 with only small shifts in migration (Fig. 7D, lane 4). Thus, the Mα and Mβ fragments in p160a likely originate from the same PMEL molecule. Given that Cys-301 is required for the formation of both the interchain disulfide bond in p250 and the intrachain disulfide bond in p160b, a disulfide bond exchange must occur during the maturation of p250 to p160.
In an attempt to identify the cysteine residue within the Mβ region of PMEL that forms a disulfide bond with Cys-301, the seven cysteine residues in this region were mutagenized individually to serine and the single point mutants analyzed for their effects on the stability and migration of p250 and p160 by non-reducing SDS-PAGE. Mutagenesis of Cys-475 resulted in disulfide-bonded PMEL species undistinguishable from those produced by wild-type PMEL, but mutagenesis of any one cysteine residue within the cysteine-rich KLD (Cys-516, Cys-525, Cys-533, Cys-541, Cys-550, or Cys-566) completely ablated formation of p160 without disrupting that of p250 (Fig. 7E). Although these data fail to isolate which cysteine residue within the Mβ region of PMEL participates in the disulfide bond with Cys-301, they suggest that an intact KLD is critical for the formation of p160.
KLD Mutagenesis Impairs PMEL Fibril Formation
The ability of KLD cysteine mutants to preferentially stabilize p250 provides a useful tool with which to investigate the function of this disulfide-bonded PMEL intermediate. Previous studies have shown that, when overexpressed in non-pigmented cells such as HeLa or Mel220, PMEL forms melanosome-like fibrillar structures within multivesicular endosomes that can be visualized and distinguished from non-fibrillar aggregates by electron microscopy (30, 31, 56, 57). In our experiments, transient expression of wild-type PMEL led to the formation of fibrillar sheets in 36 ± 4.6% (mean ± S.E.) of the multivesicular bodies present within a given HeLa cell (Fig. 9). However, following comparable expression of the C566S PMEL mutant, which forms p250 but not p160 (Fig. 7E, lane 8), many fewer multivesicular bodies (18 ± 3.5%, p < 0.01) showed evidence of fibril formation (Fig. 9). This result suggests that stabilization of p250 can impair the formation of functional amyloid fibrils and that resolution of this disulfide-bonded intermediate may be necessary for fibril formation to ensue.
FIGURE 9.

Cells expressing the C566S PMEL mutant, but not the C301S mutant, exhibit decreased fibril formation relative to those expressing wild type PMEL. HeLa cells transiently transfected with wild-type PMEL, the C301S mutant, or the C566S mutant were analyzed by transmission electron microscopy. A, example of a multivesicular body (MVB) with intraluminal vesicles (arrowheads). B, example of a multivesicular body with fibrils, intraluminal vesicles (arrowheads), and a small multilamellar structure commonly found in late endosomes and lysosomes (asterisk). C, fibril-containing organelles and multivesicular bodies without any evidence of fibril formation were quantified in at least one field of view/cell, and the percentage of fibril-containing multivesicular bodies was calculated for each cell. Box plots show the combined results of three experiments with the line in the center representing the median, the box representing the 25th and 75th percentiles, and the whiskers denoting the minimum and maximum values. Statistics were performed using a two-tailed unpaired t test comparing each mutant to wild-type PMEL. Scale bars are 500 nm.
Surprisingly, expression of the C301S variant, which fails to form both p250 and p160 (Fig. 7C, lane 5), resulted in levels of fibril formation (34 ± 3.8% of multivesicular bodies) that were not significantly different from wild-type PMEL (Fig. 9). This could have been interpreted to suggest that p250 and p160 are not required for fibril formation. However, given that p250 and p160 were detected as distinct entities under non-denaturing conditions by sedimentation and size exclusion chromatography analyses (Figs. 2 and 3), we considered the possibility that similar complexes might form non-covalently in the absence of Cys-301. Indeed, co-immunoprecipitation analyses show that Mα and Mβ remained associated in cells expressing the C301S mutant of PMEL (Fig. 10, A and B). Moreover, when transfected HeLa cells were analyzed by size exclusion chromatography, the C301S mutant generated PMEL complexes having elution volumes identical to those of wild-type PMEL (Fig. 10, C–H). Thus, although Cys-301-dependent disulfide bonds may provide additional stability to p250 and p160, the data suggest that formation of the corresponding non-covalent complexes is sufficient for function.
FIGURE 10.

Non-covalent complexes with properties similar to p250 and p160 persist despite Cys-301 mutagenesis. A and B, detergent-soluble lysates prepared from transiently transfected HeLa cells expressing wild-type PMEL (lanes 1–3) or PMEL C301S (lanes 4–6) were immunoprecipitated (IP) using αPMEL-C to the Mβ fragment (C; lanes 3 and 6 in A), NKI-beteb to the Mα fragment (N; lanes 3 and 6 in B) or matched negative (−) controls (normal rabbit serum in A, lanes 2 and 5, or the irrelevant monoclonal antibody OKT4 in B, lanes 2 and 5). The immunoprecipitated material and 3% of the inputs (I; lanes 1 and 4) were analyzed by reducing SDS-PAGE (R) and immunoblotted (IB) with antibodies to the opposite fragment: HMB45 to Mα (A) or αPMEL-C to Mβ (B). The migration of molecular weight standards are shown to the right; bands corresponding to P2, Mα, P1, and Mβ are indicated with arrows. C–F, HeLa cells transfected with wild-type PMEL (C and E) or the C301S PMEL variant (D and F) were lysed in 250 mm n-octylglucoside lysis buffer and the detergent extract fractionated by size exclusion chromatography in 25 mm n-octylglucoside running buffer. Eluted fractions were analyzed by SDS-PAGE under non-reducing (NR; C and D) or reducing (R; E and F) conditions and immunoblotted using the antibodies indicated. The migration of molecular weight standards is indicated to the right of each blot, and bands corresponding to p250, p160, Mα, and Mβ are indicated by arrows. Lanes are labeled with fraction numbers as presented in Fig. 3A. G and H, HMB45 and αPMEL-C immunoreactivity was quantified to determine the amount of Mα (G) and Mβ (H) present in each fraction. The lowest value quantified for each species was set to 0%, and the highest value was set to 100% for each experiment. Data represent the mean ± S.D. of three independent experiments.
Discussion
Very little is known about how functional amyloid formation is regulated within cells, especially in mammalian systems. Here we have shown that the pigment cell-specific amyloid protein PMEL forms a disulfide-bonded dimer in the ER or early Golgi. Resolution of this dimer to a disulfide-bonded monomer requires an intact KLD, and mutagenesis of any cysteine residue within the KLD inhibits this process and interferes with the assembly of functional amyloid fibrils. Our data are consistent with a model in which the formation of a disulfide bond-stabilized PMEL dimer in the ER or early Golgi impairs premature fibril formation early in the secretory pathway (Fig. 11). KLD-dependent disulfide bond rearrangements in the late Golgi or in a post-Golgi compartment would then resolve the dimer to monomeric forms that could be incorporated into growing amyloid fibrils.
FIGURE 11.

Model of PMEL fibril formation. The schematic diagram shows the maturation of p250 into functional amyloid fibrils. The p250 PMEL dimer is resolved to p160 via disulfide bond rearrangement with proprotein convertase cleavage occurring before, during, or after this process. p160 is then cleaved by BACE2 to Mα+Mβ′, and Mβ′ is removed to produce free Mα by an as yet unknown process. Mα then assembles into fibrils and is concomitantly or subsequently cleaved by cysteine proteases into smaller fragments including Mα′. Note that the CTF is trafficked to lysosomes and is not incorporated into functional amyloid fibrils. The fate of Mβ′ is not known. For simplicity, only one subunit of the original dimer is shown after p160. The red lines represent disulfide bonds between Cys-301 and the KLD.
The discovery of higher molecular weight disulfide-bonded p250 and p160 PMEL species that react with an antibody known to detect mature, but not immature, PMEL isoforms originally suggested to us that Golgi-modified forms of PMEL might be covalently bound to one or more unidentified partner proteins. However, several observations led us to conclude that these species contain only PMEL. First, p250 and p160 were detected in both melanocytic cells and transfected HeLa cells. Because pigment cell-specific proteins are not expressed in HeLa cells, this eliminated the possibility of covalent association with a pigment cell-specific binding partner. Second, no polypeptides other than PMEL-derived P2, Mα, and Mβ were consistently detected as components of p250 or p160 by two-dimensional PAGE of PMEL immunoprecipitated from cells metabolically labeled with [35S]methionine/cysteine for an extended period of time. This was surprising given that a binding partner would necessarily be stoichiometrically represented within the disulfide-bonded PMEL complexes. Third, no candidate binding partners of the appropriate molecular weight, relative abundance to PMEL, and subcellular localization were identified by mass spectrometry analysis of trypsin- or AspN-digested p250 and p160 bands (see Table 2 and Supplemental Table S1). Several ER chaperones were detected in moderate abundance; however, these are common contaminants of protein purification by affinity chromatography, and detailed analyses of two abundant protein disulfide isomerase family members revealed interactions with immature ER forms of PMEL but not p250 or p160.4 The mass spectrometry data effectively ruled out the possibility that a stoichiometric constituent of p250 or p160 was missed by two-dimensional PAGE due to poor labeling with [35S]methionine/cysteine. Thus, although it remains remotely possible that a poorly radiolabeled, ubiquitous binding partner might fail to generate peptides detectable by mass spectrometry using either of two proteases, we favor the more likely interpretation that p250 and p160 are composed entirely of PMEL.
With a molecular weight approximately twice that of a PMEL monomer, p250 is most likely a PMEL dimer. The exact composition of p160, however, was less obvious at first. The migration of p160 by non-reducing SDS-PAGE is considerably slower than that of a completely reduced, Golgi-modified, PMEL monomer. We initially interpreted this to suggest that p160 is composed of a full-length PMEL monomer, or linked Mα/Mβ cleavage products, in complex with an additional PMEL fragment such as Mβ. However, the migration of p160 by non-reducing SDS-PAGE was only minimally altered in the absence of Mβ production as tested using the proprotein convertase cleavage site mutant ΔCS. In addition, the naturally occurring p160b band comprising uncleaved, full-length, Golgi-modified P2 contained no additional PMEL fragments when analyzed by two-dimensional PAGE. As such, we interpreted the data to indicate that P2 migrates with a Mr of ∼160,000 when constrained by disulfide bonds but a Mr of ∼120,000 when fully reduced. We speculated that the disulfide-bonded structure of p160 limits accessibility to SDS, thus reducing the migration of this species by non-reducing SDS-PAGE.
Our results are consistent with a model in which the p250 dimer is an obligate precursor of the p160 monomer, because (i) the formation of p250 occurs prior to that of p160 by metabolic pulse-chase analysis, (ii) inhibition of intra-Golgi transport in cells treated with monensin leads to the accumulation of p250 and depletion of p160, and (iii) none of the cysteine mutants produce p160 independently of p250. Complicating this interpretation, however, was the finding that both p250 and p160 contain the full-length Golgi-modified P2 form of PMEL and the Mα/Mβ form cleaved by proprotein convertase when analyzed by two-dimensional PAGE. Because Mα and Mβ are generated by proprotein convertase cleavage of P2 in the trans-Golgi network (31) or endosomes (32), ideally p250 would contain only uncleaved P2, and p160 would contain only cleaved Mα/Mβ. Although these data raise the possibility that p250 represents an independent “dead-end” pathway for PMEL that never generates fibrils, we consider it more likely that p250 is converted to p160 at the same intracellular site but independently of proprotein convertase cleavage such that cleavage can occur before, during, or after resolution of the p250 dimer (Fig. 11).
Our data further suggest that a disulfide bond rearrangement is required to convert p250 to p160. This is because Cys-301, a cysteine residue conserved throughout vertebrate evolution (58), participates in a cross-subunit disulfide bond between two different PMEL molecules within p250 but in a disulfide bond between Mα and Mβ derived from the same PMEL precursor within p160. Although proprotein convertase cleavage of both monomers within the p250 dimer would immediately resolve p250 into a “monomeric” form in which Mα derived from one protomer is linked to Mβ from the other protomer, the observation that p160b can form without proprotein convertase cleavage suggests that the cross-subunit disulfide bond of p250 is broken to form a new disulfide bond connecting Mα and Mβ derived from the same PMEL protomer. The different relative intensity of p250 to p160 in PMEL-expressing HeLa cells as compared with MNT-1 cells further suggests that this process may occur at different rates in the two cell types. Although disulfide bonds are typically formed and remodeled in the ER (59), disulfide bond rearrangements outside the ER have been reported (40, 60). Interestingly, if our model is correct, the disulfide exchange likely occurs without the enzymatic activity of a protein disulfide isomerase; typical ER protein-disulfide isomerases such as PDIA1 and PDIA3 associate exclusively with ER forms of PMEL,4 and melanocytes do not express the only known protein disulfide isomerase of the endocytic pathway, GILT (γ-interferon-inducible lysosomal thiol reductase) (61), unless they are stimulated with γ-interferon (62).5 In addition, failure to treat cells with fresh NEM prior to lysis resulted in poor preservation of the disulfide-bonded PMEL species,4 suggesting that nearby cysteine residues rapidly reduce p250 and p160 when not blocked by alkylation. Because the integrity of the PMEL KLD is required for the resolution of p250 to p160, we speculated that cysteine residues within the KLD initiate this disulfide exchange in an autocatalytic manner. Assessment of this model would require the purification of properly folded recombinant PMEL isoforms, which are not currently available.
Many layers of regulation must exist to protect organisms against the dire consequences of inappropriate amyloid formation. Our data suggest that the KLD protects against premature PMEL fibril formation by regulating the resolution of a disulfide-stabilized PMEL dimer. The importance of an intact KLD is supported by the finding that fibril formation is impaired in HeLa cells expressing the C566S mutant, a variant of PMEL that generates p250 dimers but not p160 monomers. Given that mutagenesis of any single cysteine residue within the KLD, and even a small deletion,6 results in the same biochemical effect on p250 and p160, we predict that any disruption to the cysteine arrangement within the KLD will interfere with fibril formation. Surprisingly, all of the individual cysteine mutants pass quality control checkpoints in the ER, are cleaved by proprotein convertases in the trans-Golgi network or endosomes, and localize appropriately to late endosomes and lysosomes. This suggests that the KLD acts primarily in a post-Golgi compartment to regulate p160 formation and promote the assembly of functional amyloid fibrils. Whether the KLD can promote fibril formation independent of its effect on the disulfide-bonded PMEL species cannot be determined with the present data, but the progressive disulfide bond changes during PMEL maturation suggest that a primary function of the KLD is to regulate these changes.
Intriguingly, mutagenesis of Cys-301, the only cysteine residue integral to the formation of both p250 and p160, does not impair PMEL fibril formation. As shown by size exclusion chromatography, this is most likely because the C301S PMEL variant forms non-covalent complexes similar to the disulfide-bonded species formed by wild-type PMEL. This indicates that Cys-301-dependent disulfide bonds are not required for amyloid fibril formation but rather stabilize energetically favorable conformations at distinct stages of the PMEL biosynthetic pathway. We speculated that loss of this additional stability under physiological expression conditions might lead to premature fibril formation and consequent pathology that is not detectable in the short-term assays used here, as has been observed for several dominant PMEL mutants (63).
PMEL is not the only amyloid protein influenced by disulfide bonds. Reduction of the single disulfide bond in the functional amyloid protein somatostatin-14 in vitro results in fibrils that form faster, are more resistant to denaturation, and have decreased monomer release (64). Reduction of the intrachain and/or one of two interchain disulfide bonds in porcine insulin also accelerates fibril formation (65). Interestingly, the cysteine residues of Tau can participate in intramolecular or intermolecular disulfide bonds; the intramolecular disulfide bonds retard fibril formation by restricting the protein to conformations incompatible with the amyloid fold, whereas the intermolecular disulfide bonds accelerate amyloid fibril formation (66). However, many proteins, including insulin, have the ability to form amyloid aggregates in vitro but do not typically do so in vivo (48). Because many proteins of the secretory pathway contain disulfide bonds, we suggest that the use of disulfide bonds to stabilize pro- or anti-amyloidogenic folding intermediates might be a common mode of regulation for amyloid proteins within the endomembrane system.
Author Contributions
T. H., B. W., and M. S. M. designed the study. T. H. and B. W. generated the reagents and performed and analyzed most of the experiments; L. A. S. and S. H. S. performed the proteomic analysis. T. H. and M. S. M. drafted the manuscript, and all authors edited and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Mark A. Lemmon for helpful discussions, Dewight Williams and the University of Pennsylvania Electron Microscopy Resource Laboratory for assistance with high-pressure freezing, Nathan Roy (Children's Hospital of Philadelphia), John A. Wolf and Michael Grovola (Philadelphia Veterans Affairs Medical Center), and Frank E. Herbert and Jesse B. DeWitt (Nikon Instruments) for assistance with super-resolution microscopy (not included in the manuscript), and James Z. Hui, Andrew Tsourkas, Elizabeth A. Sweeny, and James Shorter (University of Pennsylvania) for assistance with size exclusion chromatography.
This work was supported by National Institutes of Health Grant R01 AR048155 and National Institutes of Health Training Program Grants T32 AR007465 from NIAMS, T32 AG000255 from NIA, and T32 GM007170 from NIGMS. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Table S1.
D. C. Harper and M. S. Marks, unpublished results.
T. Ho, unpublished data.
M. S. Marks, unpublished data.
T. Ho and B. Watt, unpublished data.
- PMEL
- premelanosome protein
- BACE2
- β-site APP-cleaving enzyme 2
- CTF
- C-terminal fragment
- ΔCS
- cleavage site mutant of PMEL
- ER
- endoplasmic reticulum
- KLD
- Kringle-like domain
- NEM
- N-ethylmaleimide
- RPT
- repeat domain.
References
- 1. Chiti F., and Dobson C. M. (2006) Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 [DOI] [PubMed] [Google Scholar]
- 2. Skovronsky D. M., Lee V. M., and Trojanowski J. Q. (2006) Neurodegenerative diseases: new concepts of pathogenesis and their therapeutic implications. Annu. Rev. Pathol. 1, 151–170 [DOI] [PubMed] [Google Scholar]
- 3. Fowler D. M., Koulov A. V., Balch W. E., and Kelly J. W. (2007) Functional amyloid: from bacteria to humans. Trends Biochem. Sci. 32, 217–224 [DOI] [PubMed] [Google Scholar]
- 4. Chapman M. R., Robinson L. S., Pinkner J. S., Roth R., Heuser J., Hammar M., Normark S., and Hultgren S. J. (2002) Role of Escherichia coli curli operons in directing amyloid fiber formation. Science 295, 851–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Majumdar A., Cesario W. C., White-Grindley E., Jiang H., Ren F., Khan M. R., Li L., Choi E. M., Kannan K., Guo F., Unruh J., Slaughter B., and Si K. (2012) Critical role of amyloid-like oligomers of Drosophila Orb2 in the persistence of memory. Cell 148, 515–529 [DOI] [PubMed] [Google Scholar]
- 6. Li J., McQuade T., Siemer A. B., Napetschnig J., Moriwaki K., Hsiao Y. S., Damko E., Moquin D., Walz T., McDermott A., Chan F. K., and Wu H. (2012) The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150, 339–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hammer N. D., Schmidt J. C., and Chapman M. R. (2007) The curli nucleator protein, CsgB, contains an amyloidogenic domain that directs CsgA polymerization. Proc. Natl. Acad. Sci. U.S.A. 104, 12494–12499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Evans M. L., Chorell E., Taylor J. D., Åden J., Götheson A., Li F., Koch M., Sefer L., Matthews S. J., Wittung-Stafshede P., Almqvist F., and Chapman M. R. (2015) The bacterial curli system possesses a potent and selective inhibitor of amyloid formation. Mol. Cell 57, 445–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nenninger A. A., Robinson L. S., and Hultgren S. J. (2009) Localized and efficient curli nucleation requires the chaperone-like amyloid assembly protein CsgF. Proc. Natl. Acad. Sci. U.S.A. 106, 900–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fowler D. M., Koulov A. V., Alory-Jost C., Marks M. S., Balch W. E., and Kelly J. W. (2006) Functional amyloid formation within mammalian tissue. PLos Biol. 4, e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Watt B., van Niel G., Raposo G., and Marks M. S. (2013) PMEL: a pigment cell-specific model for functional amyloid formation. Pigment Cell Melanoma Res. 26, 300–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Theos A. C., Berson J. F., Theos S. C., Herman K. E., Harper D. C., Tenza D., Sviderskaya E. V., Lamoreux M. L., Bennett D. C., Raposo G., and Marks M. S. (2006) Dual loss of ER export and endocytic signals with altered melanosome morphology in the silver mutation of Pmel17. Mol. Biol. Cell 17, 3598–3612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hellström A. R., Watt B., Fard S. S., Tenza D., Mannström P., Narfström K., Ekesten B., Ito S., Wakamatsu K., Larsson J., Ulfendahl M., Kullander K., Raposo G., Kerje S., Hallböök F., Marks M. S., and Andersson L. (2011) Inactivation of Pmel alters melanosome shape but has only a subtle effect on visible pigmentation. PLoS Genet. 7, e1002285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Burgoyne T., O'Connor M. N., Seabra M. C., Cutler D. F., and Futter C. E. (2015) Regulation of melanosome number, shape and movement in the zebrafish retinal pigment epithelium by OA1 and PMEL. J. Cell Sci. 128, 1400–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chakraborty A. K., Platt J. T., Kim K. K., Kwon B. S., Bennett D. C., and Pawelek J. M. (1996) Polymerization of 5,6-dihydroxyindole-2-carboxylic acid to melanin by the pmel 17/silver locus protein. Eur. J. Biochem. 236, 180–188 [DOI] [PubMed] [Google Scholar]
- 16. Lee Z. H., Hou L., Moellmann G., Kuklinska E., Antol K., Fraser M., Halaban R., and Kwon B. S. (1996) Characterization and subcellular localization of human Pmel 17/silver, a 110-kDa (pre)melanosomal membrane protein associated with 5,6-dihydroxyindole-2-carboxylic acid (DHICA) converting activity. J. Invest. Dermatol. 106, 605–610 [DOI] [PubMed] [Google Scholar]
- 17. Kerje S., Sharma P., Gunnarsson U., Kim H., Bagchi S., Fredriksson R., Schütz K., Jensen P., von Heijne G., Okimoto R., and Andersson L. (2004) The Dominant white, Dun and Smoky color variants in chicken are associated with insertion/deletion polymorphisms in the PMEL17 gene. Genetics 168, 1507–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schonthaler H. B., Lampert J. M., von Lintig J., Schwarz H., Geisler R., and Neuhauss S. C. (2005) A mutation in the silver gene leads to defects in melanosome biogenesis and alterations in the visual system in the zebrafish mutant fading vision. Dev. Biol. 284, 421–436 [DOI] [PubMed] [Google Scholar]
- 19. Brunberg E., Andersson L., Cothran G., Sandberg K., Mikko S., and Lindgren G. (2006) A missense mutation in PMEL17 is associated with the silver coat color in the horse. BMC Genet. 7, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Clark L. A., Wahl J. M., Rees C. A., and Murphy K. E. (2006) Retrotransposon insertion in SILV is responsible for merle patterning of the domestic dog. Proc. Natl. Acad. Sci. U.S.A. 103, 1376–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dunn L. C., and Thigpen L. W. (1930) The silver mouse: a recessive color variation. J. Hered. 21, 495–498 [Google Scholar]
- 22. Kwon B. S., Halaban R., Kim G. S., Usack L., Pomerantz S., and Haq A. K. (1987) A melanocyte-specific complementary DNA clone whose expression is inducible by melanotropin and isobutylmethyl xanthine. Mol. Biol. Med. 4, 339–355 [PubMed] [Google Scholar]
- 23. Maresh G. A., Marken J. S., Neubauer M., Aruffo A., Hellström I., Hellström K. E., and Marquardt H. (1994) Cloning and expression of the gene for the melanoma-associated ME20 antigen. DNA Cell Biol. 13, 87–95 [DOI] [PubMed] [Google Scholar]
- 24. Adema G. J., de Boer A. J., Vogel A. M., Loenen W. A., and Figdor C. G. (1994) Molecular characterization of the melanocyte lineage-specific antigen gp100. J. Biol. Chem. 269, 20126–20133 [PubMed] [Google Scholar]
- 25. Raposo G., Tenza D., Murphy D. M., Berson J. F., and Marks M. S. (2001) Distinct protein sorting and localization to premelanosomes, melanosomes, and lysosomes in pigmented melanocytic cells. J. Cell Biol. 152, 809–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Berson J. F., Harper D. C., Tenza D., Raposo G., and Marks M. S. (2001) Pmel17 initiates premelanosome morphogenesis within multivesicular bodies. Mol. Biol. Cell 12, 3451–3464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hurbain I., Geerts W. J., Boudier T., Marco S., Verkleij A. J., Marks M. S., and Raposo G. (2008) Electron tomography of early melanosomes: implications for melanogenesis and the generation of fibrillar amyloid sheets. Proc. Natl. Acad. Sci. U.S.A. 105, 19726–19731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Valencia J. C., Rouzaud F., Julien S., Chen K. G., Passeron T., Yamaguchi Y., Abu-Asab M., Tsokos M., Costin G. E., Yamaguchi H., Jenkins L. M., Nagashima K., Appella E., and Hearing V. J. (2007) Sialylated core 1 O-glycans influence the sorting of Pmel17/gp100 and determine its capacity to form fibrils. J. Biol. Chem. 282, 11266–11280 [DOI] [PubMed] [Google Scholar]
- 29. Harper D. C., Theos A. C., Herman K. E., Tenza D., Raposo G., and Marks M. S. (2008) Premelanosome amyloid-like fibrils are composed of only Golgi-processed forms of pmel17 that have been proteolytically processed in endosomes. J. Biol. Chem. 283, 2307–2322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Berson J. F., Theos A. C., Harper D. C., Tenza D., Raposo G., and Marks M. S. (2003) Proprotein convertase cleavage liberates a fibrillogenic fragment of a resident glycoprotein to initiate melanosome biogenesis. J. Cell Biol. 161, 521–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leonhardt R. M., Vigneron N., Rahner C., and Cresswell P. (2011) Proprotein convertases process Pmel17 during secretion. J. Biol. Chem. 286, 9321–9337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Theos A. C., Truschel S. T., Tenza D., Hurbain I., Harper D. C., Berson J. F., Thomas P. C., Raposo G., and Marks M. S. (2006) A lumenal domain-dependent pathway for sorting to intralumenal vesicles of multivesicular endosomes involved in organelle morphogenesis. Dev. Cell 10, 343–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rochin L., Hurbain I., Serneels L., Fort C., Watt B., Leblanc P., Marks M. S., De Strooper B., Raposo G., and van Niel G. (2013) BACE2 processes PMEL to form the melanosome amyloid matrix in pigment cells. Proc. Natl. Acad. Sci. U.S.A. 110, 10658–10663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kummer M. P., Maruyama H., Huelsmann C., Baches S., Weggen S., and Koo E. H. (2009) Formation of Pmel17 amyloid is regulated by juxtamembrane metalloproteinase cleavage, and the resulting C-terminal fragment is a substrate for γ-secretase. J. Biol. Chem. 284, 2296–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. van Niel G., Charrin S., Simoes S., Romao M., Rochin L., Saftig P., Marks M. S., Rubinstein E., and Raposo G. (2011) The tetraspanin CD63 regulates ESCRT-independent and -dependent endosomal sorting during melanogenesis. Dev. Cell 21, 708–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hoashi T., Tamaki K., and Hearing V. J. (2010) The secreted form of a melanocyte membrane-bound glycoprotein (Pmel17/gp100) is released by ectodomain shedding. FASEB J. 24, 916–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Watt B., van Niel G., Fowler D. M., Hurbain I., Luk K. C., Stayrook S. E., Lemmon M. A., Raposo G., Shorter J., Kelly J. W., and Marks M. S. (2009) N-terminal domains elicit formation of functional Pmel17 amyloid fibrils. J. Biol. Chem. 284, 35543–35555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kushimoto T., Basrur V., Valencia J., Matsunaga J., Vieira W. D., Ferrans V. J., Muller J., Appella E., and Hearing V. J. (2001) A model for melanosome biogenesis based on the purification and analysis of early melanosomes. Proc. Natl. Acad. Sci. U.S.A. 98, 10698–10703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maresh G. A., Wang W. C., Beam K. S., Malacko A. R., Hellström I., Hellström K. E., and Marquardt H. (1994) Differential processing and secretion of the melanoma-associated ME20 antigen. Arch. Biochem. Biophys. 311, 95–102 [DOI] [PubMed] [Google Scholar]
- 40. Chng S. S., Xue M., Garner R. A., Kadokura H., Boyd D., Beckwith J., and Kahne D. (2012) Disulfide rearrangement triggered by translocon assembly controls lipopolysaccharide export. Science 337, 1665–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nichols S. E., Harper D. C., Berson J. F., and Marks M. S. (2003) A novel splice variant of Pmel17 expressed by human melanocytes and melanoma cells lacking some of the internal repeats. J. Invest. Dermatol. 121, 821–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Higuchi R., Krummel B., and Saiki R. K. (1988) A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 16, 7351–7367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Marks M. S. (2001) Determination of molecular size by zonal sedimentation analysis on sucrose density gradients. Curr. Protoc. Cell Biol. Chapter 5, Unit 5.3 [DOI] [PubMed] [Google Scholar]
- 44. Tanford C., Nozaki Y., Reynolds J. A., and Makino S. (1974) Molecular characterization of proteins in detergent solutions. Biochemistry 13, 2369–2376 [DOI] [PubMed] [Google Scholar]
- 45. Lorber B., Bishop J. B., and DeLucas L. J. (1990) Purification of octyl β-d-glucopyranoside and re-estimation of its micellar size. Biochim. Biophys. Acta 1023, 254–265 [DOI] [PubMed] [Google Scholar]
- 46. Reynolds E. S. (1963) The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J. Cell Biol. 17, 208–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chiamenti A. M., Vella F., Bonetti F., Pea M., Ferrari S., Martignoni G., Benedetti A., and Suzuki H. (1996) Anti-melanoma monoclonal antibody HMB-45 on enhanced chemiluminescence-Western blotting recognizes a 30–35 kDa melanosome-associated sialated glycoprotein. Melanoma Res. 6, 291–298 [DOI] [PubMed] [Google Scholar]
- 48. Stefani M., and Dobson C. M. (2003) Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. (Berl.) 81, 678–699 [DOI] [PubMed] [Google Scholar]
- 49. Nishigai M., Osada T., and Ikai A. (1985) Structural changes in α-2- and ovomacroglobulins studied by gel chromatography and electron microscopy. Biochim. Biophys. Acta 831, 236–241 [DOI] [PubMed] [Google Scholar]
- 50. VanAken T., Foxall-VanAken S., Castleman S., and Ferguson-Miller S. (1986) Alkyl glycoside detergents: synthesis and applications to the study of membrane proteins. Methods Enzymol. 125, 27–35 [DOI] [PubMed] [Google Scholar]
- 51. Biaselle C. J., and Millar D. B. (1975) Studies on Triton X-100 detergent micelles. Biophys. Chem. 3, 355–361 [DOI] [PubMed] [Google Scholar]
- 52. Vennegoor C., Hageman P., Van Nouhuijs H., Ruiter D. J., Calafat J., Ringens P. J., and Rümke P. (1988) A monoclonal antibody specific for cells of the melanocyte lineage. Am. J. Pathol. 130, 179–192 [PMC free article] [PubMed] [Google Scholar]
- 53. Tartakoff A. M. (1983) Perturbation of vesicular traffic with the carboxylic ionophore monensin. Cell 32, 1026–1028 [DOI] [PubMed] [Google Scholar]
- 54. Stachel S. J., Coburn C. A., Steele T. G., Jones K. G., Loutzenhiser E. F., Gregro A. R., Rajapakse H. A., Lai M. T., Crouthamel M. C., Xu M., Tugusheva K., Lineberger J. E., Pietrak B. L., Espeseth A. S., Shi X. P., Chen-Dodson E., Holloway M. K., Munshi S., Simon A. J., Kuo L., and Vacca J. P. (2004) Structure-based design of potent and selective cell-permeable inhibitors of human β-secretase (BACE-1). J. Med. Chem. 47, 6447–6450 [DOI] [PubMed] [Google Scholar]
- 55. Kawaguchi M., Hozumi Y., and Suzuki T. (2015) ADAM protease inhibitors reduce melanogenesis by regulating PMEL17 processing in human melanocytes. J. Dermatol. Sci. 78, 133–142 [DOI] [PubMed] [Google Scholar]
- 56. Leonhardt R. M., Vigneron N., Hee J. S., Graham M., and Cresswell P. (2013) Critical residues in the PMEL/Pmel17 N-terminus direct the hierarchical assembly of melanosomal fibrils. Mol. Biol. Cell 24, 964–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Leonhardt R. M., Vigneron N., Rahner C., Van den Eynde B. J., and Cresswell P. (2010) Endoplasmic reticulum export, subcellular distribution, and fibril formation by Pmel17 require an intact N-terminal domain junction. J. Biol. Chem. 285, 16166–16183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Theos A. C., Truschel S. T., Raposo G., and Marks M. S. (2005) The Silver locus product Pmel17/gp100/Silv/ME20: controversial in name and in function. Pigment Cell Res. 18, 322–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Oka O. B., and Bulleid N. J. (2013) Forming disulfides in the endoplasmic reticulum. Biochim. Biophys. Acta 1833, 2425–2429 [DOI] [PubMed] [Google Scholar]
- 60. Liu Y. D., Chen X., Enk J. Z., Plant M., Dillon T. M., and Flynn G. C. (2008) Human IgG2 antibody disulfide rearrangement in vivo. J. Biol. Chem. 283, 29266–29272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. West L. C., and Cresswell P. (2013) Expanding roles for GILT in immunity. Curr. Opin. Immunol. 25, 103–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. O'Donnell P. W., Haque A., Klemsz M. J., Kaplan M. H., and Blum J. S. (2004) Cutting edge: induction of the antigen-processing enzyme IFN-γ-inducible lysosomal thiol reductase in melanoma cells Is STAT1-dependent but CIITA-independent. J. Immunol. 173, 731–735 [DOI] [PubMed] [Google Scholar]
- 63. Watt B., Tenza D., Lemmon M. A., Kerje S., Raposo G., Andersson L., and Marks M. S. (2011) Mutations in or near the transmembrane domain alter PMEL amyloid formation from functional to pathogenic. PLoS Genet. 7, e1002286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Anoop A., Ranganathan S., Das Dhaked B., Jha N. N., Pratihar S., Ghosh S., Sahay S., Kumar S., Das S., Kombrabail M., Agarwal K., Jacob R. S., Singru P., Bhaumik P., Padinhateeri R., Kumar A., and Maji S. K. (2014) Elucidating the role of disulfide bond on amyloid formation and fibril reversibility of somatostatin-14: relevance to its storage and secretion. J. Biol. Chem. 289, 16884–16903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Li Y., Gong H., Sun Y., Yan J., Cheng B., Zhang X., Huang J., Yu M., Guo Y., Zheng L., and Huang K. (2012) Dissecting the role of disulfide bonds on the amyloid formation of insulin. Biochem. Biophys. Res. Commun. 423, 373–378 [DOI] [PubMed] [Google Scholar]
- 66. Walker S., Ullman O., and Stultz C. M. (2012) Using intramolecular disulfide bonds in Tau protein to deduce structural features of aggregation-resistant conformations. J. Biol. Chem. 287, 9591–9600 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.