

Abstract
Acylcarnitine metabolites have gained attention as biomarkers of nutrient stress, but their physiological relevance and metabolic purpose remain poorly understood. Short chain carnitine conjugates, including acetylcarnitine, derive from their corresponding acyl-CoA precursors via the action of carnitine acetyltransferase (CrAT), a bidirectional mitochondrial matrix enzyme. We show here that contractile activity reverses acetylcarnitine flux in muscle, from net production and efflux at rest to net uptake and consumption during exercise. Disruption of this switch in mice with muscle-specific CrAT deficiency resulted in acetyl-CoA deficit, perturbed energy charge and diminished exercise tolerance, whereas acetylcarnitine supplementation produced opposite outcomes in a CrAT-dependent manner. Likewise, in exercise-trained compared to untrained humans, post-exercise phosphocreatine recovery rates were positively associated with CrAT activity and coincided with dramatic shifts in muscle acetylcarnitine dynamics. These findings show acetylcarnitine serves as a critical acetyl buffer for working muscles and provide insight into potential therapeutic strategies for combatting exercise intolerance.
Graphical Abstract
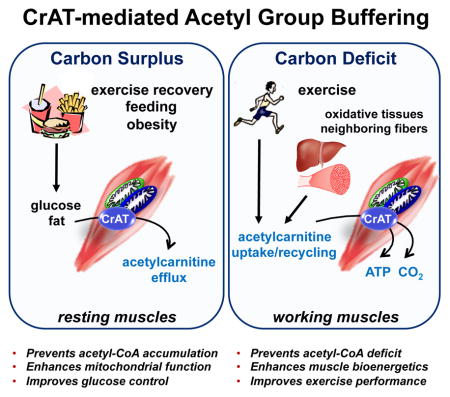
Introduction
Habitual exercise not only improves physical fitness and muscle strength, but also promotes metabolic health and mitigates a wide range of medical conditions (Pattyn et al., 2013). Importantly however, aging and other chronic disorders are often accompanied by exercise intolerance, leading to a vicious cycle of inactivity and accelerated cardiometabolic decay. In light of an aging population and the growing worldwide prevalence of metabolic disease, efforts to understand and modify exercise fatigue have become increasingly relevant to global health.
At the most fundamental level, exercise tolerance depends on the ability of working muscles to resynthesize ATP at a rate that matches the costs of contractile activity. Sustained regeneration of ATP is driven principally by mitochondrial oxidative phosphorylation (OXPHOS), a less powerful but more efficient and higher capacity system than glycolysis. Whereas muscle mitochondrial content and respiratory potential are well-recognized determinants of exercise tolerance, several studies have shown that performance during both short and long term activities can also be influenced by the degree to which rates of OXPHOS lag during the first several minutes of exercise and/or during stepwise increments in workload before achieving a new steady-state (reviewed in (Poole and Jones, 2012). This slow component of oxygen uptake kinetics in response to increasing power output is thought to reflect metabolic capacitance conferred by the creatine energy transfer system and has been linked to metabolic instability and muscle contractile inefficiency (Poole and Jones, 2012).
Rapid adjustments in the rate of OXPHOS depend on shifts in muscle energy charge, availability of oxygen as the final electron acceptor and a steady supply of electron donors, in the form of NADH and FADH2, to fuel the electron transport chain (ETC). Historically, two general theories have been investigated to account for the foregoing lag in whole body oxygen consumption during abrupt increases in work rate. The first centers on potential limitations at the level of bulk oxygen delivery to working muscles, whereas an alternative explanation proposes intramuscular processes involving delayed enzyme activation and carbon flux through oxidative metabolic machinery, known as metabolic inertia (Greenhaff et al., 2002; Murias et al., 2014; Poole and Jones, 2012). Whereas limitations in oxygen delivery have been identified under some circumstances (Murias et al., 2014; Raper et al., 2014), considerable evidence suggests a more prominent role for metabolic inertia (Poole and Jones, 2012). Among potential factors contributing to sluggish ramping of oxidative metabolism is a deficit in acetyl group availability (Greenhaff et al., 2002). Acetyl-CoA holds a prominent position in exercise bioenergetics as the universal two-carbon intermediate of glucose, fatty acid and amino acid catabolism. Because acetyl-CoA fuels the tricarboxylic acid cycle (TCAC), which in turn provides reducing power for OXPHOS, a deficit in acetyl-CoA supply would be predicted to limit the rate of oxidative ATP production.
The metabolic inertia theory draws attention to nutritional and pharmacological maneuvers that might augment the provision of specific acetyl group precursors (Raper et al., 2014). Relevant to this idea is emergent evidence that mitochondrial acetyl-CoA balance can be nutritionally regulated via the carnitine-dependent enzyme, carnitine acetyltransferase (CrAT) (Muoio et al., 2012). This enzyme, which is highly enriched in muscle and heart (Noland et al., 2009) and localized to the mitochondrial matrix, converts short chain acyl-CoAs to their membrane permeant acylcarnitine counterparts, thereby permitting intracellular trafficking of acyl moieties. Results from loss- and gain-of-function studies show that CrAT-mediated acetylcarnitine production and efflux promotes glucose homeostasis and alleviates product inhibition of pyruvate dehydrogenase (PDH), the mitochondrial enzyme complex that connects glycolysis to glucose oxidation (Muoio et al., 2012; Noland et al., 2009). Moreover, dietary supplementation with L-carnitine increased tissue acetylcarnitine efflux and enhanced glucose tolerance and oxidation when administered to obese rodents and humans (Muoio et al., 2012; Noland et al., 2009). These findings raised the question of why muscle tissues express such high amounts of an enzyme that essentially siphons acetyl-CoA from the TCA cycle. The answer probably relates to the freely reversible nature of CrAT, which interconverts acetyl-CoA and acetylcarnitine with an equilibrium constant of ~1.5 (Pieklik and Guynn, 1975). Thus, CrAT is positioned to play a key role in buffering the mitochondrial acetyl group pool during transitions from low to high energy demand, and vice versa.
Short chain acylcarnitines are present in tissues and blood and have gained increasing attention as biomarkers of metabolic stress. Although previous studies have reported that intramuscular and circulating levels of acetylcarnitine increase during and/or immediately after exercise (Hiatt et al., 1989; Putman et al., 1993; Sahlin, 1990), still unclear is whether these metabolites are dispensable metabolic byproducts or, alternatively, if they serve an important physiologic purpose. Herein, we sought to determine whether acetylcarnitine metabolism is required for optimal exercise performance. To this end, we examined the impact of skeletal muscle-specific CrAT deficiency on muscle bioenergetics, acetyl group balance and exercise stamina. Our findings not only establish important roles for CrAT and acetylcarnitine in modulating muscle energy economy during contraction, but also suggest that nutritional and/or pharmacological strategies aimed at promoting CrAT activity could prove useful for offsetting metabolic inertia, delaying muscle fatigue and combatting exercise intolerance.
Results and Discussion
CrAT fiber type distribution and regulation by exercise
To characterize the relationship between CrAT and muscle function we examined enzyme abundance in different muscle types as well as its regulation in response to exercise. In general, CrAT protein abundance was highest in the most oxidative/red muscles as compared to those comprised of a more glycolytic/white fiber types (Figure 1A, S. Figure 1). This expression pattern was only partly attributable to total mitochondrial content, as CrAT abundance was also dissimilar among isolated mitochondria preparations harvested from different rat muscles (Figure 1B, S. Figure 1). Mitochondrial abundance of CrAT tracked most closely with muscle enrichment of type IIa (intermediate) myofibers. Skeletal muscle mRNA levels of Crat increased 4-fold in response to an acute 90 minute mid-intensity exercise bout (Figure 1C). This was evident immediately and up to 24 h after exercise as compared to the rested controls and was accompanied by upregulation of the exercise-inducible transcriptional co-activator, Pgc1α (Koves et al., 2013). Likewise, muscles from MCK-Pgc1α transgenic mice had increased Crat gene expression (Figure 1D), and adenoviral-mediated overexpression of mPgc1α in primary human skeletal myocytes grown in culture produced a similar stimulatory effect (Figure 1E), suggesting a role for this enzyme in exercise adaptation. We next used a tissue incubation bath system coupled with mass spectrometry-based [13C]glucose tracer methodology to estimate contraction-induced changes in net acetylcarnitine flux in isolated soleus muscles. Notably, electrical stimulation appeared to reverse acetylcarnitine flux, promoting net muscle uptake of the metabolite, whereas the recovery period was characterized by elevated rates of efflux as compared to either the contracted or rested states (Figure 1F). In aggregate, these results implied a key role for CrAT in maintaining mitochondrial acetyl group balance during and immediately after exercise.
Figure 1. CrAT fiber type distribution and regulation by exercise.

(A) CrAT protein abundance was determined in three-month-old rat heart, red quadriceps (RQ), red gastrocnemius (RG), tibialis anterior (TA), soleus (SOL), plantaris (Plant), white gastrocnemius (WG), white quadriceps (WQ), extensor digitorum longus (EDL) and liver tissue homogenates and (B) isolated mitochondria. Values are expressed as arbitrary units (A.U.). CrAT mRNA expression normalized to 18S was determined in (C) mouse tibialis anterior muscle at rest and 15 min, 3 h and 24 h after a 90 min exercise bout, (D) tibialis anterior muscle from MCK-Pgc1α transgenic and non-transgenic (NT) control mice, and (E) primary human skeletal myocytes treated with recombinant adenovirus encoding β-galactosidase or rat Pgc1α. (F) [13C2]Acetylcarnitine content (pmol/muscle) of the incubation buffer was measured after isolated mouse soleus muscles were incubated in KHB buffer supplemented with 5 mM [U-13C] glucose and 2 mM [U-13C pyruvate] for 30 min of rest (R), followed by electrically-stimulated contraction (CONT) and recovery. Data are means ± SEM (n=3–5). * p<0.05.
Skeletal muscle-specific loss of CrAT compromised exercise performance despite increased fat oxidation
To further explore the functional importance of CrAT during exercise we used the Lox-P/Cre system to generate mice lacking Crat specifically in skeletal muscle (CrATSM−/−). This strategy resulted in 80–90% loss of CrAT activity in skeletal muscles without affecting the heart (S. Figure 2D). Residual activity was highest in soleus and red quadriceps (S. Figure 2E). Muscle quantities of acetyl CoA, the principal substrate of CrAT, were increased 1.8-fold in whole quadriceps from CrATSM−/− as compared to control mice (S. Figure 1F), whereas the muscle pool of acetylcarnitine was only 27% lower (S. Figure 2F), possibly due to residual CrAT activity and/or exogenous supply of the metabolite.
We next subjected CrATSM−/− mice and littermate controls to two different treadmill protocols (Figure 2A), both designed to challenge the capacity of the muscle to cope with several abrupt increments in energy demand. The first was an endurance protocol with high intensity ramping during the latter half of the run. Time and distance to exhaustion during the ramping period were decreased 18% and 29%, respectively in CrATSM−/− compared CrATfl/fl mice (Figure 2B). During a second, higher intensity test the frequency and magnitude of the increments were increased and whole body respiration was monitored by indirect calorimetry. Again, the CrATSM−/− mice performed poorly and completed only 66% of the distance covered by the control group (Figure 2C). CrATSM−/− mice tended to have higher O2 consumption and lower CO2 production throughout the exercise, as well as a lower peak RER of 0.931 compared to 0.992 in the CrATfl/fl controls (Figure 2D–F).
Figure 2. CrAT deficiency compromises exercise performance despite increased fat oxidation.

(A) Twelve week old male and female mice ran to exhaustion in two separate experiments using two distinct graded treadmill protocols (n=11, 6 M and 5 F mice per group). (B) Time and distance to exhaustion measured during the ramping period of the endurance protocol. (C) Time and distance to exhaustion measured during high intensity exercise. Indirect calorimetry was performed using an enclosed metabolic treadmill and (D) VO2, (E) VCO2, and (F) respiratory exchange ratio (RER) were calculated. (G) Mitochondria isolated from gastrocnemius muscles of CrATfl/fl and CrATSM−/− littermates were used to assess state 3 respiration in the presence of 2 mM malate, 1 mM carnitine and 5, 10 or 20 μM palmitoylcarnitine (n = 5 M mice). (H) mRNA expression normalized to 18S was determined for carnitine palmitoyltransferase 1 (Cpt1), carnitine acylcarnitine translocase (Cact), carnitine palmitoyltransferase 2 (Cpt2), very long chain acyl-CoA dehydrogenase (Vlcad) and medium chain acyl-CoA dehydrogenase (Mcad) in CrATfl/fl and CrATSM−/− mouse gastrocnemius muscle (n=6 M mice). Data are means ± SEM. * Genotype effect (p<0.05).
Because a reduction in RER typically reflects a substrate shift in favor of fat oxidation, we examined possible changes in β-oxidation potential using mitochondria isolated from gastrocnemius muscles. Consistent with the whole body measures, maximal rates of palmitoylcarnitine-supported respiration were increased 38% in mitochondria harvested from CrAT deficient versus control muscles (Figure 2G). This was accompanied by 1.5–2.0-fold increases in muscle mRNA expression of several β-oxidation enzymes (Figure 2H). We suspect that upregulation of the β-oxidation machinery might be due in part to elevated tissue levels of long chain acyl-CoAs (Muoio et al., 2012) and consequent activation of lipid-responsive transcription factors such as PPARα and PPARδ (Muoio et al., 2002). Collectively, these results revealed an atypical physiologic phenotype wherein enhanced capacity for fat oxidation was accompanied by reduced exercise tolerance. Since heavy reliance on fat oxidation diminishes respiratory efficiency (lower P/O ratio), the failure of CrATSM−/− mice to fully shift to carbohydrate oxidation at higher workloads might have contributed to early fatigue.
CrAT deficiency disrupted acetyl-CoA buffering capacity and exercise bioenergetics
To gain insight into the biochemical underpinnings of premature fatigue in CrATSM−/− mice we evaluated muscle metabolite levels prior to exhaustion, using tissues collected from a second set of mice that were subjected to only 12 min of high intensity treadmill running. Metabolites were measured in quadriceps muscles harvested 10 min or 1 h after exercise cessation and compared against those from rested controls. In CrATfl/fl control mice, muscle concentrations of both acetylcarnitine (Figure 3A) and acetyl-CoA (Figure 3B) were remarkably stable at these time points. By contrast, acetylcarnitine/acetyl-CoA balance fluctuated dramatically in muscles from the CrATSM−/− mice (Figure 3C). Acetylcarnitine content in muscles from CrATSM−/− trended lower in the rested state (Figure 3A), but then increased to levels comparable to the control group during the post-exercise period, which might reflect contraction-stimulated uptake of acetylcarnitine as suggested by results shown in Figure 1F. Conversely, acetyl-CoA concentrations in muscles from CrATSM−/− versus CrATfl/fl mice were elevated 2-fold at rest but then fell dramatically in response to exercise, implying a connection between acetyl group deficit and early fatigue. In CrATSM−/− mice, acetyl-CoA levels were partly restored during the 1 h recovery period, whereas acetylcarnitine remained elevated. Because muscle concentrations of free carnitine and CoA were similar between genotypes and unaffected by exercise at the time points measured, the dramatic shifts in the acetylcarnitine/acetyl CoA ratio in the knockout mice (Figure 3C) reflect a directionally similar shift in the disequilibrium ratio (ρ), such that mass action immediately after exercise favored the reverse CrAT reaction (i.e. acetyl CoA production). This observation is consistent with the premise that during intense exercise CrAT catalyzes net flux in the direction of acetyl CoA.
Figure 3. CrAT deficiency disrupts muscle acetyl-CoA balance and bioenergetics during and after exercise.

Quadriceps muscles were harvested from male CrATfl/fl and CrATSM−/− littermates at rest or 10 min and 1 h after 12 min of high intensity exercise. Tandem mass spectrometry was used to measure muscle concentrations of (A) acetylcarnitine, (B) acetyl-CoA and (C) the ratio of acetylcarnitine:acetyl-CoA. The same muscles were used to measure (D) phosphocreatine (PCr), (E) glycogen, and (F) exercise-induced glycogen breakdown, calculated as the difference between pre and post exercise. (G) Quantified plantaris muscle fiber type composition data (triplicate sections counted). (H) Representative fiber type images from plantaris; blue (type 1), red (2a), green (2b), unstained (2x). Data are means ± SEM, n=4–8 per group. ^Exercise effect, * Genotype effect (p<0.05).
Abrupt fluctuations in muscle energy demand are immediately buffered by phosphocreatine (PCr), which donates its high-energy phosphoryl group to ADP in a freely reversible phosphotransfer reaction catalyzed by creatine kinase (CK). Muscle contraction favors CK flux in the forward direction to generate ATP and Cr, whereas the opposite holds true during exercise recovery. Because the limited myocellular storage pools of PCr can provide for only a few seconds of maximal activity, continuous regeneration of PCr is critical to muscle performance and recovery. Within 10 minutes of exercise cessation, the PCr pool in muscles from control mice was not only fully replenished, but was actually elevated compared to the rested state (Figure 3D, S. Table 1). This post-exercise overshoot is typical of a short duration, high intensity activity and indicative of robust cardiometabolic fitness (Korzeniewski and Zoladz, 2005; Sahlin et al., 1997). Notably, CrAT deficient muscles appeared to lose capacity to quickly stockpile PCr during the same recovery period (Figure 3D, S. Table 1). Deficiencies in oxidative ATP production increase reliance on glycolysis and thus muscle glycogen reserves. Interestingly, in the rested state muscle glycogen levels were elevated in CrATSM−/− mice, perhaps due to the foregoing shift in substrate selection (Figure 3E). However, despite elevated fat oxidation during the treadmill test (Figure 3E), exercise-induced depletion of muscle glycogen was 3.5-fold greater in the CrATSM−/− mice (Figure 3F). Blood lactate levels increased from 3.8±0.39 mM at rest to 12.0±1.1 mM at 5 min post exercise, but were surprisingly similar between genotypes. CrAT deletion did not affect muscle fiber type composition (Figures 3G and H).
In aggregate, these findings support previous studies pointing to substrate deficit as a factor that limits mitochondrial flux at the onset of exercise. Much of the earlier work on this topic centered on exercise-induced activation of PDH, the mitochondrial enzyme complex that converts pyruvate to acetyl-CoA. PDH activity increases upon dephosphorylation of the complex, which depends on the balance of upstream kinase and phosphatase enzymes that receive input from a number of metabolic cues. Delayed activation of PDH at the onset of exercise is presumed to limit acetyl-CoA availability and TCAC flux, thereby restricting OXPHOS. Evidence favoring this hypothesis comes from several studies showing that administration of a pharmacological PDH activator, dichloroacetate (DCA), immediately prior to moderately intense exercise (~65% VO2 max) enhances oxidative metabolism and diminishes reliance on substrate phosphorylation from the PCr/CK and glycogenolytic/glycolytic reactions (Howlett et al., 1999; Timmons et al., 2004). Interestingly, this maneuver also resulted in rapid expansion of the muscle acetylcarnitine pool and improved exercise performance (Durkot et al., 1995; Ludvik et al., 1993)(Timmons et al., 1998; Timmons et al., 1996), implying that substrate availability plays a role in optimizing aerobic ATP production during transitions of escalating workload. Accordingly, we performed Western blot analysis of PDH phosphorylation at serines 293 and 300, which inactivates the enzyme. The results revealed a trend towards increased pPDH in the knockout mice, consistent with a possible limitation in pyruvate flux (S. Figure 3). Taken together, these findings provide strong support for the acetyl group deficit theory of muscle fatigue and offer the first conclusive evidence that CrAT is essential for optimizing acetyl-CoA balance and muscle energy economy during and after exercise.
Exogenously supplied acetylcarnitine delayed fatigue and improved energy economy in a CrAT-dependent manner
Results of the exercise tolerance studies suggested that CrAT-derived acetylcarnitine supports mitochondrial respiration when production of acetyl-CoA from other fuel sources becomes limiting. This hypothesis was subsequently tested in isolated mitochondria, which maintained high rates of acetylcarnitine-supported respiration in a CrAT-dependent manner (Figure 4A). Using mitochondria from wildtype mice, we measured respiration in the presence of saturating levels of malate and ADP along with increasing doses of either palmitoylcarnitine or acetylcarnitine. Acetylcarnitine permitted higher maximal rates of respiration than palmitoylcarnitine, although the concentration required for saturation was much greater (Figure 4B). In a subsequent experiment, addition of submaximal concentrations of acetylcarnitine to mitochondria respiring on a saturating dose of palmitoylcarnitine further increased rates of oxygen consumption (Figure 4C). By contrast, addition of free carnitine, which promotes acetylcarnitine efflux, lowered respiration.
Figure 4. Exogenously supplied acetylcarnitine delayed fatigue and improved energy economy in a CrAT-dependent manner.

Mitochondria isolated from gastrocnemius muscles of CrATfl/fl and CrATSM−/− male mice were used for Seahorse-based assessment of state 3 oxygen consumption rates (OCR) in the presence of (A) 1.0–10 mM acetylcarnitine, or (B) saturating doses of palmitoylcarnitine (PC) compared to acetylcarnitine (AC) (20 μM and 10 mM, respectively), or (C) a saturating dose of PC followed by addition of vehicle, 5 mM AC or L-carnitine (n=5); and then rotenone and antimycin A (AntA). (D) Contraction-induced oxidation [14C]Acetylcarnitine to [14C]CO2 was assessed in intact EDL muscles from CrATfl/fl and CrATSM−/− mice at rest and after 4 and 10 min electrical stimulation at 12.5 volts per chamber (n=4). Time to half maximal force generation (t1/2) was determined using EDL muscles from (E) CrATfl/fl and (F) CrATSM−/− mice in the absence or presence of 5 mM AC stimulated at 20 volts per chamber (n=6). In separate experiments, EDL muscles were flash frozen after a 4 min stimulation ± 5mM AC and used for measuring the ratios of (G) PCr/Creatine (H) AMP/ATP. Data are means ±SE, n=5–10 per group.* effect of AC (p<0.05).
Results presented in Figures 1F and 3A suggested that exercise might stimulate muscle acetylcarnitine uptake. To test this possibility we used the muscle bath incubation system to examine contraction-induced changes in the oxidation rates of exogenously supplied [14C]acetylcarnitine. EDL muscles were selected for this analysis based on their fiber type properties, high level of mitochondrial CrAT content (Figure 1B) and low residual activity in the knockout mice. As predicted, electrical stimulation applied during a 10 min incubation caused a marked 5-fold increase in acetylcarnitine catabolism to [14C]CO2 as compared to rested/unstimulated muscles (Figure 4D). This outcome was fully dependent on CrAT activity. Moreover, in control muscles, provision of 5 mM acetylcarnitine diminished the rate of fatigue, such that time to half maximal force generation during a high intensity regimen increased by 50% (Figure 4E). By contrast, acetylcarnitine had no effect on the force-fatigue properties of EDL from CrATSM−/− mice (Figure 4F).
We repeated these experiments and assessed muscle energy charge after four minutes of contraction. As expected, pilot experiments performed with control mice confirmed robust contraction-induced shifts in muscle content of high-energy phosphagens (S. Table 2). In control but not CrAT-deficient muscles, exogenously supplied acetylcarnitine defended the muscle PCr pool, resulting in a near two-fold increase in the post-contraction PCr/Cr ratio (Figure 4G, S. Table 3). This observation is functionally relevant because a decline in the PCr/Cr ratio limits forward flux through the CK reaction and thereby diminishes maximal power output during exercise. In addition to the CK reaction, adenylate kinase also plays a key role in exercise bioenergetics by catalyzing phosphoryl group transfer from one ADP to another, thereby regenerating one ATP along with AMP. The latter molecule can then be deaminated to produce IMP as a critical step in the adenine nucleotide cycle. When acetylcarnitine was provided to control muscles, post-contraction levels of the adenine nucleotides reflected a favorable shift in energy charge. Thus, acetylcarnitine lowered both the AMP/ATP and IMP/ATP ratios (Figure 4H and S. Table 3), consistent with improved metabolic stability. By contrast, these same indices of energy stress were completely unresponsive to the acetylcarnitine treatment in muscles from CrATSM−/− mice.
Carnitine supplementation enhanced exercise performance in control but not CrATSM−/− mice
To determine whether the benefits of enhanced acetyl group buffering capacity would take effect at a whole body level, we performed the same two graded treadmill tests described in Figure 2, but after two and four weeks of dietary L-carnitine supplementation. Consistent with previous reports (Makowski et al., 2009; Noland et al., 2009), administration of L-carnitine in the drinking water for two weeks increased resting blood levels of free carnitine and acetylcarnitine by 30% and 20%, respectively, regardless of genotype (Figure 5A and B). In control mice, two and four weeks of L-carnitine supplementation improved distance to exhaustion by 30% and 27% during the endurance with ramping and the high intensity regimens, respectively. This carnitine-mediated improvement of exercise performance was absent in the CrATSM−/− mice (Figure 5C and D). Serum acetylcarnitine levels measured 10 min after the second treadmill test were increased 34–38% in both carnitine supplemented groups (Figure 5E). Thus, in line with results of the muscle incubation experiments (Figure 4E and 4F), increased supply of acetylcarnitine to working muscles was accompanied by enhanced exercise tolerance in CrATfl/fl mice but not their CrATSM−/− counterparts. Despite the robust impact on performance, carnitine supplementation did not alter intramuscular levels of free, esterified or total carnitine measured 10 min after exercise (Figure 5F), suggesting rapid equilibration of the muscle carnitine pool upon exercise cessation due to consumption and/or efflux of acetylcarnitine during recovery (Figure 1F).
Figure 5. Dietary L-Carnitine supplementation enhanced exercise performance in control but not CrATSM−/− mice.

Male mice were randomly selected to receive supplementation with pH-neutralized drinking water ±1.0 mg/ml L-carnitine for four weeks. Tandem mass spectrometry was used to assess (A) free carnitine and (B) acetylcarnitine in blood spots taken from the tail vein after 2 weeks of treatment. Running time and distance to exhaustion were determined in two separate experiments using treadmill protocols consisting of (C) endurance exercise with high intensity ramping (data shown for the ramping period only) or (D) high intensity exercise after two and four weeks of supplementation, respectively. In a separate endpoint experiment, plasma and quadriceps muscles were collected after 90 min of endurance running with ramping (10′ PostEx) and used for measurement of (E) plasma acetylcarnitine and (F) total carnitine (free carnitine + acetylcarnitine) in muscle. Data are means ±SEM, n=6 per group. * carnitine effect, ^ genotype effect (p<0.05).
Exercise alters acetylcarnitine dynamics in humans
Lastly, we sought to evaluate the translational relevance of these findings in humans. To this end, CrAT protein abundance was found to be elevated in skeletal muscle biopsy specimens from trained athletes as compared to age-matched untrained subjects, and trended lower in muscle from older, type 2 diabetic subjects (Figure 6A). Protein abundance correlated strongly with muscle CrAT activity (Figure 6B), and enzyme activity was positively associated with mitochondrial function, assessed in vivo by determining PCr recovery kinetics after exercise using magnetic resonance spectroscopy (MRS) (Figure 6C), as recently described (Lindeboom et al., 2014). We then conducted a small pilot study in a separate cohort of trained and untrained subjects in which a newly developed MRS technique was used to examine in vivo muscle acetylcarnitine content before and muscle content and kinetics after 30 min on a cycle ergometer at 50% maximal workload. A dramatic between-group difference was observed in exercise-induced acetylcarnitine metabolism. Whereas the cycle exercise increased tissue acetylcarnitine content in both groups, peak accumulation immediately after exercise was markedly lower in the trained group, suggesting increased consumption (Figure 6D). Moreover, during the 40 min recovery period the acetylcarnitine signal decayed rapidly in the trained group but continued to rise in the untrained subjects.
Figure 6. CrAT expression and acetylcarnitine levels in human medial vastus lateralis muscle.

A) CrAT protein abundance measured in muscle biopsy specimens collected from exercise trained (T), untrained (UT) and type 2 diabetic (T2D) subjects (n=6–11). (B) Relationship between CrAT protein abundance and CrAT activity. (C) Relationship between CrAT activity and phosphocreatine recovery rates measured by 31P-MRS after 5 min of exercise. (D) Muscle levels of acetylcarnitine measured in trained and untrained subjects by 1H-MRS before, 15 min after and during recovery from 30 min of cycle ergometer exercise performed at 50% of maximal workload (n=4–5).
Although further studies are necessary to better understand whole body acetylcarnitine flux in humans, these findings align with the notion that training-induced upregulation of CrAT in humans influences muscle bioenergetics and acetyl group balance during and after exercise. Conversely, low CrAT activity, which has been observed in the context of aging, obesity and diabetes, might exacerbate muscle fatigue (Muoio et al., 2012; Noland et al., 2009; Seiler et al., 2014). Also noteworthy is that previous studies in humans using either proton MRS (Ren et al., 2013) or biochemical methods (Stephens et al., 2007) showed that mid- to high-intensity muscle contraction shifts the tissue carnitine pool from a principally non-esterified to a mostly esterified state, suggesting that availability of free carnitine could limit CrAT activity under these circumstances. Moreover, systemic carnitine homeostasis can be compromised by aging, obesity, cardiometabolic disease, hepatic steatosis and use of drugs that form carnitine conjugates (Costell et al., 1989; Evangeliou and Vlassopoulos, 2003). Together with current study, these findings imply that nutritional or pharmacological strategies aimed at increasing acetyl-CoA buffering capacity have the potential to augment energy efficiency an delay muscle fatigue, and might prove beneficial for treating some patients with exercise intolerance (Brevetti et al., 1991; Wall et al., 2011).
Working model of CrAT flux during exercise
Whereas our results provide strong evidence that CrAT activity influences muscle energetics and contractile performance, delineating the biophysical basis of this finding is not as straightforward. We consider three explanations as illustrated in Figure 7. First, acetylcarnitine derived from both local and circulating pools might serve as an important acetyl group donor during brief periods when rates of OXPHOS exceed acetyl-CoA production from PDH and beta-oxidation. This possibility is supported by our evidence that muscle clears and oxidizes exogenously supplied acetylcarnitine during contractile activity, and that provision of acetylcarnitine delayed fatigue in isolated working skeletal muscles. However, because blood concentrations of acetylcarnitine are in the low μM range, we presume that acetylcarnitine supply from the periphery to working muscles accounts for only a small fraction of total ATP demand. Additionally, acetylcarnitine metabolites might be transferred between neighboring myofibers. For example, perhaps highly oxidative type I myofibers with elevated mitochondrial content but lower force-generating potential shuttle surplus acetylcarnitine to more powerful, ATP-consuming type II fibers. To this point, we found that CrAT protein levels were most abundant in isolated mitochondria from muscles enriched in type IIa fibers (Bloemberg and Quadrilatero, 2012). Thus, mitochondria resident in type IIa myofibers might have a greater need for acetylcarnitine substrate to meet the energetic demands of mid to high intensity workloads.
Figure 7. Proposed role of CrAT in mitochondrial acetyl-CoA buffering and acetyl group transfer during exercise.

Acetyl-CoA is the universal catabolic intermediate that fuels the tricarboxylic acid cycle (TCAC), which in turn serves as the main source of reducing equivalents (NADH) that support oxidative regeneration of ATP and phosphocreatine (PCr) by the electron transport chain (ETC) and mitochondrial creatine kinase (CK). During transitions from low to high exercise workloads, TCAC flux must increase to keep pace with the high ATP demands of muscle contraction. A shortfall in acetyl-CoA provision will force heavy reliance on substrate level phosphorylation, resulting in depletion of PCr and muscle glycogen reserves, along with production of lactic acid and other deleterious metabolic byproducts. CrAT functions to sustain high rates oxidative ATP regeneration via three proposed mechanisms. First, contraction-induced recycling and/or import of acetylcarnitine (AcCarn) supplies a readily available source of acetyl group donors to buffer transient deficits in glucose, amino acid and fatty acid catabolism. Secondly, by residing in close proximity to the various enzymatic sources of acetyl-CoA (e.g. pyruvate dehydrogenase (PDH) and ketothiolase (KT)), CrAT alleviates product inhibition of metabolic enzymes while also regenerating essential cofactor (CoA) necessary for continued catabolic flux. Thirdly, CrAT permits rapid and efficient delivery of acetyl groups from their site of production to the TCAC (blue arrows). Thus, by acting as a conduit for acetyl group transfer, CrAT overcomes the thermodynamic inefficiencies of diffusional flux, which requires an energetically unfavorable gradient profile (black triangle).
Secondly, by residing in close proximity to various enzymatic sources of acetyl-CoA, CrAT could function to alleviate kinetic hindrances due to product inhibition, while also regenerating essential cofactors (CoA) necessary for continued catabolic flux. For example, acetyl-CoA inhibits PDH by activating one or more of its four known PDH kinases (PDK1-4), which phosphorylate and inactivate the E1a subunit of the complex (Sugden and Holness, 2003). Acetyl-CoA and free CoA compete for binding to the complex and thus small shifts in the local acetyl-CoA/CoA ratio can contribute to substantive changes in enzyme activity. Accordingly, provision of L-carnitine was previously shown to increase both PDH activity and pyruvate-supported respiration in isolated mitochondria from control but not CrAT deficient muscles (Fisher-Wellman et al., 2015; Muoio et al., 2012).
Lastly, a third model positions CrAT as a conduit for acetyl group transfer. This possibility emerges from studies that used hyperpolarized 13C MRS to examine acetylcarnitine dynamics in perfused working heart. Investigators estimated that under the conditions of these experiments, 50% of pyruvate-derived citrate cycles through the acetylcarnitine pool before entering the TCAC (Schroeder et al., 2012). Similarly, another study that used a mass spectrometry-based tracer approach concluded that pyruvate-derived acetyl-CoA is preferentially channeled to CrAT (Li et al., 2015). Based on these and the current results, we envision a key role for CrAT in metabolic channeling. Thus, flux through the CrAT reaction during exercise might permit rapid and efficient delivery of acetyl groups from their site of production to citrate synthase (Figure 7). By acting as a shuttle system for acetyl group transfer, CrAT would be predicted to overcome the thermodynamic inefficiencies of diffusional flux, which requires an energetically unfavorable gradient profile (Aliev et al., 2011). This paradigm of substrate transfer through a series of near-equilibrium reactions is conceptually analogous to the phospho-energy transfer networks driven by the adenylate kinase and creatine kinase systems (Dzeja and Terzic, 2003; Sweeney, 1994).
The foregoing mechanisms could act simultaneously if acetylcarnitine produced by the PDH complex under high flux conditions is efficiently recycled, while exogenously supplied and/or the local storage pool of acetylcarnitine also contribute to citrate synthase flux. Notably, the affinities of CrAT, PDH and citrate synthase for acetyl-CoA align with this possibility. For example, the Km of citrate synthase for acetyl CoA is approximately 10-fold lower than CrAT, whereas the Ki of acetyl CoA for PDH and the PDKs is roughly equivalent to its Km for CrAT. This suggests that CrAT has the potential to modulate PDH flux without competing with citrate synthase. We therefore propose that CrAT runs in the forward direction in the vicinity of PDH, while running in the reverse direction near citrate synthase, such that net flux through CrAT during exercise favors acetyl-CoA production, at least in some fibers. This model is supported by results of (Schroeder et al., 2012) showing that the CrAT reaction continues to run in the reverse direction even during supraphysiological supply of acetyl-CoA via PDH, and that the total acetylcarnitine pool in heart decreases upon increasing cardiac workload.
In sum, we have shown that CrAT-mediated acetyl group buffering is critical for muscle contractile performance and fatigue resistance and that acetylcarnitine has a metabolic purpose during exercise. The findings also imply that CrAT maintains the acetyl-CoA microenvironment of specific mitochondrial enzyme complexes in a manner the permits optimal TCAC flux and aerobic power. This work speaks to longstanding questions on the importance of metabolic inertia and acetyl deficit as limiting factors during exercise and advances the concept of metabolic channeling.
Materials and Methods
Animals
Animal studies were approved by the Duke University Institutional Animal Care and Use Committee. Generation of the Crat homozygous floxed control (Cratfl/fl) mice has been described (Muoio et al., 2012). Skeletal muscle specific knockout mice (CratSKM−/−) were generated by breeding Cratfl/fl mice with transgenic mice expressing Cre recombinase under control of the mouse myogenin promoter and MEF2C (myo-CreTg/0), which express Cre recombinase exclusively in skeletal muscle but not heart. All lines were backcrossed >6 generations to the C57BL/6 background. Animals were housed in a temperature-controlled environment with a 12-hour light:12-hour dark cycle and allowed ad libitum access to standard chow (Purina Rodent Chow no. 5001, Purina Mills, St. Louis, MO, USA) and water. Studies were performed in male animals unless otherwise noted in the Figure legends. We found no interactions between sex and genotype.
Mitochondrial Isolation
Skeletal muscle mitochondria were prepared according to (DeBalsi et al., 2014).
Carnitine acetyltransferase activity
Tissues were ground into powder and processed in CelLytic MT buffer (Sigma Chemicals, St. Louis, MO) by freeze fracturing three times and sonication at five times one second pulses on setting five. CrAT activity was assessed according to (Seiler et al., 2014).
Western blots
Protein was isolated using CelLytic MT lysis buffer (Sigma Chemicals, St. Louis, MO) and quantified by using a bicinchoninic acid (BCA) kit (Sigma Chemicals, St. Louis, MO). Protein (50μg from cell/tissue lysates) was separated by SDS-PAGE, transferred to nitrocellulose, and incubated with antibodies prepared with fish gelatin blocking buffer (0.5% fish gelatin, 1X PBS, 1 mg/ml casein and 15 mM NaN3). IgG-congugated AlexFluor fluorescent secondary antibodies (Invitrogen) were used with the Li-COR (Li-COR Biosciences) to visualize bands. Protein expression was normalized to total protein as determined by MemCode staining (Thermo Scientific). The primary CrAT antibody was a generous gift from the laboratory of Dr. Fausto Hegardt.
Metabolite Analysis
Tissue and plasma samples were processed and analysed as described in (Muoio et al., 2012; Noland et al., 2009). Detailed methods for the mass spectrometry- and MRS-based measurements of metabolites in mouse and human muscles are provided in the Supplementary Materials. PCr and creatine analysis was performed according to (Harris et al., 1974; HU, 1974).
Glycogen Content
Five hundred microliters of 1 N HCl was added to 20 mg of powdered skeletal muscle in a 2.0-ml screw top tube and homogenized at 25K RPM for 30 seconds. Samples were boiled for 90 min, tubes were cooled to room temperature and 0.5 ml of 1 N NaOH was added. Glycogen content was assessed at 340 nm after addition of glucose assay reagent (Sigma).
Fibertype Analysis
Plantaris muscles were embedded in OCT and tragacanth before being frozen in liquid nitrogen cooled 2-Methylbutane and stored at −80°C. 8 μm sections were prepared and fixed in cold acetone for 2 minutes, washed in 1X PBS and blocked for 1 hour at room temperature in 10% normal goat serum before incubation in the following antibodies: BA-F8 MHC type 1 (1:100), BF-F3 MHC 2b (1:200) and SC-71 MHC-2a (Developmental Studies Hybridoma Bank, University of Iowa); Alexa Fluor conjugated secondary antibody cocktail (1:100) 555, 647, 488 (Invitrogen) at room temperature for 30 minutes in the dark. Slides were mounted in FluorSave Reagent (Calbiochem), imaged on a Zeiss Axioplan 2 and fiber type composition quantified using ImageJ.
Exercise Studies
Exercise studies were carried out in a blinded fashion. Three days prior to the experiment, mice were acclimated via exposure to a 3 minute run with one minute at each of the first three speeds in the respective protocol being tested. Exhaustion was defined as remaining on the shock grid for more than 10 seconds with nudging. High Intensity: Exercise capacity during high intensity running was determined using an enclosed treadmill (Columbus instruments) attached to a Comprehensive Laboratory Animal Monitoring System (CLAMS) with a fixed incline of 10°. Mice began running at 14 m/min and speed increased 3 m/min every 3 minutes until exhaustion. Measurements were collected every 30 seconds. Airflow was set to 0.6 l/min. Blood blood lactate was measured with a Lactate Plus lactometer (Nova Biomedical, Waltham, MA). Endurance with High Intensity Ramping: The transition from endurance to high intensity running was completed with a fixed slope of 10°. Mice began running at 8 m/min and speed increased 2 m/min every 15 minutes for 1 hour before transitioning to 20 m/min for 1 min, then 23 m/min for 10 min, with an increase of 3.5 m/min every 10 min until exhaustion. Distance and time to exhaustion presented reflect the ramp up period only.
For the carnitine supplementation experiment, mice were randomly selected to receive two weeks of supplementation with pH-neutralized drinking water ±1.0 mg/ml L-carnitine. After exercise, mice were anesthetized using an intraperitoneal injection of 100 mg/kg body weight Nembutal. Tissue samples harvested approximately 10 min after cessation of exercise, frozen immediately in liquid nitrogen and stored at −80°C.
Ex-vivo muscle Stimulation and Acetylcarnitine Oxidation
Braided silk suture loops were attached to muscle tendons and excised from the mouse. Muscles were placed in pre-warmed KHB buffer (pH 7.4) containing (in mmol/l) 118 NaCl, 4.7 KCl, 2.52 CaCl2, 1.64 MgSO4, 24.88 NaCO3, 1.18 KH2PO4, 5.5 glucose and 2.0 Na-pyruvate. After placement in a Radnoti 2 mL tissue bath system, muscles were tensed to 1 gram and allowed to relax for 15 min. Muscles were then re-tensed to 1 gram and allowed to rest 5 min, which was repeated 3 times.. KHB buffer was used in muscle transport and tensing, whereas low glucose KHB (2 mM glucose, no pyruvate) was used during stimulation to mimic an energy stressed state. The following stimulation protocol was used unless otherwise stated: stimulation rate: 60 pps, delay: 2000 ms, duration: 300 ms, volts: 20 per chamber. O2 flow and temperature (25°C) were kept constant throughout stretching and stimulation. Acetylcarnitine (Sigma) was made up at 1 M in H2O and added at a final concentration of 5 mM to stimulated muscles 30 seconds after initiation of contraction. This allowed for normalization of contracting muscles prior to adding acetylcarnitine. Acetylcarnitine oxidation was assessed by capturing media [14C]CO2 from 200 μM [1-14C]acetylcarnitine (ARC 1615) during rest or stimulation. Oxidation studies were done in low glucose KHB buffer with 3 mg/mL HEPES added. Acetylcarnitine efflux experiments were conducted with isolated soleus muscles from wildtype mice incubated in KHB buffer supplemented with 3 mg/mL HEPES, 5 mM [U-13C] glucose and 2 mM [U-13C] pyruvate. Buffer was sampled before, during and after contraction using the following stimulation protocol: stimulation rate: 60 pps, delay: 2000 ms, duration: 300 ms, volts: 12.5 per chamber, time: 10 min. [13C2]acetylcarnitine was quantified by standard isotope dilution methodology using D3 acetylcarnitine as the internal standard after correcting for the natural abundance of the M+1peak of the [2-13C]acetylcarnitine peak. This correction was determined empirically from a spectrum of [2-13C]acetylcarnitine with no added internal standard.
Statistics
Statistical analyses were performed using SigmaStat (SysStat Software, Inc., Point Richmond, CA) or the Microsoft Excel statistical package. Within-group responses to experimental manipulations were evaluated using a paired t test, where appropriate. All data are presented as mean±S.E., and the level of significance was established a priori at p less than or equal to 0.05.
Supplementary Material
Acknowledgments
The authors are supported by grants from the United States Public Health Service: R01-DK089312 (DMM), 2P01-DK058398 (DMM), R01-HL101189R01 (DMM), F32-DK-094573 (KEW), F32-DK093256 (MND) and K01-GM1093200-01 (JRG) and the American Heart Association (KLD). P. Schrauwen is supported by a VICI (grant 918.96.618) for innovative research from the Netherlands Organization for Scientific Research (NWO). V.B. Schrauwen-Hinderling is supported by a VENI (grant 91611136) for innovative research from the Netherlands Organization for Scientific Research (NWO). We thank Dr. Randy Mynatt at the Pennington Biomedical Research Center for providing the Cratfl/fl mice.
Footnotes
Conflict of interest statement: The authors have no conflict of interest to declare.
Author Contributions: Conceptualization: D.M.M., T.R.K.; Methodology: T.R.K., D.M.M., P.S., V.B.S.H; Investigation: S.E.S., T.R.K., J.R.G., R.S.S., O.R.I., K.E.W., A.H.W., K.L.D., M.N.D., L.L., V.B.S.H.; Writing - Original Draft: S.E.S., V.B.S.H. T.R.K., D.M.M; Writing – Review & Editing: T.R.K., D.M.M., P.S., V.B.S.H.; Visualization: T.R.K., D.M.M; Resources: D.M.M., P.S., V.B.S.H.; Funding Acquisition: D.M.M., P.S., V.B.S.H.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aliev M, Guzun R, Karu-Varikmaa M, Kaambre T, Wallimann T, Saks V. Molecular system bioenergics of the heart: experimental studies of metabolic compartmentation and energy fluxes versus computer modeling. Int J Mol Sci. 2011;12:9296–9331. doi: 10.3390/ijms12129296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloemberg D, Quadrilatero J. Rapid determination of myosin heavy chain expression in rat, mouse, and human skeletal muscle using multicolor immunofluorescence analysis. PLoS One. 2012;7:e35273. doi: 10.1371/journal.pone.0035273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brevetti G, Angelini C, Rosa M, Carrozzo R, Perna S, Corsi M, Matarazzo A, Marcialis A. Muscle carnitine deficiency in patients with severe peripheral vascular disease. Circulation. 1991;84:1490–1495. doi: 10.1161/01.cir.84.4.1490. [DOI] [PubMed] [Google Scholar]
- Costell M, O’Connor JE, Grisolia S. Age-dependent decrease of carnitine content in muscle of mice and humans. Biochem Biophys Res Commun. 1989;161:1135–1143. doi: 10.1016/0006-291x(89)91360-0. [DOI] [PubMed] [Google Scholar]
- DeBalsi KL, Wong KE, Koves TR, Slentz DH, Seiler SE, Wittmann AH, Ilkayeva OR, Stevens RD, Perry CG, Lark DS, et al. Targeted metabolomics connects thioredoxin-interacting protein (TXNIP) to mitochondrial fuel selection and regulation of specific oxidoreductase enzymes in skeletal muscle. The Journal of biological chemistry. 2014;289:8106–8120. doi: 10.1074/jbc.M113.511535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durkot MJ, De Garavilla L, Caretti D, Francesconi R. The effects of dichloroacetate on lactate accumulation and endurance in an exercising rat model. Int J Sports Med. 1995;16:167–171. doi: 10.1055/s-2007-972986. [DOI] [PubMed] [Google Scholar]
- Dzeja PP, Terzic A. Phosphotransfer networks and cellular energetics. J Exp Biol. 2003;206:2039–2047. doi: 10.1242/jeb.00426. [DOI] [PubMed] [Google Scholar]
- Evangeliou A, Vlassopoulos D. Carnitine metabolism and deficit--when supplementation is necessary? Curr Pharm Biotechnol. 2003;4:211–219. doi: 10.2174/1389201033489829. [DOI] [PubMed] [Google Scholar]
- Fisher-Wellman KH, Lin CT, Ryan TE, Reese LR, Gilliam LA, Cathey BL, Lark DS, Smith CD, Muoio DM, Neufer PD. Pyruvate dehydrogenase complex and nicotinamide nucleotide transhydrogenase constitute an energy consuming redox circuit. The Biochemical journal. 2015 doi: 10.1042/BJ20141447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhaff PL, Campbell-O’Sullivan SP, Constantin-Teodosiu D, Poucher SM, Roberts PA, Timmons JA. An acetyl group deficit limits mitochondrial ATP production at the onset of exercise. Biochem Soc Trans. 2002;30:275–280. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- Harris RC, Hultman E, Nordesjo LO. Glycogen, glycolytic intermediates and high-energy phosphates determined in biopsy samples of musculus quadriceps femoris of man at rest. Methods and variance of values. Scand J Clin Lab Invest. 1974;33:109–120. [PubMed] [Google Scholar]
- Hiatt WR, Regensteiner JG, Wolfel EE, Ruff L, Brass EP. Carnitine and acylcarnitine metabolism during exercise in humans. Dependence on skeletal muscle metabolic state. The Journal of clinical investigation. 1989;84:1167–1173. doi: 10.1172/JCI114281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett RA, Heigenhauser GJ, Hultman E, Hollidge-Horvat MG, Spriet LL. Effects of dichloroacetate infusion on human skeletal muscle metabolism at the onset of exercise. The American journal of physiology. 1999;277:E18–25. doi: 10.1152/ajpendo.1999.277.1.E18. [DOI] [PubMed] [Google Scholar]
- HUB . Methods of Enzymatic Analysis. New York: Academic; 1974. [Google Scholar]
- Korzeniewski B, Zoladz JA. Some factors determining the PCr recovery overshoot in skeletal muscle. Biophys Chem. 2005;116:129–136. doi: 10.1016/j.bpc.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Koves TR, Sparks LM, Kovalik JP, Mosedale M, Arumugam R, DeBalsi KL, Everingham K, Thorne L, Phielix E, Meex RC, et al. PPARgamma coactivator-1alpha contributes to exercise-induced regulation of intramuscular lipid droplet programming in mice and humans. Journal of lipid research. 2013;54:522–534. doi: 10.1194/jlr.P028910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Deng S, Ibarra RA, Anderson VA, Brunengraber H, Zhang GF. Multiple mass isotopomer tracing of acetyl-CoA metabolism in langendorff-perfused rat hearts: Channeling of acetyl-CoA from pyruvate dehydrogenase to carnitine acetyltransferase. The Journal of biological chemistry. 2015 doi: 10.1074/jbc.M114.631549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeboom L, Nabuurs CI, Hoeks J, Brouwers B, Phielix E, Kooi ME, Hesselink MK, Wildberger JE, Stevens RD, Koves T, et al. Long-echo time MR spectroscopy for skeletal muscle acetylcarnitine detection. The Journal of clinical investigation. 2014;124:4915–4925. doi: 10.1172/JCI74830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludvik B, Mayer G, Stifter S, Putz D, Barnas U, Graf H. Effects of dichloroacetate on exercise performance in healthy volunteers. Pflugers Arch. 1993;423:251–254. doi: 10.1007/BF00374403. [DOI] [PubMed] [Google Scholar]
- Makowski L, Noland RC, Koves TR, Xing W, Ilkayeva OR, Muehlbauer MJ, Stevens RD, Muoio DM. Metabolic profiling of PPARalpha−/− mice reveals defects in carnitine and amino acid homeostasis that are partially reversed by oral carnitine supplementation. FASEB J. 2009;23:586–604. doi: 10.1096/fj.08-119420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muoio DM, MacLean PS, Lang DB, Li S, Houmard JA, Way JM, Winegar DA, Corton JC, Dohm GL, Kraus WE. Fatty acid homeostasis and induction of lipid regulatory genes in skeletal muscles of peroxisome proliferator-activated receptor (PPAR) alpha knock-out mice. Evidence for compensatory regulation by PPAR delta. The Journal of biological chemistry. 2002;277:26089–26097. doi: 10.1074/jbc.M203997200. [DOI] [PubMed] [Google Scholar]
- Muoio DM, Noland RC, Kovalik JP, Seiler SE, Davies MN, DeBalsi KL, Ilkayeva OR, Stevens RD, Kheterpal I, Zhang J, et al. Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metab. 2012;15:764–777. doi: 10.1016/j.cmet.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murias JM, Spencer MD, Paterson DH. The critical role of O2 provision in the dynamic adjustment of oxidative phosphorylation. Exercise and sport sciences reviews. 2014;42:4–11. doi: 10.1249/JES.0000000000000005. [DOI] [PubMed] [Google Scholar]
- Noland RC, Koves TR, Seiler SE, Lum H, Lust RM, Ilkayeva O, Stevens RD, Hegardt FG, Muoio DM. Carnitine insufficiency caused by aging and overnutrition compromises mitochondrial performance and metabolic control. The Journal of biological chemistry. 2009;284:22840–22852. doi: 10.1074/jbc.M109.032888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattyn N, Cornelissen VA, Eshghi SR, Vanhees L. The effect of exercise on the cardiovascular risk factors constituting the metabolic syndrome: a meta-analysis of controlled trials. Sports Med. 2013;43:121–133. doi: 10.1007/s40279-012-0003-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieklik JR, Guynn RW. Equilibrium constants of the reactions of choline acetyltransferase, carnitine acetyltransferase, and acetylcholinesterase under physiological conditions. The Journal of biological chemistry. 1975;250:4445–4450. [PubMed] [Google Scholar]
- Poole DC, Jones AM. Oxygen uptake kinetics. Comprehensive Physiology. 2012;2:933–996. doi: 10.1002/cphy.c100072. [DOI] [PubMed] [Google Scholar]
- Putman CT, Spriet LL, Hultman E, Lindinger MI, Lands LC, McKelvie RS, Cederblad G, Jones NL, Heigenhauser GJ. Pyruvate dehydrogenase activity and acetyl group accumulation during exercise after different diets. The American journal of physiology. 1993;265:E752–760. doi: 10.1152/ajpendo.1993.265.5.E752. [DOI] [PubMed] [Google Scholar]
- Raper JA, Love LK, Paterson DH, Peters SJ, Heigenhauser GJ, Kowalchuk JM. Effect of high-fat and high-carbohydrate diets on pulmonary O2 uptake kinetics during the transition to moderate-intensity exercise. J Appl Physiol (1985) 2014;117:1371–1379. doi: 10.1152/japplphysiol.00456.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Lakoski S, Haller RG, Sherry AD, Malloy CR. Dynamic monitoring of carnitine and acetylcarnitine in the trimethylamine signal after exercise in human skeletal muscle by 7T 1H-MRS. Magn Reson Med. 2013;69:7–17. doi: 10.1002/mrm.24249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahlin K. Muscle carnitine metabolism during incremental dynamic exercise in humans. Acta physiologica Scandinavica. 1990;138:259–262. doi: 10.1111/j.1748-1716.1990.tb08845.x. [DOI] [PubMed] [Google Scholar]
- Sahlin K, Soderlund K, Tonkonogi M, Hirakoba K. Phosphocreatine content in single fibers of human muscle after sustained submaximal exercise. The American journal of physiology. 1997;273:C172–178. doi: 10.1152/ajpcell.1997.273.1.C172. [DOI] [PubMed] [Google Scholar]
- Schroeder MA, Atherton HJ, Dodd MS, Lee P, Cochlin LE, Radda GK, Clarke K, Tyler DJ. The cycling of acetyl-coenzyme A through acetylcarnitine buffers cardiac substrate supply: a hyperpolarized 13C magnetic resonance study. Circ Cardiovasc Imaging. 2012;5:201–209. doi: 10.1161/CIRCIMAGING.111.969451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler SE, Martin OJ, Noland RC, Slentz DH, Debalsi KL, Ilkayeva OR, An J, Newgard CB, Koves TR, Muoio DM. Obesity and lipid stress inhibit carnitine acetyltransferase activity. Journal of lipid research. 2014;55:635–644. doi: 10.1194/jlr.M043448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens FB, Constantin-Teodosiu D, Greenhaff PL. New insights concerning the role of carnitine in the regulation of fuel metabolism in skeletal muscle. The Journal of physiology. 2007;581:431–444. doi: 10.1113/jphysiol.2006.125799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugden MC, Holness MJ. Recent advances in mechanisms regulating glucose oxidation at the level of the pyruvate dehydrogenase complex by PDKs. American journal of physiology Endocrinology and metabolism. 2003;284:E855–862. doi: 10.1152/ajpendo.00526.2002. [DOI] [PubMed] [Google Scholar]
- Sweeney HL. The importance of the creatine kinase reaction: the concept of metabolic capacitance. Medicine and science in sports and exercise. 1994;26:30–36. [PubMed] [Google Scholar]
- Timmons JA, Constantin-Teodosiu D, Poucher SM, Greenhaff PL. Acetyl group availability influences phosphocreatine degradation even during intense muscle contraction. The Journal of physiology. 2004;561:851–859. doi: 10.1113/jphysiol.2004.069419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons JA, Gustafsson T, Sundberg CJ, Jansson E, Hultman E, Kaijser L, Chwalbinska-Moneta J, Constantin-Teodosiu D, Macdonald IA, Greenhaff PL. Substrate availability limits human skeletal muscle oxidative ATP regeneration at the onset of ischemic exercise. The Journal of clinical investigation. 1998;101:79–85. doi: 10.1172/JCI1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons JA, Poucher SM, Constantin-Teodosiu D, Worrall V, Macdonald IA, Greenhaff PL. Increased acetyl group availability enhances contractile function of canine skeletal muscle during ischemia. The Journal of clinical investigation. 1996;97:879–883. doi: 10.1172/JCI118490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall BT, Stephens FB, Constantin-Teodosiu D, Marimuthu K, Macdonald IA, Greenhaff PL. Chronic oral ingestion of L-carnitine and carbohydrate increases muscle carnitine content and alters muscle fuel metabolism during exercise in humans. The Journal of physiology. 2011;589:963–973. doi: 10.1113/jphysiol.2010.201343. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.