

ABSTRACT
The modified nucleotides (p)ppGpp play an important role in bacterial physiology. While the accumulation of the nucleotides is vital for adaptation to various kinds of stress, changes in the basal level modulates growth rate and vice versa. Studying the phenotypes unique to the strain lacking (p)ppGpp (ppGpp0) under overtly unstressed growth conditions may be useful to understand functions regulated by basal levels of (p)ppGpp and its physiological significance. In this study, we show that the ppGpp0 strain, unlike the wild type, requires the Lon protease for cell division and viability in LB. Our results indicate the decrease in FtsZ concentration in the ppGpp0 strain makes cell division vulnerable to SulA inhibition. We did not find evidence for SOS induction contributing to the cell division defect in the ppGpp0 Δlon strain. Based on the results, we propose that basal levels of (p)ppGpp are required to sustain normal cell division in Escherichia coli during growth in rich medium and that the basal SulA level set by Lon protease is important for insulating cell division against a decrease in FtsZ concentration and conditions that can increase the susceptibility of FtsZ to SulA.
IMPORTANCE The physiology of the stringent response has been the subject of investigation for more than 4 decades, with the majority of the work carried out using the bacterial model organism Escherichia coli. These studies have revealed that the accumulation of (p)ppGpp, the effector of the stringent response, is associated with growth retardation and changes in gene expression that vary with the intracellular concentration of (p)ppGpp. By studying a synthetic lethal phenotype, we have uncovered a function modulated by the basal levels of (p)ppGpp and studied its physiological significance. Our results show that (p)ppGpp and Lon protease contribute to the robustness of the cell division machinery in E. coli during growth in rich medium.
INTRODUCTION
Adaptation in response to stress is a universal trait among living organisms. In eubacteria and plants, two modified nucleotides, namely, guanosine tetraphosphate (ppGpp) and guanosine pentaphosphate (pppGpp), collectively referred to as (p)ppGpp, have been implicated in adaptation to various nutritional and environmental stresses (1–5). By far the best-studied stress response mediated by (p)ppGpp is the stringent response in the bacterial model organism Escherichia coli, and it is characterized by the repression of rRNA transcription and the activation of amino acid biosynthetic genes. This adaptation requires the RelA-mediated synthesis of (p)ppGpp in response to amino acid starvation and results in the extensive reprogramming of gene expression globally (6, 7). More commonly, the stringent response also refers to responses caused by elevating (p)ppGpp by other means. In E. coli, relA and spoT genes are involved in (p)ppGpp metabolism and are part of the RSH (Rel-Spo homolog) superfamily of genes conserved in eubacteria and plants (3, 8). (p)ppGpp is synthesized in response to amino acid starvation when uncharged tRNA binds to the ribosomal A site and activates the ribosome-bound RelA protein (9, 10). RelA synthesizes pppGpp/ppGpp using GTP and GDP through the transfer of pyrophosphate from ATP. The amount of (p)ppGpp made following amino acid starvation is much higher than the amount of ribosome-bound RelA protein in the cell, indicating multiple rounds of catalysis by each RelA molecule. Based on in vitro and in vivo studies, some mechanistic models have been proposed for how this could be achieved (10–13). SpoT-dependent (p)ppGpp synthesis has been documented in response to deprivations such as iron, phosphorus, fatty acid, and carbon sources (14–19). SpoT is a bifunctional protein with (p)ppGpp synthetase and pyrophosphohydrolase activities; the latter activity generates GTP and GDP from (p)ppGpp, predominates during normal growth conditions, and is reported to be an essential function (20). Based on biochemical and structural studies, models have been proposed for the regulation of the opposing catalytic activities within the protein (21–24).
Consistent with the profound alteration in the gene expression profile induced by (p)ppGpp, RNA polymerase (RNAP) is a primary target of (p)ppGpp (25–28). The regulation of transcription has been shown to occur at the level of initiation, elongation, or competition of sigma factors for the RNA polymerase core (2, 3, 29, 30). Effects of (p)ppGpp on transcription are potentiated by DksA, especially that of the negative and positive regulation of rRNA and amino acid biosynthetic genes, respectively (31–33). DksA is a structural homolog of transcription elongation factors GreA/GreB and regulates transcription through the secondary channel of RNAP (34, 35). However, DksA also functions independently of (p)ppGpp and exhibits complex interplay with its structural homologs GreA and GreB (35, 36).
Intracellular ppGpp content is inversely proportional to growth rate; this is called basal regulation, and it is primarily SpoT and growth rate dependent (37–39). Mutations in SpoT that progressively increase the cellular ppGpp content are associated with a proportionate decrease in growth rate (40). One way to identify the physiological functions modulated by basal (p)ppGpp would be to compare the phenotype of a wild-type or relA mutant strain during growth in rich medium such as LB to that of a relA spoT double mutant that makes no detectable (p)ppGpp (called ppGpp0). Some of the known phenotypes of the ppGpp0 strain are auxotrophy for multiple amino acids, filamentation of cells during growth in minimal media, adhesion, motility defects, and decreased virulence in pathogenic bacteria (20, 36, 41). To facilitate the identification of functions regulated by basal (p)ppGpp, we have been studying synthetic phenotypes specific to the ppGpp0 background (42). This approach in general identifies the functions redundantly regulated by (p)ppGpp and is useful to understand the altered physiological state of cells lacking (p)ppGpp that makes ordinarily nonessential functions important for viability.
In this study, we have characterized the synthetic lethality arising from the deletion of lon, coding for the Lon protease in a ppGpp0 strain during growth in LB. Lon protein of E. coli is an ATP-dependent protease that functions as an oligomer of identical subunits; crystallization of the carboxy-terminal domain containing the proteolytic active site shows that Lon forms a ring-shaped hexamer (43). Lon degrades proteins involved in a variety of biological processes and plays an important role in protein quality control by degrading misfolded proteins prone to aggregation (44). Lon is dispensable for the viability of E. coli, but lon mutants show sensitivity to DNA-damaging agents. The molecular basis of the phenotype is the stabilization of SulA, a substrate of Lon, and a constituent of the SOS regulon, whose expression increases upon DNA damage, leading to an inhibition of cell division by inactivation of FtsZ (45). In a wild-type strain, inhibition of cell division following DNA damage is transient, since sulA expression decreases following the repair of DNA damage and SulA protein is degraded by Lon. Overexpression of Lon has been reported to confer lethality from increased protein degradation, indicating an appropriate amount of the protease is required to ensure the normal progression of cellular processes (46, 47). In this study, we show that Lon is essential for the viability of a ppGpp0 strain, and the ppGpp0 Δlon synthetic lethality arises from the inhibition of the cell division protein FtsZ by SulA. Our data show that the inhibition is not a consequence of SOS induction but is due to a reduction in FtsZ level in the ppGpp0 strain. The results are consistent with the idea that the maintenance of the basal SulA level by Lon protease is important for sustaining cell division when FtsZ concentration decreases.
MATERIALS AND METHODS
Growth conditions and strains.
The Luria-Bertani (LB) medium had 1% tryptone, 0.5% yeast extract, and 1% NaCl. Isopropyl β-d-thiogalactopyranoside (IPTG) and 5-bromo-4-chloro 3-indolyl-b-d-galactoside (X-Gal) were used at a final concentration of 1 mM and 50 μg ml−1, respectively. Unless otherwise indicated, the final concentrations of antibiotics used were the following: ampicillin (Amp), 50 μg/ml for low-copy-number plasmids and 100 μg/ml for high-copy-number plasmids; kanamycin (Kan), 25 μg/ml; spectinomycin (Sp), 12.5 μg/ml; tetracycline (Tet), 10 μg/ml; and chloramphenicol (Cm), 15 μg/ml. The growth temperature was 37°C. The ppGpp0 strain was always maintained in the presence of pRCspoT shelter. Strains used in this study are derivatives of E. coli K-12 and are listed in Table S3 in the supplemental material.
Genetic procedures and plasmid constructions.
Plasmids and primers used in the study are listed in Tables S3 and S4 in the supplemental material. Standard protocols were followed for experiments involving recombinant DNA and plasmid manipulations (48). Transductions, P1 lysate preparations, and transformations were performed by standard procedures (49). pHR53 was constructed from pJK537 (50) by PvuII digestion followed by religation of the plasmid, leading to the truncation of DksA. This plasmid is expected to be identical to pJK533 (50). pRC7 is a single-copy, Ampr, unstable mini-F derivative of pFZY1 (51). spoT and dksA genes were cloned into the EcoRI and HindIII sites of this plasmid following PCR amplification from the MG1655 genome using primer pairs JGOpRC7spoTFP/JGOpRC7spoTRP and JGOpRC7dksAFP/JGOpRC7dksARP to generate pRCspoT and pRCdksA, respectively. The genes show IPTG-dependent expression. An Spr derivative, pRCSp-spoT, was made from pRCspoT by replacing the bla gene with aadA by recombineering using primers JGO_spec_pRChom_FP and JGO_spec_pRChom_RP and published protocols (52). The transcriptional lac fusion at the native sulA gene was made using published procedures (53). Following the introduction of the marked deletions from the Keio collection, the excision of the cassette encoding antibiotic resistance was done using pCP20 (54).
β-Galactosidase assay.
Cultures were grown to an A600 of 0.4 to 0.6 from a 1:100 dilution of overnight cultures, and β-galactosidase activity was assayed and the values reported in Miller units (49). Values reported are the means from at least three independent experiments rounded to the nearest whole number, and the standard deviations of the means are given as error bars.
Plasmid loss and viability measurements.
Overnight cultures of chromosomally Δlac strains grown with selection for the appropriate plasmid(s) in the presence of 1 mM IPTG were washed, and serial dilutions were made in minimal A medium with 10 mM MgSO4. Suitable dilutions were spread on LB agar plates with 1 mM IPTG and X-Gal and scored for blue and white colonies, representing the retention and loss of plasmid, respectively. Plates were scored after 24 h at 37°C, and when white colonies were not observed, incubation was continued for a further 36 h. Whenever a second plasmid was present (along with pRCspoT or pRCSp-spoT), an appropriate antibiotic was used to select for it. Viability was scored by spotting 5-μl aliquots of serial 10-fold dilutions from cultures on appropriate plates, and CFU were scored after 24 to 36 h at 37°C.
Microscopy.
Fresh overnight cultures in LB containing appropriate antibiotics to select for plasmids were diluted 1:100 and taken for microscopy at the exponential phase of growth. For the ppGpp0/pRCspoT strain AN120 and ppGpp0 Δlon Δlac/pRCspoT strain AN519 (Fig. 1B), the overnight culture grown in the presence of Amp and 1 mM IPTG was diluted (1:500) into two flasks, one with LB containing Amp and 1 mM IPTG and the other with LB containing Amp. Following growth to saturation, the cultures were diluted 1:10,000 into the respective medium and cells were taken from exponential phase for microscopy. A similar procedure, but in the presence of appropriate antibiotic, was followed to culture ppGpp0 Δlon Δlac/pRCSp-spoT/pdksA′ (AN688) and ppGpp0 Δlon Δlac/pRCspoT/pCL1920 (AN677) strains. Slides for microscopy were prepared as previously described (55), with some modifications. Briefly, 200 μl of 1% molten agarose was layered on glass slides between two strips of tape, and a clean coverslip was placed on it to obtain a level surface. The coverslip was removed and 5 μl of culture placed on an agarose surface and covered with a fresh coverslip. Microscopy was performed using a 100× oil immersion objective on a Nikon Eclipse 80i microscope, and the differential interference contrast (DIC) images of the cells were captured using NIS-Elements D3.0 software, which also was used for determining mean cell size using at least 100 randomly selected cells. Fluorescence images were captured on a Zeiss LSM 710 Meta inverted confocal microscope.
FIG 1.

Analyses of ppGpp0 Δlon mutant synthetic lethality and its suppression. (A) Retention or loss of pRCspoT plasmid was assessed by blue and white colony color in LB agar plates containing X-Gal and IPTG at 37°C. All strains carried pRCspoT, and their relevant genotypes are indicated above each image. The fraction of white colonies obtained and the actual number of white colonies and total colonies used for calculation are shown in parentheses below each image. (B) Growth of ppGpp0 Δlon/pRCspoT strain (AN519) in LB Amp medium with or without IPTG. Arrows indicate time points at which aliquots were taken for microscopic analysis of cells (shown in Fig. 3 and Table 1). Details about growth conditions are given in the microscopy section in Materials and Methods.
Gel electrophoresis and immunoblotting for FtsZ.
Cultures in mid-exponential phase were normalized using A600 data, solubilized in 1× sample buffer at 99°C for 5 min, and subjected to electrophoresis on 12% sodium dodecyl sulfate (SDS)-polyacrylamide gels. Cell extracts equivalent to an A600 of 0.04 and 0.02 were loaded and run using Tris-glycine-SDS buffer. Separated proteins were electrotransferred to polyvinylidene difluoride (PVDF) membrane (Amersham) and probed using anti-FtsZ primary antibody (rabbit polyclonal), washed, and probed with anti-rabbit IgG conjugated to horseradish peroxidase (HRP) as described previously (48). Membranes were developed with chemiluminescence reagent (Amersham ECL Prime) and visualized with the aid of a chemiluminescence detection system according to the manufacturer's protocol (Sigma Chemical Co., St. Louis, MO). The quantification of band intensity and subtraction of background was done using Image Quant software.
RESULTS
Deletion of lon is synthetically lethal in a ΔrelA ΔspoT (ppGpp0) strain.
In an ongoing study to understand the regulation of the process of trans translation by (p)ppGpp, we studied the effect of deleting the clpP and lon genes that encode proteases that target proteins tagged with SsrA peptide for degradation (56). In cells deleted for relA and spoT genes (referred to as ppGpp0), we were unable to introduce lon deletion by phage P1 transduction and suspected that the ppGpp0 Δlon genetic combination is lethal. To demonstrate synthetic lethality, we made use of the unstable plasmid pRC7, derived from a mini-F based replicon (51), and cloned the spoT gene such that its expression is IPTG dependent (pRCspoT). We confirmed the functionality and IPTG-dependent expression of the spoT gene from its ability to rescue the multiple-amino-acid auxotrophy of the ppGpp0 strain (20) in an IPTG-dependent manner (data not shown). Since the plasmid confers a lac+ phenotype, plasmid loss can be visualized in chromosomally Δlac strains as white colonies on LB agar plates with IPTG and X-Gal. When a wild-type strain carrying the plasmid is cultured overnight with selection for the plasmid and subsequently washed and dilutions spread on plates, typically 20 to 30% of colonies on LB X-Gal IPTG are white, reflective of the extent of pRC7 plasmid instability. If the introduction of the lon deletion in a ppGpp0 strain confers synthetic lethality, we had two expectations: first, the ppGpp0 Δlac Δlon/pRCspoT strain would show IPTG-dependent growth, and second, since plasmid loss would lead to growth inhibition, white colonies would not be recovered in the absence of selection for the plasmid. As shown in Fig. 1Avi and B, these expectations were met, showing that ppGpp0 Δlon genetic combination is lethal; the segregation of white colonies was seen as expected in the wild type; ΔrelA, ppGpp0, Δlon, and Δlon ΔrelA strains also gave rise to white colonies, indicating the loss of pRCspoT does not impair the growth of these strains (Fig. 1Ai to v). To further verify that the ppGpp0 Δlon combination is lethal, the cotransduction frequency of the Δlon::Kan allele with the linked tsx::Tn10 marker was studied by phage P1 transduction. Our results show that the Δlon::Kan allele could be recovered in the wild-type or ΔrelA strain but only in the presence of pRCspoT in the ppGpp0 strain (see Table S1 in the supplemental material). Growth of the Δlon strain is marginally affected in the presence of ΔrelA mutation but completely inhibited in the presence of ΔrelA and ΔspoT mutations (see Fig. S1). The results indicate basal (p)ppGpp levels set by SpoT function are required for the viability of the Δlon strain in LB medium.
The protease and ATPase activity of Lon are essential for the viability of ppGpp0 strain.
In the Lon protease, ATPase and proteolytic activity are part of a single polypeptide chain, and the mutations that distinguish these functions have been studied (57). To study the contribution of each activity to the viability of a ppGpp0 strain, pBADlon constructs were used to express the wild-type, ATPase-deficient (K362Q), protease-deficient (S679A), and ATPase/protease double-deficient alleles of lon; arabinose-dependent expression of the lon alleles in these constructs has been reported (57). Each plasmid was introduced into the ppGpp0 Δlon/pRCSp-spoT strain, and the strains obtained were tested for the segregation of blue and white colonies and plating efficiency in the presence or absence of 0.2% arabinose. We observed arabinose-dependent segregation of white colonies and at least a 103-fold improvement in plating efficiency in the presence of plon+ but not the vector (Fig. 2Ai and ii and B). When the mutant alleles of lon were tested similarly, no arabinose-dependent segregation of white colonies or rescue of plating efficiency was observed (Fig. 2Aiii to v and B). These results show that both the ATPase and protease activity of Lon are required for the viability of the ppGpp0 strain.
FIG 2.

Analyses of Lon function necessary for survival of ppGpp0 strain. (A) Strains carrying the plasmid indicated were scored for retention or the loss of pRCSp-spoT as described in the legend to Fig. 1 in the presence or absence of 0.2% arabinose (ara) in LB Amp IPTG X-Gal plates. Other relevant genotypes of the strains also are indicated. (B) Plating efficiency was determined for the same set of strains in LB containing Amp and Sp in the presence or absence of arabinose.
Deletion of sulA rescues ppGpp0 Δlon lethality.
Two well-studied regulatory proteins that are substrates of Lon are SulA, the inhibitor of the cell division protein FtsZ (58), and RcsA (59), the activator of capsular polysaccharide biosynthesis. The phenotypes of UV sensitivity and mucoidy in a lon mutant arise from the stabilization of SulA and RcsA, respectively; SulA inhibits cell division while RcsA activates capsule synthesis (cps) genes in an RcsB-dependent manner. We tested if the inviability conferred by lon deletion in the ppGpp0 strain is related to the stabilization of these proteins. The sulA and rcsB gene deletions sourced from the Keio collection (60) were individually introduced into the ppGpp0 Δlon/pRCspoT strain by phage P1 transduction, and strains obtained were scored for the ability to survive the loss of pRCspoT plasmid by screening for segregation of white colonies on LB IPTG X-Gal plates. As shown in Fig. 1Avii, segregation of white colonies was observed in the presence of sulA deletion, indicating that it suppressed the synthetic lethality of the ppGpp0Δlon strain. The ppGpp0 Δlon ΔrcsB/pRCspoT strain behaved identically to the isogenic rcsB+ parent and could not survive the loss of pRCspoT (Fig. 1Aviii). The results indicate that the stabilization of SulA but not RcsB is a requirement for ppGpp0 Δlon strain synthetic lethality. Since a well-studied target of SulA is FtsZ, to which it binds and inhibits cell septation, we examined cells microscopically for cell division defects.
ppGpp0 Δlon mutant shows extensive filamentation that is suppressed by sulA deletion.
lon mutants form filaments following UV irradiation (61), and filamentation has been reported previously for the ppGpp0 strain during growth in synthetic medium (20). To understand the cause of growth inhibition in the ppGpp0 Δlon strain in LB, we compared the cell morphology of the wild-type, ppGpp0, Δlon, and ppGpp0 Δlon/pRCspoT strains grown under permissive and nonpermissive conditions; the mean cell size for each strain was calculated from the captured images. As seen from Fig. 3A to E and the mean cell size quantified in Table 1, cells of the ppGpp0 and Δlon mutant strains were longer than those of the wild-type or ΔrelA strains; the cell size of the ΔrelA Δlon mutant was similar to that of the Δlon mutant. In the ppGpp0 strain carrying pRCspoT, induction of spoT expression results in a cell size that is similar to that of the wild type, indicating that the expression of SpoT is sufficient (that is, even in the absence of RelA function) to restore cell size under these growth conditions (Fig. 3A, F, and G and Table 1). In the ppGpp0 Δlon mutant cultured under permissive conditions, the cell size is more similar to that of the wild type than that of the ppGpp0 strain (Fig. 3A, C, and H and Table 1); however, when cultured under nonpermissive conditions, extensive filamentation was observed (Fig. 3I) associated with the cessation of growth (Fig. 1B). Filaments in the ppGpp0 Δlon strain are not uniform in their morphology; some filaments seem to be disintegrating and appear as a string of vesicles (indicated with an arrow in Fig. 3I). We examined the effect of sulA deletion on filamentation. The morphology and size of the ΔsulA mutant were comparable to those of the wild-type strain (Fig. 3A and J and Table 1). When the ΔsulA allele was introduced into the ppGpp0 Δlon background, it suppressed filamentation (Fig. 3L and Table 1); the cell size and morphology of the ppGpp0 Δlon ΔsulA strain were similar to those of the ppGpp0 or the ppGpp0 ΔsulA strain (Fig. 3C, K, and L and Table 1). These results indicate that (i) inhibition of cell septation by SulA was responsible for the growth arrest in the ppGpp0 Δlon strain, and (ii) SulA function may not be solely responsible for the larger size of the ppGpp0 cells. Although the average cell size for the ppGpp0 Δlon strain (Fig. 3I) was calculated to be ∼50 μm (Table 1), this is an underestimate, because long filaments that could not be captured in one field were frequently observed and not used for calculation of mean cell size. Since some of the filaments were found to disintegrate (Fig. 3I), this also should contribute to the growth defect of the ppGpp0 Δlon mutant.
FIG 3.

DIC microscopy. Images of cells from different strains are shown with their relevant genotypes and strain numbers indicated above. All strains were grown in LB; whether Amp or Amp and IPTG also were present is indicated below each image. Bars, 10 μm. See the text for details.
TABLE 1.
Mean cell sizes of selected strains used in this study
| Figure | Relevant genotype (strain) | Medium | Mean cell size ± SD (μm)a |
|---|---|---|---|
| 3A | Wild type (MG1655) | LB | 2.6 ± 0.3 |
| 3B | ΔrelA (CF12510) | LB | 2.52 ± 0.47 |
| 3C | ΔrelA ΔspoT (HR202) | LB | 6.2 ± 3.3 |
| 3D | Δlac::FRT Δlon::Kan (AN207b) | LB | 3.9 ± 1.6 |
| 3E | ΔrelA Δlon::Kan (AN844b) | LB | 4.2 ± 2.9 |
| 3F | ΔrelA ΔspoT/pRCspoT (AN120) | LB Amp IPTG | 2.7 ± 0.3 |
| 3G | ΔrelA ΔspoT/pRCspoT (AN120) | LB Amp | 6.1 ± 1.7 |
| 3H | ΔrelA ΔspoT Δlon::Kan/pRCspoT (AN519) | LB Amp IPTG | 3.6 ± 0.6 |
| 3I | ΔrelA ΔspoT Δlon::Kan/pRCspoT (AN519) | LB Amp | 55.8 ± 34.1 |
| 3J | Δlac::FRT ΔsulA::Kan (AN803) | LB | 2.6 ± 0.48 |
| 3K | ΔrelA ΔspoT ΔsulA::FRT (AN565b) | LB | 4.5 ± 2.7 |
| 3L | ΔrelA ΔspoT ΔsulA::FRT Δlon::Kan (AN567b) | LB | 5.0 ± 3.3 |
| 3M | ΔrelA ΔspoT Δlon::Kan lexA3/pRCspoT (AN648) | LB Amp | 41.5 ± 32.9 |
| 5Ci | ΔrelA ΔspoT Δlon::Kan/pRCSp-spoT/pdksA′ (AN688) | LB Sp Amp | 51.5 ± 29.4 |
| 5Cii | ΔrelA ΔspoT Δlon::Kan/pdksA (AN689b) | LB Amp | 4.0 ± 1.1 |
| 5Ciii | ΔrelA ΔspoT Δlon::Kan/pdksANN (AN690b) | LB Amp | 4.6 ± 0.6 |
| 6Ci | ΔrelA ΔspoT Δlon::Kan/pRCspoT/pCL1920 (AN677) | LB Amp Sp | 52.8 ± 37.6 |
| 6Cii | ΔrelA ΔspoT Δlon::Kan/pCLftsQAZ (AN678b) | LB Sp | 3.1 ± 0.4 |
| 6Ciii | ΔrelA ΔspoT Δlon::Kan/pCLsdiA (AN679b) | LB Sp | 3.7 ± 0.6 |
Size was calculated using at least 100 cells.
A white colony obtained from the strain was used.
SOS induction is not a feature of ppGpp0 or ppGpp0 Δlon ΔsulA strains.
Since SulA function was required for growth inhibition in the ppGpp0 Δlon strain, we tested if an increase in cellular SulA expression is associated with growth inhibition. A chronic SOS induction in the ppGpp0 strain could elevate the cellular SulA concentration sufficiently to make cell division contingent on the presence of functional Lon. This has been reported to be the basis for synthetic lethality observed in lon mutants with mutations that induce SOS (62). To monitor SOS induction, we generated a sulA-lacZ transcriptional fusion at the native sulA locus and used it to monitor expression in different genetic backgrounds. As expected, in the presence of sublethal concentrations of mitomycin C (which causes DNA damage), we observed a 2.5-fold induction of β-galactosidase activity with the fusion (Fig. 4). No induction of β-galactosidase expression was observed in the ppGpp0 or Δlon strain compared to that of the wild type during growth in LB, indicating that there is no SOS induction in these strains under our growth conditions and that the expression of sulA is not regulated by (p)ppGpp. We also tested if SOS induction is associated with the absence of (p)ppGpp and Lon function using the ppGpp0 Δlon sulA-lacZ strain, which is viable since it lacks functional SulA. The induction of sulA-lacZ was not observed in this strain either (Fig. 4), and it showed the least expression among the strains tested. The results indicate the inhibition of cell septation by SulA due to chronic SOS induction is unlikely to account for the essentiality of Lon function in the ppGpp0 strain and the longer cells in the ppGpp0 and ppGpp0 Δlon ΔsulA strains.
FIG 4.

Measurement of β-galactosidase activity from sulA-lacZ transcriptional fusion. Strains whose relevant genotypes are indicated were grown to mid-exponential phase in LB, and β-galactosidase activity was measured. The values were calculated from at least three independent experiments. Error bars indicate standard deviations of the means. Mitomycin C (MC) was added to a final concentration of 0.5 μg/ml. WT (AN658), srl300::Tn10 (AN837), recA srl300::Tn10 (AN838), lexA3 (AN839), ppGpp0 (AN660), Δlon (AN774), and ppGpp0 Δlon (AN743) strains were used.
Since SOS induction requires functional RecA and LexA cleavage (63), to further rule out a role for SOS, the recA deletion and the lexA3 (Ind−) allele, encoding the noninducible form of the repressor, were introduced separately by phage P1 transduction into the ppGpp0 Δlon/pRCspoT strain. The ability of the ΔrecA or lexA3 derivative to survive the loss of pRCspoT was similar to that of the parent strain (see Table S2 in the supplemental material, compare AN519, AN648, and AN750). The ppGpp0 Δlon lexA3/pRCspoT strain showed IPTG-dependent growth (see Fig. S2A) and filamentation during growth in the absence of IPTG (Fig. 3M and Table 1), indicating that spoT expression was essential for viability and that the loss of viability was associated with filamentation. Unlike the case in the ppGpp0 Δlon lexA3 strain, pRCspoT plasmid was unstable in the ppGpp0 lexA3 mutant, indicating that lexA3 did not inhibit plasmid loss in any fashion (see AN647 in Table S2 and Fig. S2B). recA or lexA3 mutation did not alter the expression of sulA-lac fusion under our growth conditions (Fig. 4). From these results we rule out a role for SOS induction in SulA-mediated growth inhibition observed in the ppGpp0 Δlon strain and that the lexA3 or recA mutation does not alter sulA expression under our growth conditions.
Overexpression of DksA or DksANN and stringent RNA polymerase mutations restores viability in ppGpp0 Δlon mutant.
As mentioned in the introduction, RNA polymerase is a primary target for (p)ppGpp. DksA regulates transcription synergistically with (p)ppGpp through its interaction with the RNA polymerase (RNAP) secondary channel and potentiates both positive and negative regulatory functions mediated by (p)ppGpp. We tested if a ΔdksA Δlon strain exhibited a synthetic phenotype similar to that of the ppGpp0 Δlon strain. Following a strategy similar to that described above to study the ppGpp0 Δlon synthetic lethality, we cloned DksA in plasmid pRC7 to construct plasmid pRCdksA and verified the functionality of DksA by using it to complement a ΔdksA mutant for growth in minimal glucose in the presence of IPTG (data not shown). The lon deletion was introduced into the ΔdksA/pRCdksA strain by phage P1 transduction, and the ΔdksA Δlon::Kan/pRCdksA strain obtained was maintained in the presence of Amp and IPTG. We studied the ability of this strain to survive the loss of pRCdksA by scoring for segregation of white colonies. As shown in Fig. 5Ai, the segregation of white colonies was observed and there was no drop in plating efficiency in the absence of IPTG (data not shown). We conclude that unlike the case for a ppGpp0 strain, lon deletion is not lethal in the ΔdksA mutant.
FIG 5.

Suppression of ppGpp0 Δlon synthetic lethality by overexpression of DksA or DksANN and stringent RNAP mutations. (A) Retention or loss of the indicated pRC7-derived plasmids was assessed as described in the legend to Fig. 1. Relevant genotypes of the strains and the plasmids, when present, are indicated. The medium used was LB IPTG X-Gal alone (i) or with Amp included (ii to iv). (B) Plating efficiency of the indicated strains in LB containing Amp and Sp with or without IPTG. (C) DIC images of cells cultured in LB Amp; relevant genotypes, plasmids, and strain numbers are indicated above the panels. A superscript a indicates that a white colony from the strain was used for scoring cell morphology. Bars, 10 μm. (D) Plating efficiency in LB containing Amp in the presence or absence of IPTG. Strain numbers and their relevant genotypes are indicated.
Since overexpression of DksA can partially compensate for (p)ppGpp-regulated functions in the absence of (p)ppGpp (32, 33), we studied the effect of DksA overexpression using the multicopy plasmid pJK537 (50). pJK537 was introduced into the ppGpp0 Δlon/pRCSp-spoT strain, and subsequently pRCSp-spoT plasmid loss and plating efficiency were examined. As seen from Fig. 5A to C and Table 1, segregation of white colonies, increase in plating efficiency, and inhibition of filamentation were observed in the presence of plasmid pJK537. Plasmid pHR53, a derivative of pJK537 that is equivalent to pJK533 (50), wherein the DksA protein is truncated after the 36th amino acid and did not support the segregation of white colonies, failed to suppress the growth defect or filamentation. The results show that overexpression of DksA restored cell division and growth, probably by compensating for functions missing or altered in the ppGpp0 Δlon strain. DksANN is an allele of DksA with mutations in two conserved acidic amino acid residues (D71N and D74N) located at the tip of the coiled-coil domain, and it interacts with RNAP residues through the secondary channel (31). The D74 residue has been shown to be vital for the modulation of RNAP function by DksA (31, 64). When DksANN was expressed from a multicopy plasmid in the ppGpp0 Δlon/pRCSp-spoT strain, segregation of white colonies, a 4-log increase in plating efficiency, and suppression of filamentation were observed that were similar to those seen with wild-type DksA (Fig. 5A to C and Table 1). The results indicate functions associated with the acidic amino acid residues are not important for suppression of the ppGpp0Δlon lethality by DksA. DksANN overexpression can rescue the amino acid auxotrophy of the ppGpp0 strain (65), bradytrophic growth of the ΔdksA mutant in minimal glucose (data not shown), and the replication fork progression defect in the ΔdksA strain (65, 66).
Genetic evidence suggesting that RNAP was the target of (p)ppGpp came from the discovery of mutations that rescued the multiple-amino-acid auxotrophy (called the M− phenotype) of the ppGpp0 strain. The mutants were generically referred to as M+ mutants, and the few tested mimicked in vitro and in vivo the physiology and transcription regulation conferred by (p)ppGpp even in its absence (2, 67). We chose two mutations, one isolated in the Cashel laboratory, rpoBL571P (68), and another isolated by McGlynn and Lloyd, rpoB*35, which additionally rescued the UV sensitivity of the ppGpp0 ΔruvAC strain (69), and tested their ability to rescue the growth defect of the ppGpp0 Δlon strain. As expected, both mutations improved the plating efficiency of the ppGpp0 Δlon/pRCspoT strain in the absence of IPTG (Fig. 5D) and allowed the segregation of white colonies on LB IPTG X-Gal plates (data not shown).
Rescue of ppGpp0 Δlon synthetic lethality by FtsZ overexpression and absence of FtsZ ring in the ppGpp0 Δlon filaments.
Evidence from previous studies has indicated the positive regulation of cell division by (p)ppGpp (70–72). The expression of the ftsQAZ operon and (p)ppGpp concentration have been reported to show regulation that is the inverse of the growth rate (40, 73), which is suggestive of a positive regulatory role for (p)ppGpp in ftsQAZ expression. However, the increase in FtsZ concentration with the reduction in growth rate could not be reproduced in a later study (74). Studies have shown that SulA inhibits cell division by sequestration of FtsZ monomers and thereby inhibits FtsZ ring formation (55, 75). Biochemical and biophysical studies indicate that the affinity of an FtsZ monomer to SulA is similar to that for the ends of a growing FtsZ protofilament (75). We reasoned that if sequestration of FtsZ by SulA is facilitated due to a decrease in the cellular FtsZ level in the absence of (p)ppGpp and an increase in the basal SulA level due to the absence of Lon protease, it could result in the inhibition of cell division. If this was the case, increasing the concentration of FtsZ should be able to suppress the extensive filamentation observed in the ppGpp0 Δlon mutant. To test this, we used low-copy-number pSC101 plasmids, namely, pCLftsQAZ (76) and pSCftsQAZ (77), which have been shown to increase cellular levels of FtsQ, FtsA, and FtsZ, and we asked if the ppGpp0 Δlon synthetic lethality can be suppressed in the presence of each plasmid. As shown in Fig. 6A and B, both plasmids allowed survival in the absence of pRCspoT and rescued the plating defect of the ppGpp0 Δlon strain. Similarly, the overexpression of SdiA, a known positive regulator of the ftsQAZ operon (78), using plasmid pCLsdiA (76) also allowed the segregation of white colonies and rescued the plating defect, indicating that positive regulation by SdiA is independent of (p)ppGpp. The results show that coordinately increasing the concentration of FtsQ, FtsA, and FtsZ proteins using plasmids or the induction of the chromosomal genes using a transcriptional activator results in the suppression of ppGpp0 Δlon synthetic lethality. Suppression was tested following the coordinate increase in the expression of the ftsQAZ genes, since maintaining the ratio of these proteins is important for the progression of cell division (79, 80). However, we also tested if an increase in FtsZ concentration is sufficient for suppression using pCA24NftsZ plasmid from the ASKA library (81) that allows IPTG-regulated expression of FtsZ from Ptac. The presence of the plasmid but not the vector supported survival in the absence of pRCspoT, rescued the plating defect in the absence of IPTG (Fig. 6A and B), and inhibited growth in the presence of IPTG, probably due to FtsZ overexpression (Fig. 7B) (82). The suppression of the synthetic lethality in the absence of IPTG indicates the basal-level expression of FtsZ from a multicopy plasmid could sufficiently elevate the FtsZ concentration to allow the suppression of the ppGpp0 Δlon synthetic lethality and yet not perturb the ratio of FtsZ with respect to the other cell division proteins so as to inhibit cell septation. The results support the idea that the sequestration of FtsZ monomers by SulA and the consequent decrease in FtsZ levels below the critical concentration required for the assembly of Z rings is the cause of synthetic lethality in the ppGpp0 Δlon mutant; the overexpression of FtsZ inhibits the sequestration of FtsZ monomers by SulA.
FIG 6.

Rescue of ppGpp0 Δlon synthetic lethality by FtsZ overexpression and absence of FtsZ ring from ppGpp0 Δlon filaments. (A) Retention or loss of plasmid pRCspoT was assessed as described in the legend to Fig. 1. Strain numbers, relevant genotypes, and plasmids are indicated. Media used included LB containing IPTG and X-Gal with Sp (i to iii) or Tet (iv), LB containing X-Gal and Cm (v and vi), or X-Gal and 0.1 mM IPTG (vii). (B) Plating efficiency of strains used in panel A with or without IPTG. Appropriate antibiotics were included to select for the plasmids. (C) DIC image of cells cultured in LB containing Amp and Sp (i) or Sp alone (ii and iii); a superscript a indicates that a white colony obtained from the indicated strain was studied. Bars, 10 μm. (D) Fluorescence microscopy of ppGpp0 Δlon attλ::P208-ftsZ-gfp/pRCSp-spoT strain (AN840) following growth in LB containing 0.1 mM IPTG with (i) or without (ii and iii) Sp. Relevant genotypes, plasmids, and strain numbers are indicated above each image.
FIG 7.
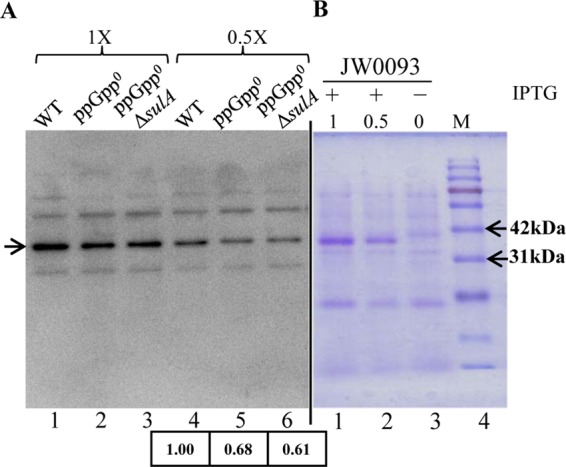
Reduction in FtsZ level in the ppGpp0 strain. (A) Samples were harvested during exponential growth in LB from strains whose relevant genotypes are indicated. Extracts from cells equivalent to an A600 of 0.04 (1×) and 0.02 (0.5×) were loaded, and immunoblotting was performed using anti-FtsZ antibody. The band corresponding to FtsZ is indicated by an arrow, and the relative FtsZ levels, calculated by quantification of band intensity and normalized to the wild type, is indicated below the lanes. (B) Coomassie-stained gel showing IPTG-dependent expression of FtsZ from pCA24NftsZ. The molecular masses of two marker bands are indicated for reference. WT (white colony from AN131), ppGpp0 (white colony from AN120), and ppGpp0 ΔsulA (white colony from AN565) strains were used.
To ask if filamentation in the ppGpp0 Δlon strain is accompanied by the loss of FtsZ ring formation, ftsZ-gfp integrated at the λ-att site and expressed from an attenuated Ptrc promoter, made from the P208-ftsZ-gfp construct of Weiss et al. (83), was obtained from Manjula Reddy and introduced into the ppGpp0 Δlon/pRCSp-spoT mutant by phage P1 transduction. Segregation of blue and white colonies was observed in the strain in the presence of 0.1 mM IPTG (Fig. 6Avii; also see Table S2 in the supplemental material), indicating that the increased expression of FtsZ-green fluorescent protein (FtsZ-GFP) also could suppress the ppGpp0 Δlon synthetic lethality. We examined cells by fluorescence microscopy to look for the GFP associated with the FtsZ ring. In the presence of pRCSp-spoT, cell division appears normal, and in cells the GFP signal is localized with the position being consistent with that of the cell septum (Fig. 6Di). When cultured in the absence of selection for pRCSp-spoT, we observed both normal and filamentous cells (Fig. 6Dii and iii), reflecting the partial suppression of the ppGpp0 Δlon synthetic phenotype and consistent with the slower growth of the white colonies relative to that of the blue colonies shown in Fig. 6Avii. Some of the filamentous cells did not have GFP signal while others had faint or intense GFP signal distributed throughout the filament, but none showed localized GFP expression. Assuming the filamentous cells observed here and in the ppGpp0 Δlon strain arise from the same molecular defect, we propose that the decrease in FtsZ concentration and consequent inability to form FtsZ rings leads to cell filamentation and the loss of viability in the ppGpp0 Δlon strain.
Reduction in FtsZ protein level in the ppGpp0 strain.
By immunoblotting using anti-FtsZ antibody, we compared cellular FtsZ concentrations in the extracts of wild-type, ppGpp0, and ppGpp0 ΔsulA strains normalized using A600 measurements (Fig. 7A). A band migrating at a position corresponding to a size of FtsZ (40 kDa) showed decreased intensity in the cell extracts of ppGpp0 and ppGpp0 ΔsulA mutants compared to that of the wild-type extract. This band also comigrates with the band that appears following IPTG induction in the cell extract of JW0093 carrying plasmid pCA24NftsZ (Fig. 7B), where ftsZ is induced from the Tac promoter (pCA24NftsZ could suppress the ppGpp0 Δlon synthetic lethality in the absence of IPTG) (Fig. 6B). Quantification of the band intensity showed 32% and 39% reduction in FtsZ concentration in the ppGpp0 and ppGpp0 ΔsulA strains, respectively. That the FtsZ concentration was similarly low in the ppGpp0 and ppGpp0 ΔsulA strains suggests the suppression of ppGpp0 Δlon mutant lethality by ΔsulA is not due to the restoration of FtsZ concentration and that cell septation is restored despite the reduction in FtsZ concentration.
The 30% reduction in FtsZ content in ppGpp0 strain is consistent with the longer mean length of the cells (Table 1 and Fig. 3C). Increasing the expression of FtsZ in the ppGpp0 Δlon strain decreases the mean cell length, and the cells are significantly smaller than the ppGpp0 cells (Table 1, HR202 and AN678, and compare Fig. 3C to 6Cii). This indicates that the lower level of FtsZ already impairs cell division in the ppGpp0 strain and leaves it poised for filamentation.
Why is lon mutation required for filamentation in the ppGpp0 strain? We think the lon deletion could increase basal SulA levels and facilitate the sequestration of FtsZ, whose levels are low in the ppGpp0 strain, and tip the balance in favor of filamentation. This would imply that the Lon protease, in addition to ensuring the restoration of cell division when the SOS signal recedes, can buffer cell division under suboptimal FtsZ concentrations.
DISCUSSION
Filamentation observed in the ppGpp0 cells during growth in minimal medium with glucose and amino acids suggests a division-promoting role for the nucleotide (20). We find that during growth in LB, the ppGpp0 cells are significantly longer than those of the wild type, and this is associated with a ∼30% reduction in the cellular FtsZ concentration (Fig. 7). Since a 30 to 40% reduction in FtsZ levels can lead to filamentation and growth inhibition (84), we suggest that the ppGpp0 strain is poised for filamentation. The growth arrest and filamentation in the ppGpp0 Δlon strain and the suppression of these phenotypes in the ppGpp0 Δlon ΔsulA mutant indicate cell division is inhibited by SulA in the ppGpp0 strain in the absence of the Lon protease. Since we did not observe an increase in the activity of sulA-lacZ fusion in the ppGpp0 or ppGpp0 Δlon strains (Fig. 4), we believe the inhibition of cell division by SulA is not due to an increase in sulA expression but mainly due to the reduction in the FtsZ concentration. Based on the rescue of cell division in the ppGpp0 Δlon mutant by sulA inactivation or overexpression of FtsZ, and taking into consideration the proposed mechanism of inhibition of cell division by SulA (55, 75, 85), we propose the following model. The level of FtsZ in a ppGpp0 background is low but enough to sustain growth; however, the reduction in the FtsZ concentration increases its susceptibility to inhibition by SulA. An increase in the basal SulA level in the absence of Lon protease and (p)ppGpp leads to the sequestration of FtsZ monomers and pushes the monomer concentration below the threshold required to maintain the protofilament assembly and cell division.
Previous studies have shown that conditions that increase cellular (p)ppGpp concentrations confer the suppression of cell division defects, specifically, (i) inhibition of cell division in the spherical cells that arise following the inhibition of penicillin binding protein (PBP2) can be restored by increasing (p)ppGpp concentration (86, 87), and (ii) an increase in the (p)ppGpp pool suppressed the temperature sensitivity of ftsZ84(Ts) strains (71). However, the mechanism of restoration of cell division by (p)ppGpp is not clear. Although RNAP is a primary target of (p)ppGpp, in studies that looked at the effect of the molecule on transcription, no increase in transcription from some of the promoters characterized for ftsQAZ genes could be documented (71, 88). The suppression of ftsZ84 temperature sensitivity by (p)ppGpp was associated with a 2- to 3-fold decrease in transcription from Qp1 or Qp2 promoters and no changes in transcription from the pZ set of promoters (71). The authors had speculated that the suppression of the division defect by (p)ppGpp could arise from an increase in the ratio of FtsZ to FtsA and/or FtsQ or due to the effect of (p)ppGpp on transcription from the multiple promoters upstream of FtsZ, resulting in the alleviation of promoter occlusion (89). Despite the lack of evidence for transcription activation, the suppression of the temperature sensitivity of ftsZ84 was associated with a 4-fold increase in FtsZ84 (71); however, no increase in the FtsZ content was evident in the strains where the basal (p)ppGpp level was elevated 2-fold and sufficient to restore division in the absence of PBP2 (88). That is, the effect was restricted to the mutant.
An inverse correlation between ftsQAZ transcription (and FtsZ protein content) and growth rate (73) suggested a positive regulatory role for (p)ppGpp, whose concentration shows a similar correlation with growth rate (37, 39). However, later studies have not reproduced the growth rate-dependent increase in FtsZ protein content in E. coli or B. subtilis (74). Since the size of the cell reduces with a decrease in growth rate (90), how does this relate to FtsZ? From studies on mutants with altered cell size, it has been postulated that despite the amount of FtsZ being constant, cell size is controlled by the amount of FtsZ available for the assembly of Z rings and division (91).
lon mutation exhibits incompatibility/sickness, with mutations that induce SOS, and is rescued by inactivation of sulA or the lexA3 mutation, which abrogates SOS induction (62), indicating that the Lon protease is essential to prevent the inhibition of cell division following a moderate increase in sulA expression. However, we do not find evidence for SOS induction in the ppGpp0 or ppGpp0 Δlon mutants (Fig. 4), and as would be expected, viability was not restored by recA or lexA3 mutation (see Fig. S2B and Table S2 in the supplemental material), which also did not alter sulA expression under our growth conditions (Fig. 4). In the absence of SOS induction, the steady-state level of SulA in lon mutants can be higher than that in a wild-type strain, since SulA is rapidly degraded by Lon. To our knowledge, the extent of increase in basal SulA has not been documented under different growth conditions. It is possible that the SulA level in the ppGpp0 Δlon mutant is higher than that in the lon mutant and contributes to the division inhibition. It has been reported that the protease-deficient lon mutant (S679A), unlike the ATPase-deficient mutant (K362Q), traps SulA following UV exposure and partially restores cell division (57). In this study, the behavior of the two mutant alleles was similar (Fig. 2B). One explanation is the absence of an increase in SulA expression under our experimental conditions. High concentrations of ppGpp or polyphosphate (850 μM) inhibit Lon activity in vitro (92). Since the intracellular (p)ppGpp concentration during growth in rich medium is low (2), direct modulation of Lon activity or indirect modulation through polyphosphate is less likely to contribute to the regulation of cell division by (p)ppGpp in rich medium.
Although Lon protease functions to keep the inhibition of cell division transient during DNA damage through the degradation of SulA, if the cell division is allowed to continue after DNA damage in the Δlon ΔsulA mutant, viability remains unaffected and therefore the physiological relevance of cell division arrest is not apparent (45). It is possible that a physiologically relevant function of Lon with respect to cell division is the regulation of basal levels of SulA so as to buffer cell division against the transient reduction in the FtsZ level or conditions unfavorable for FtsZ polymerization that make it vulnerable to sequestration by SulA. Studies have shown that the lon mutant does not adapt well to the shift-up from poor to rich media and transiently filaments (93, 94). The filamentation of the lon mutant during shift-up is not seen in the presence of sfiB103 (an ftsZ allele resistant to SulA) (95). Although the role of sulA was not explored, a simple model to explain the observations is that the basal level of SulA is elevated in the lon mutant and, following shift-up, the FtsZ concentration drops and is sequestered by SulA, leading to the inhibition of septation. Consistent with this model, the inactivation of sulA did not affect cell size in a lon+ strain following shift-up (96), implying that the low basal level of SulA in a lon+ mutant cannot inhibit cell division even if the FtsZ concentration decreases during shift-up. These studies show that the regulation of SulA level by Lon is important even under growth conditions where there is no DNA damage but possibly a drop in FtsZ. We think a drop in FtsZ concentration following shift-up could be related to the decrease in (p)ppGpp concentration (2) and is being investigated.
HslUV has been reported as a secondary protease that can target SulA and, at higher growth temperatures, can alleviate the UV sensitivity of lon mutants (97, 98). Mutations that reduce sulA expression, sulA null mutation, or ftsZ alleles that confer resistance to SulA can suppress the growth defect of strains lacking all known cytoplasmic AAA+ proteases in LB, indicating that SOS induction is not always a requirement for the inhibition of cell division by SulA (97). Further studies to understand physiological conditions that render FtsZ vulnerable to SulA inhibition and the role of (p)ppGpp therein are being pursued.
Supplementary Material
ACKNOWLEDGMENTS
We thank Manjula Reddy, Tom Bernhardt, Susan Gottesman, and Mike Cashel for kind gifts of strains, plasmids, and FtsZ antibody used in this study. We gratefully acknowledge strains used from the Keio collection (NBRP, Japan). We thank members of the Laboratory of Bacterial Genetics for suggestions.
This work was funded primarily by a Center of Excellence in Microbial Biology research grant of the Department of Biotechnology, Government of India. A.N. is a recipient of a CSIR fellowship.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00693-15.
REFERENCES
- 1.Braeken K, Moris M, Daniels R, Vanderleyden J, Michiels J. 2006. New horizons for (p)ppGpp in bacterial and plant physiology. Trends Microbiol 14:45–54. doi: 10.1016/j.tim.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 2.Cashel M, Gentry D, Hernandez VJ, Vinella D. 1996. The stringent response, p 1458–1496. In Neidhardt FC, Curtiss R III, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE (ed), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed, vol 1 ASM Press, Washington, DC. [Google Scholar]
- 3.Potrykus K, Cashel M. 2008. (p)ppGpp: still magical? Annu Rev Microbiol 62:35–51. doi: 10.1146/annurev.micro.62.081307.162903. [DOI] [PubMed] [Google Scholar]
- 4.Tozawa Y, Nomura Y. 2011. Signalling by the global regulatory molecule ppGpp in bacteria and chloroplasts of land plants. Plant Biol 13:699–709. doi: 10.1111/j.1438-8677.2011.00484.x. [DOI] [PubMed] [Google Scholar]
- 5.van der Biezen EA, Sun J, Coleman MJ, Bibb MJ, Jones JD. 2000. Arabidopsis RelA/SpoT homologs implicate (p) ppGpp in plant signaling. Proc Natl Acad Sci U S A 97:3747–3752. doi: 10.1073/pnas.97.7.3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Durfee T, Hansen AM, Zhi H, Blattner FR, Jin DJ. 2008. Transcription profiling of the stringent response in Escherichia coli. J Bacteriol 190:1084–1096. doi: 10.1128/JB.01092-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Traxler MF, Summers SM, Nguyen HT, Zacharia VM, Hightower GA, Smith JT, Conway T. 2008. The global, ppGpp-mediated stringent response to amino acid starvation in Escherichia coli. Mol Microbiol 68:1128–1148. doi: 10.1111/j.1365-2958.2008.06229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Atkinson GC, Tenson T, Hauryliuk V. 2011. The RelA/SpoT homolog (RSH) superfamily: distribution and functional evolution of ppGpp synthetases and hydrolases across the tree of life. PLoS One 6:e23479. doi: 10.1371/journal.pone.0023479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haseltine WA, Block R. 1973. Synthesis of guanosine tetra- and pentaphosphate requires the presence of a codon-specific, uncharged transfer ribonucleic acid in the acceptor site of ribosomes. Proc Natl Acad Sci U S A 70:1564–1568. doi: 10.1073/pnas.70.5.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wendrich TM, Blaha G, Wilson DN, Marahiel MA, Nierhaus KH. 2002. Dissection of the mechanism for the stringent factor RelA. Mol Cell 10:779–788. doi: 10.1016/S1097-2765(02)00656-1. [DOI] [PubMed] [Google Scholar]
- 11.Agirrezabala X, Fernandez IS, Kelley AC, Carton DG, Ramakrishnan V, Valle M. 2013. The ribosome triggers the stringent response by RelA via a highly distorted tRNA. EMBO Rep 14:811–816. doi: 10.1038/embor.2013.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.English BP, Hauryliuk V, Sanamrad A, Tankov S, Dekker NH, Elf J. 2011. Single-molecule investigations of the stringent response machinery in living bacterial cells. Proc Natl Acad Sci U S A 108:E365–E373. doi: 10.1073/pnas.1102255108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shyp V, Tankov S, Ermakov A, Kudrin P, English BP, Ehrenberg M, Tenson T, Elf J, Hauryliuk V. 2012. Positive allosteric feedback regulation of the stringent response enzyme RelA by its product. EMBO Rep 13:835–839. doi: 10.1038/embor.2012.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Battesti A, Bouveret E. 2006. Acyl carrier protein/SpoT interaction, the switch linking SpoT-dependent stress response to fatty acid metabolism. Mol Microbiol 62:1048–1063. doi: 10.1111/j.1365-2958.2006.05442.x. [DOI] [PubMed] [Google Scholar]
- 15.Gentry DR, Cashel M. 1996. Mutational analysis of the Escherichia coli spoT gene identifies distinct but overlapping regions involved in ppGpp synthesis and degradation. Mol Microbiol 19:1373–1384. doi: 10.1111/j.1365-2958.1996.tb02480.x. [DOI] [PubMed] [Google Scholar]
- 16.Seyfzadeh M, Keener J, Nomura M. 1993. spoT-dependent accumulation of guanosine tetraphosphate in response to fatty acid starvation in Escherichia coli. Proc Natl Acad Sci U S A 90:11004–11008. doi: 10.1073/pnas.90.23.11004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spira B, Silberstein N, Yagil E. 1995. Guanosine 3′,5′-bispyrophosphate (ppGpp) synthesis in cells of Escherichia coli starved for Pi. J Bacteriol 177:4053–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Traxler MF, Chang DE, Conway T. 2006. Guanosine 3′,5′-bispyrophosphate coordinates global gene expression during glucose-lactose diauxie in Escherichia coli. Proc Natl Acad Sci U S A 103:2374–2379. doi: 10.1073/pnas.0510995103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vinella D, Albrecht C, Cashel M, D'Ari R. 2005. Iron limitation induces SpoT-dependent accumulation of ppGpp in Escherichia coli. Mol Microbiol 56:958–970. doi: 10.1111/j.1365-2958.2005.04601.x. [DOI] [PubMed] [Google Scholar]
- 20.Xiao H, Kalman M, Ikehara K, Zemel S, Glaser G, Cashel M. 1991. Residual guanosine 3′,5′-bispyrophosphate synthetic activity of relA null mutants can be eliminated by spoT null mutations. J Biol Chem 266:5980–5990. [PubMed] [Google Scholar]
- 21.Avarbock A, Avarbock D, Teh JS, Buckstein M, Wang ZM, Rubin H. 2005. Functional regulation of the opposing (p)ppGpp synthetase/hydrolase activities of RelMtb from Mycobacterium tuberculosis. Biochemistry 44:9913–9923. doi: 10.1021/bi0505316. [DOI] [PubMed] [Google Scholar]
- 22.Avarbock D, Avarbock A, Rubin H. 2000. Differential regulation of opposing RelMtb activities by the aminoacylation state of a tRNA.ribosome.mRNA. RelMtb complex. Biochemistry 39:11640–11648. [DOI] [PubMed] [Google Scholar]
- 23.Hogg T, Mechold U, Malke H, Cashel M, Hilgenfeld R. 2004. Conformational antagonism between opposing active sites in a bifunctional RelA/SpoT homolog modulates (p)ppGpp metabolism during the stringent response. Cell 117:57–68. doi: 10.1016/S0092-8674(04)00260-0. [DOI] [PubMed] [Google Scholar]
- 24.Mechold U, Murphy H, Brown L, Cashel M. 2002. Intramolecular regulation of the opposing (p)ppGpp catalytic activities of Rel(Seq), the Rel/Spo enzyme from Streptococcus equisimilis. J Bacteriol 184:2878–2888. doi: 10.1128/JB.184.11.2878-2888.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mechold U, Potrykus K, Murphy H, Murakami KS, Cashel M. 2013. Differential regulation by ppGpp versus pppGpp in Escherichia coli. Nucleic Acids Res 41:6175–6189. doi: 10.1093/nar/gkt302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reddy PS, Raghavan A, Chatterji D. 1995. Evidence for a ppGpp-binding site on Escherichia coli RNA polymerase: proximity relationship with the rifampicin-binding domain. Mol Microbiol 15:255–265. doi: 10.1111/j.1365-2958.1995.tb02240.x. [DOI] [PubMed] [Google Scholar]
- 27.Ross W, Vrentas CE, Sanchez-Vazquez P, Gaal T, Gourse RL. 2013. The magic spot: a ppGpp binding site on E. coli RNA polymerase responsible for regulation of transcription initiation. Mol Cell 50:420–429. doi: 10.1016/j.molcel.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zuo Y, Wang Y, Steitz TA. 2013. The mechanism of E. coli RNA polymerase regulation by ppGpp is suggested by the structure of their complex. Mol Cell 50:430–436. doi: 10.1016/j.molcel.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jishage M, Kvint K, Shingler V, Nystrom T. 2002. Regulation of sigma factor competition by the alarmone ppGpp. Genes Dev 16:1260–1270. doi: 10.1101/gad.227902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Magnusson LU, Farewell A, Nystrom T. 2005. ppGpp: a global regulator in Escherichia coli. Trends Microbiol 13:236–242. doi: 10.1016/j.tim.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 31.Lennon CW, Ross W, Martin-Tumasz S, Toulokhonov I, Vrentas CE, Rutherford ST, Lee JH, Butcher SE, Gourse RL. 2012. Direct interactions between the coiled-coil tip of DksA and the trigger loop of RNA polymerase mediate transcriptional regulation. Genes Dev 26:2634–2646. doi: 10.1101/gad.204693.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paul BJ, Barker MM, Ross W, Schneider DA, Webb C, Foster JW, Gourse RL. 2004. DksA: a critical component of the transcription initiation machinery that potentiates the regulation of rRNA promoters by ppGpp and the initiating NTP. Cell 118:311–322. doi: 10.1016/j.cell.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 33.Paul BJ, Berkmen MB, Gourse RL. 2005. DksA potentiates direct activation of amino acid promoters by ppGpp. Proc Natl Acad Sci U S A 102:7823–7828. doi: 10.1073/pnas.0501170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nickels BE, Hochschild A. 2004. Regulation of RNA polymerase through the secondary channel. Cell 118:281–284. doi: 10.1016/j.cell.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 35.Perederina A, Svetlov V, Vassylyeva MN, Tahirov TH, Yokoyama S, Artsimovitch I, Vassylyev DG. 2004. Regulation through the secondary channel—structural framework for ppGpp-DksA synergism during transcription. Cell 118:297–309. doi: 10.1016/j.cell.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 36.Magnusson LU, Gummesson B, Joksimovic P, Farewell A, Nystrom T. 2007. Identical, independent, and opposing roles of ppGpp and DksA in Escherichia coli. J Bacteriol 189:5193–5202. doi: 10.1128/JB.00330-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Friesen JD, Fiil NP, von Meyenburg K. 1975. Synthesis and turnover of basal level guanosine tetraphosphate in Escherichia coli. J Biol Chem 250:304–309. [PubMed] [Google Scholar]
- 38.Lazzarini RA, Cashel M, Gallant J. 1971. On the regulation of guanosine tetraphosphate levels in stringent and relaxed strains of Escherichia coli. J Biol Chem 246:4381–4385. [PubMed] [Google Scholar]
- 39.Sokawa Y, Sokawa J, Kaziro Y. 1975. Regulation of stable RNA synthesis and ppGpp levels in growing cells of Escherichia coli. Cell 5:69–74. doi: 10.1016/0092-8674(75)90093-8. [DOI] [PubMed] [Google Scholar]
- 40.Sarubbi E, Rudd KE, Cashel M. 1988. Basal ppGpp level adjustment shown by new spoT mutants affect steady state growth rates and rrnA ribosomal promoter regulation in Escherichia coli. Mol Gen Genet 213:214–222. doi: 10.1007/BF00339584. [DOI] [PubMed] [Google Scholar]
- 41.Dalebroux ZD, Svensson SL, Gaynor EC, Swanson MS. 2010. ppGpp conjures bacterial virulence. Microbiol Mol Biol Rev 74:171–199. doi: 10.1128/MMBR.00046-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harinarayanan R, Murphy H, Cashel M. 2008. Synthetic growth phenotypes of Escherichia coli lacking ppGpp and transketolase A (tktA) are due to ppGpp-mediated transcriptional regulation of tktB. Mol Microbiol 69:882–894. doi: 10.1111/j.1365-2958.2008.06317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Botos I, Melnikov EE, Cherry S, Tropea JE, Khalatova AG, Rasulova F, Dauter Z, Maurizi MR, Rotanova TV, Wlodawer A, Gustchina A. 2004. The catalytic domain of Escherichia coli Lon protease has a unique fold and a Ser-Lys dyad in the active site. J Biol Chem 279:8140–8148. doi: 10.1074/jbc.M312243200. [DOI] [PubMed] [Google Scholar]
- 44.Tsilibaris V, Maenhaut-Michel G, Van Melderen L. 2006. Biological roles of the Lon ATP-dependent protease. Res Microbiol 157:701–713. doi: 10.1016/j.resmic.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 45.Gottesman S. 1989. Genetics of proteolysis in Escherichia coli. Annu Rev Genet 23:163–198. doi: 10.1146/annurev.ge.23.120189.001115. [DOI] [PubMed] [Google Scholar]
- 46.Christensen SK, Maenhaut-Michel G, Mine N, Gottesman S, Gerdes K, Van Melderen L. 2004. Overproduction of the Lon protease triggers inhibition of translation in Escherichia coli: involvement of the yefM-yoeB toxin-antitoxin system. Mol Microbiol 51:1705–1717. doi: 10.1046/j.1365-2958.2003.03941.x. [DOI] [PubMed] [Google Scholar]
- 47.Goff SA, Goldberg AL. 1987. An increased content of protease La, the lon gene product, increases protein degradation and blocks growth in Escherichia coli. J Biol Chem 262:4508–4515. [PubMed] [Google Scholar]
- 48.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 49.Miller JH. 1992. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia Coli and related bacteria. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 50.Kang PJ, Craig EA. 1990. Identification and characterization of a new Escherichia coli gene that is a dosage-dependent suppressor of a dnaK deletion mutation. J Bacteriol 172:2055–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bernhardt TG, de Boer PA. 2004. Screening for synthetic lethal mutants in Escherichia coli and identification of EnvC (YibP) as a periplasmic septal ring factor with murein hydrolase activity. Mol Microbiol 52:1255–1269. doi: 10.1111/j.1365-2958.2004.04063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. 2000. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci U S A 97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ellermeier CD, Janakiraman A, Slauch JM. 2002. Construction of targeted single copy lac fusions using lambda Red and FLP-mediated site-specific recombination in bacteria. Gene 290:153–161. doi: 10.1016/S0378-1119(02)00551-6. [DOI] [PubMed] [Google Scholar]
- 54.Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. doi: 10.1016/0378-1119(95)00193-A. [DOI] [PubMed] [Google Scholar]
- 55.Dajkovic A, Mukherjee A, Lutkenhaus J. 2008. Investigation of regulation of FtsZ assembly by SulA and development of a model for FtsZ polymerization. J Bacteriol 190:2513–2526. doi: 10.1128/JB.01612-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lies M, Maurizi MR. 2008. Turnover of endogenous SsrA-tagged proteins mediated by ATP-dependent proteases in Escherichia coli. J Biol Chem 283:22918–22929. doi: 10.1074/jbc.M801692200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Van Melderen L, Gottesman S. 1999. Substrate sequestration by a proteolytically inactive Lon mutant. Proc Natl Acad Sci U S A 96:6064–6071. doi: 10.1073/pnas.96.11.6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mizusawa S, Gottesman S. 1983. Protein degradation in Escherichia coli: the lon gene controls the stability of sulA protein. Proc Natl Acad Sci U S A 80:358–362. doi: 10.1073/pnas.80.2.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Majdalani N, Gottesman S. 2005. The Rcs phosphorelay: a complex signal transduction system. Annu Rev Microbiol 59:379–405. doi: 10.1146/annurev.micro.59.050405.101230. [DOI] [PubMed] [Google Scholar]
- 60.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Howard-Flanders P, Simson E, Theriot L. 1964. A locus that controls filament formation and sensitivity to radiation in Escherichia coli K-12. Genetics 49:237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.SaiSree L, Reddy M, Gowrishankar J. 2000. lon incompatibility associated with mutations causing SOS induction: null uvrD alleles induce an SOS response in Escherichia coli. J Bacteriol 182:3151–3157. doi: 10.1128/JB.182.11.3151-3157.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kuzminov A. 1999. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol Mol Biol Rev 63:751–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee JH, Lennon CW, Ross W, Gourse RL. 2012. Role of the coiled-coil tip of Escherichia coli DksA in promoter control. J Mol Biol 416:503–517. doi: 10.1016/j.jmb.2011.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vinella D, Potrykus K, Murphy H, Cashel M. 2012. Effects on growth by changes of the balance between GreA, GreB, and DksA suggest mutual competition and functional redundancy in Escherichia coli. J Bacteriol 194:261–273. doi: 10.1128/JB.06238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tehranchi AK, Blankschien MD, Zhang Y, Halliday JA, Srivatsan A, Peng J, Herman C, Wang JD. 2010. The transcription factor DksA prevents conflicts between DNA replication and transcription machinery. Cell 141:595–605. doi: 10.1016/j.cell.2010.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhou YN, Jin DJ. 1998. The rpoB mutants destabilizing initiation complexes at stringently controlled promoters behave like “stringent” RNA polymerases in Escherichia coli. Proc Natl Acad Sci U S A 95:2908–2913. doi: 10.1073/pnas.95.6.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Murphy H, Cashel M. 2003. Isolation of RNA polymerase suppressors of a (p)ppGpp deficiency. Methods Enzymol 371:596–601. doi: 10.1016/S0076-6879(03)71044-1. [DOI] [PubMed] [Google Scholar]
- 69.McGlynn P, Lloyd RG. 2000. Modulation of RNA polymerase by (p)ppGpp reveals a RecG-dependent mechanism for replication fork progression. Cell 101:35–45. doi: 10.1016/S0092-8674(00)80621-2. [DOI] [PubMed] [Google Scholar]
- 70.Joseleau-Petit D, Vinella D, D'Ari R. 1999. Metabolic alarms and cell division in Escherichia coli. J Bacteriol 181:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Powell BS, Court DL. 1998. Control of ftsZ expression, cell division, and glutamine metabolism in Luria-Bertani medium by the alarmone ppGpp in Escherichia coli. J Bacteriol 180:1053–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vinella D, Joseleau-Petit D, Thevenet D, Bouloc P, D'Ari R. 1993. Penicillin-binding protein 2 inactivation in Escherichia coli results in cell division inhibition, which is relieved by FtsZ overexpression. J Bacteriol 175:6704–6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aldea M, Garrido T, Pla J, Vicente M. 1990. Division genes in Escherichia coli are expressed coordinately to cell septum requirements by gearbox promoters. EMBO J 9:3787–3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Weart RB, Levin PA. 2003. Growth rate-dependent regulation of medial FtsZ ring formation. J Bacteriol 185:2826–2834. doi: 10.1128/JB.185.9.2826-2834.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen Y, Milam SL, Erickson HP. 2012. SulA inhibits assembly of FtsZ by a simple sequestration mechanism. Biochemistry 51:3100–3109. doi: 10.1021/bi201669d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reddy M. 2007. Role of FtsEX in cell division of Escherichia coli: viability of ftsEX mutants is dependent on functional SufI or high osmotic strength. J Bacteriol 189:98–108. doi: 10.1128/JB.01347-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bernhardt TG, de Boer PA. 2005. SlmA, a nucleoid-associated, FtsZ binding protein required for blocking septal ring assembly over chromosomes in E. coli. Mol Cell 18:555–564. doi: 10.1016/j.molcel.2005.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.de Boer PA, Crossley RE, Rothfield LI. 1989. A division inhibitor and a topological specificity factor coded for by the minicell locus determine proper placement of the division septum in E. coli. Cell 56:641–649. doi: 10.1016/0092-8674(89)90586-2. [DOI] [PubMed] [Google Scholar]
- 79.Dai K, Lutkenhaus J. 1992. The proper ratio of FtsZ to FtsA is required for cell division to occur in Escherichia coli. J Bacteriol 174:6145–6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lutkenhaus J, Mukherjee A. 1996. Cell division, p 1615–1626. In Neidhardt FC, Curtiss R III, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE (ed), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed, vol 1 ASM Press, Washington, DC. [Google Scholar]
- 81.Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. 2005. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res 12:291–299. [DOI] [PubMed] [Google Scholar]
- 82.Ward JE Jr, Lutkenhaus J. 1985. Overproduction of FtsZ induces minicell formation in E. coli. Cell 42:941–949. doi: 10.1016/0092-8674(85)90290-9. [DOI] [PubMed] [Google Scholar]
- 83.Weiss DS, Chen JC, Ghigo JM, Boyd D, Beckwith J. 1999. Localization of FtsI (PBP3) to the septal ring requires its membrane anchor, the Z ring, FtsA, FtsQ, and FtsL. J Bacteriol 181:508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dai K, Lutkenhaus J. 1991. ftsZ is an essential cell division gene in Escherichia coli. J Bacteriol 173:3500–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Erickson HP, Anderson DE, Osawa M. 2010. FtsZ in bacterial cytokinesis: cytoskeleton and force generator all in one. Microbiol Mol Biol Rev 74:504–528. doi: 10.1128/MMBR.00021-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Joseleau-Petit D, Thevenet D, D'Ari R. 1994. ppGpp concentration, growth without PBP2 activity, and growth-rate control in Escherichia coli. Mol Microbiol 13:911–917. doi: 10.1111/j.1365-2958.1994.tb00482.x. [DOI] [PubMed] [Google Scholar]
- 87.Vinella D, D'Ari R, Jaffe A, Bouloc P. 1992. Penicillin binding protein 2 is dispensable in Escherichia coli when ppGpp synthesis is induced. EMBO J 11:1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Navarro F, Robin A, D'Ari R, Joseleau-Petit D. 1998. Analysis of the effect of ppGpp on the ftsQAZ operon in Escherichia coli. Mol Microbiol 29:815–823. doi: 10.1046/j.1365-2958.1998.00974.x. [DOI] [PubMed] [Google Scholar]
- 89.Adhya S, Gottesman M. 1982. Promoter occlusion: transcription through a promoter may inhibit its activity. Cell 29:939–944. doi: 10.1016/0092-8674(82)90456-1. [DOI] [PubMed] [Google Scholar]
- 90.Schaechter M, Maaloe O, Kjeldgaard NO. 1958. Dependency on medium and temperature of cell size and chemical composition during balanced grown of Salmonella typhimurium. J Gen Microbiol 19:592–606. doi: 10.1099/00221287-19-3-592. [DOI] [PubMed] [Google Scholar]
- 91.Chien AC, Hill NS, Levin PA. 2012. Cell size control in bacteria. Curr Biol 22:R340–R349. doi: 10.1016/j.cub.2012.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Osbourne DO, Soo VW, Konieczny I, Wood TK. 2014. Polyphosphate, cyclic AMP, guanosine tetraphosphate, and c-di-GMP reduce in vitro Lon activity. Bioengineered 5:264–268. doi: 10.4161/bioe.29261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gayda RC, Yamamoto LT, Markovitz A. 1976. Second-site mutations in capR (lon) strains of Escherichia coli K-12 that prevent radiation sensitivity and allow bacteriophage lambda to lysogenize. J Bacteriol 127:1208–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Walker JR, Smith JA. 1970. Cell division of the Escherichia coli lon- mutant. Mol Gen Genet 108:249–257. [DOI] [PubMed] [Google Scholar]
- 95.Huisman O, D'Ari R, George J. 1980. Inducible sfi dependent division inhibition in Escherichia coli. Mol Gen Genet 177:629–636. [DOI] [PubMed] [Google Scholar]
- 96.Huisman O, Jacques M, D'Ari R, Caro L. 1983. Role of the sfiA-dependent cell division regulation system in Escherichia coli. J Bacteriol 153:1072–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kanemori M, Yanagi H, Yura T. 1999. The ATP-dependent HslVU/ClpQY protease participates in turnover of cell division inhibitor SulA in Escherichia coli. J Bacteriol 181:3674–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wu WF, Zhou Y, Gottesman S. 1999. Redundant in vivo proteolytic activities of Escherichia coli Lon and the ClpYQ (HslUV) protease. J Bacteriol 181:3681–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.