

Abstract
Objective
Axial spondyloarthritis (SpA) is a chronic inflammatory disease characterized by back pain and stiffness. The objective of this study was to determine whether golimumab is superior to placebo in patients with nonradiographic axial SpA.
Methods
This phase III, double‐blind, randomized, placebo‐controlled trial was performed to evaluate subcutaneous golimumab (50 mg) versus placebo in patients ages ≥18 years to ≤45 years who had active nonradiographic axial SpA according to the Assessment of SpondyloArthritis international Society (ASAS) criteria for ≤5 years since diagnosis, high disease activity, and an inadequate response to or intolerance of nonsteroidal antiinflammatory drugs. Patients were randomized 1:1 to receive golimumab or placebo subcutaneously every 4 weeks. The primary end point was 20% improvement according to the ASAS criteria (ASAS20) at week 16. Key secondary end points were an ASAS40 response, ASAS partial remission, 50% improvement in the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), and change in the Spondyloarthritis Research Consortium of Canada (SPARCC) magnetic resonance imaging (MRI) index for sacroiliac (SI) joint inflammation (SPARCC score).
Results
Of the 198 patients randomized, 197 were treated (97 received golimumab, and 100 received placebo). The mean age of the patients was 31 years, and 57.1% were male. At baseline, the mean ± SD BASDAI was 6.5 ± 1.5, the mean ± SD ASDAS was 3.5 ± 0.9, and the mean ± SD SPARCC score was 11.3 ± 14.0. The primary end point, an ASAS20 response, was achieved by significantly more patients in the golimumab group compared with the placebo group (71.1% versus 40.0%; P < 0.0001). An ASAS40 response was also achieved by significantly more patients in the golimumab group compared with the placebo group (56.7% versus 23.0%; P < 0.0001). The incidence of adverse events did not differ meaningfully between groups.
Conclusion
Patients with active nonradiographic axial SpA treated with golimumab had significantly greater improvement in symptoms compared with patients treated with placebo. Golimumab was well tolerated and had a favorable risk/benefit profile.
Axial spondyloarthritis (SpA) is a chronic inflammatory rheumatic disease characterized by inflammation of the sacroiliac (SI) joints and spine 1, 2. Patients with axial SpA experience chronic back pain and spinal stiffness as well as a reduction in mobility and quality of life (QoL) 3. Over time, permanent damage to spinal mobility and function can occur due to new bone formation in the spine 3.
The term axial SpA encompasses patients with evident radiographic changes in the SI joints according to the modified New York criteria 4, also termed ankylosing spondylitis (AS), and patients who have no evident radiographic signs of structural damage but who may have evidence of sacroiliitis visible by magnetic resonance imaging (MRI) and/or share other features with AS such as spinal inflammation, chronic back pain, HLA–B27 positivity, and other nonarticular symptoms 5, 6. This latter group is described as having nonradiographic axial SpA, and nonradiographic axial SpA was recently classified by the Assessment of SpondyloArthritis international Society (ASAS) as part of axial SpA 2, 7. A late diagnosis of axial SpA frequently leads to delays in treatment 8. Adoption of the ASAS criteria has the potential to lead to earlier identification of patients with axial SpA 6, early in the disease course for many, and more timely therapeutic intervention.
The current standard of care for axial SpA is nonsteroidal antiinflammatory drugs (NSAIDs) 9, 10, 11, 12, 13. If there is an insufficient response to or intolerance of NSAIDs the next line of treatment is tumor necrosis factor (TNF)–targeted therapies, which have demonstrated efficacy in recent trials in patients with nonradiographic axial SpA 14, 15, 16, 17, 18, 19. TNF‐blocking agents have already been approved for this indication in the European Union (EU) and other countries but not yet in the US.
In this randomized, double‐blind, placebo‐controlled clinical trial (GO‐AHEAD), we investigated the effect of treatment with golimumab, a fully human anti‐TNF antibody, administered subcutaneously every 4 weeks at a dose of 50 mg over 16 weeks, in patients with active nonradiographic axial SpA. The primary end point was 20% improvement in disease activity according to the ASAS criteria (ASAS20) 20 at week 16.
PATIENTS AND METHODS
Study design and patients
The GO‐AHEAD study (Protocol 06; ClinicalTrials.gov identifier: NCT01453725) is a 2‐part phase III, multicenter, randomized, parallel‐group, double‐blind, placebo‐controlled trial evaluating the safety and efficacy of golimumab monotherapy for the treatment of patients with active nonradiographic axial SpA. The first part of the study was performed from February 2012 through May 2014. Patients were randomized in 52 centers in 13 countries (Czech Republic, Denmark, Finland, Germany, Greece, Ireland, Italy, Russia, Slovakia, Spain, Turkey, the UK, and the US). The study was conducted in accordance with the Principles of Good Clinical Practice and standards for protection of human patients participating in biomedical research and was approved by institutional review boards and regulatory agencies. All patients provided written informed consent before any study procedures were performed.
Eligible patients were randomized using an interactive voice response system and interactive web response system (IVRS/IWRS; Almac) and were stratified according to evidence of sacroiliitis on MRI (≤50% patients did not have evidence of sacroiliitis) and C‐reactive protein (CRP) level (≤60% patients had a normal CRP level at the time of screening). All MRIs were read by a central reader at Synarc who reported the presence of inflammation of the SI joints, which is highly suggestive of sacroiliitis 2, 7, or the absence of SI joint inflammation. All investigators, site personnel, patients, and sponsor personnel were blinded with regard to treatment allocation.
Patients were ≥18 to ≤45 years of age with a physician's diagnosis of active nonradiographic axial SpA, a disease duration since diagnosis of ≤5 years, and chronic back pain of ≥3 months' duration. All patients were required to meet either the ASAS classification criterion for the presence of sacroiliitis by MRI and have 1 SpA feature or be HLA–B27 positive and have ≥2 SpA features. Patients had active disease at both screening and baseline, as defined by a spinal pain score of ≥4 and a Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) 21 score of ≥4.0 on a 10‐cm visual analog scale (VAS). An additional inclusion requirement was an inadequate response to or intolerance of at least 1 NSAID or the inability to tolerate a maximal dose of NSAIDs for 30 days.
Key exclusion criteria included radiographic evidence of grade II sacroiliitis bilaterally or grade III or IV sacroiliitis unilaterally at screening (radiographs were read by a central reader at Synarc), any systemic inflammatory rheumatic condition between screening and baseline other than nonradiographic axial SpA, a serious infection within 2 months prior to the first injection of study medication, a history of chronic or recurrent infectious disease, lymphoproliferative disease, current active malignancy or malignancy within the previous 5 years, or chronic heart failure.
Additional exclusion criteria included previous treatment with TNF‐targeted therapies, any biologic agent or cytotoxic drug (e.g., chlorambucil or cyclophosphamide), disease‐modifying antirheumatic drugs or any investigational medications within 30 days of screening, live vaccinations within 3 months of screening or Bacille Calmette‐Guérin vaccination within 12 months, and evidence of untreated latent or active tuberculosis (TB) prior to screening.
This was a 2‐part study that includes a preplanned 44‐week open‐label extension phase (part 2) to evaluate the long‐term treatment effect and safety of golimumab at a dose of 50 mg. The second part of the study will be reported separately.
Study treatment
Patients were randomized at a 1:1 ratio to receive subcutaneous golimumab 50 mg or placebo at weeks 0, 4, 8, and 12 (Figure 1A). Trained personnel at the study site administered injections using prefilled syringes. Patients who successfully completed part 1 of the GO‐AHEAD study (weeks 0–16) were eligible to participate in part 2 (weeks 16–60) of the trial.
Figure 1.
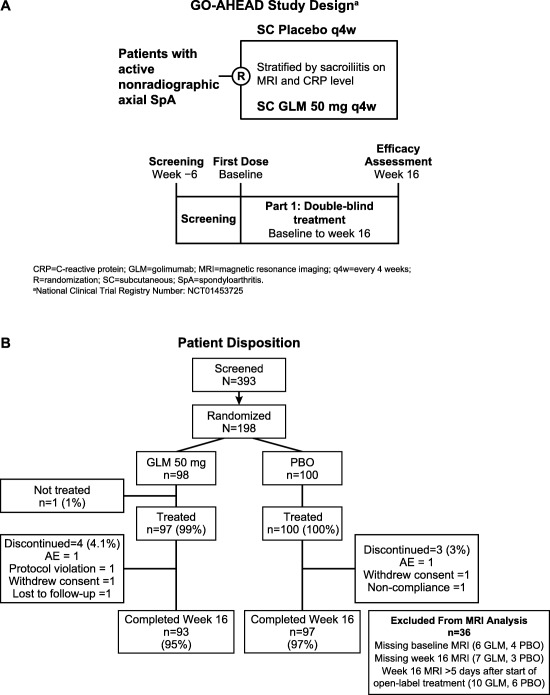
A, GO‐AHEAD study design. B, Patient disposition. PBO = placebo; AE = adverse event.
Outcome measures
The primary efficacy measure evaluated the effect of golimumab 50 mg compared with placebo for the treatment of active nonradiographic axial SpA, as measured by the proportion of patients who achieved an ASAS20 response at week 16. Key secondary end points were an ASAS40 response, ASAS partial remission, 50% improvement in the BASDAI (BASDAI 50 response), and change in the Spondyloarthritis Research Consortium of Canada (SPARCC) MRI index for SI joint inflammation 22, all at 16 weeks.
Other secondary end points included efficacy as evaluated by measuring the change in the BASDAI, the AS Disease Activity Score (ASDAS) 23 using the CRP level, the Bath Ankylosing Spondylitis Functional Index (BASFI) 24, the Bath Ankylosing Spondylitis Metrology Index (BASMI) 25, total back pain (as measured using a 10‐cm VAS), the CRP level, the swollen joint count (SJC) and tender joint count (TJC), and the Maastricht Ankylosing Spondylitis Enthesitis Score (MASES) 26. Several patient‐reported QoL outcomes were also evaluated, including the Short Form 36 (SF‐36) 27, the AS Quality of Life (ASQoL) questionnaire 28, and the EuroQol 5‐domain (EQ‐5D) questionnaire 29.
Subgroup analyses of the primary end point, an ASAS20 response at week 16, were conducted for several prespecified demographic and baseline factors. These factors, which included sex, age, weight, HLA–B27 status, geographic region, MRI status (positive or negative for SI), CRP status (level above or below the upper limit of normal [ULN]), MRI negative and CRP level above the ULN, MRI negative and CRP level lower than the ULN, MRI positive and CRP level above the ULN, and MRI positive and CRP level lower than the ULN, were assessed to determine the consistency of the treatment effect.
Safety measurement
Clinical evaluations and routine laboratory measurements, including serum chemistry and hematology testing, were performed at screening, on day 1, and at weeks 4 and 16 or the time of early termination. Vital signs were measured at screening, on day 1, and at weeks 4, 8, 12, and 16 or the time of early termination. Laboratory measurements were performed at a certified Quest Diagnostics Clinical Trials laboratory. Physical examination, determination of height and weight, hepatitis B virus screening, and a tuberculin skin test or QuantiFeron‐TB Gold test (Qiagen) were performed at the screening visit. Serum specimens were collected on day 1 and at week 16 to assess for anti‐golimumab antibodies and golimumab concentrations. Assessments of adverse events (AEs) were performed throughout the study. AEs of clinical interest were monitored throughout the study, including an elevated aspartate aminotransferase or alanine aminotransferase level (≥3‐fold the ULN), elevated bilirubin level (≥2‐fold the ULN) with an alkaline phosphatase level <2‐fold the ULN, clinically significant opportunistic infections, TB, and hypersensitivity reactions.
Statistical analysis
This study was expected to randomize ∼200 patients. For the primary hypothesis, with a sample size of 100 patients per group, there was at least 95% power to detect a 26% treatment difference between golimumab 50 mg and placebo (overall α = 0.05 by 2‐sided test, assuming a response rate for placebo of 25% [based on estimates from pivotal anti‐TNF trials in AS]).
The full analysis set, consisting of all treated patients, was used for all efficacy analyses except that for the MRI SI joint score. The MRI SI joint score analysis was performed using the SPARCC method 22 in patients with complete MRI data (i.e., at baseline and week 16). For the primary efficacy end point, the proportion of ASAS20 responders at week 16 was compared between patients receiving golimumab and those receiving placebo, according to the stratified method described by Miettinen and Nurminen 30, using baseline evidence of sacroiliitis on MRI and the CRP level at the screening visit as the stratification factors. To test the robustness of the primary analysis, a sensitivity analysis in which patients who discontinued treatment due to an AE were considered nonresponders was also performed.
Key secondary end points were evaluated in the same manner as the primary end point. The change from baseline to week 16 in the SPARCC MRI SI joint score was compared between patients in the golimumab group and those in the placebo group, using the Mann‐Whitney test. Continuous end points were evaluated using a repeated‐measures model, specifically the constrained longitudinal data analysis approach proposed by Liang and Zeger 31, with MRI stratum, CRP stratum, and treatment‐by‐time as fixed effects and subject as a random effect. Type I error was controlled for using a closed testing procedure in which the primary end point was tested first and, if the result was significant, was followed by tests of the key secondary end points in a step‐down procedure in the following order: ASAS40, BASDAI 50, ASAS partial remission, and SPARCC MRI SI joint score.
For the ASAS‐related end points at week 16 (ASAS20, ASAS40, and ASAS partial remission), patients for whom all of the ASAS component values were missing at week 16 were considered not to have achieved the primary end point (nonresponder imputation). If some of the ASAS component values were missing at week 16, then the following imputation rules applied: 1) if the component value was missing from all visits (baseline through week 16), then 0% improvement was imputed; 2) if the component value was missing at week 16 but was provided at an earlier visit, imputation was done using the last observation carried forward method; and 3) if a baseline value was missing but postbaseline measures were available, the baseline value was imputed using the median baseline component value for all patients in the same strata.
For the BASDAI 50, if the patient had no observation at week 16, then the last nonmissing observation prior to week 16, including the baseline value, was carried forward to week 16. The BASDAI was calculated if at least 3 of the 5 component values were available; otherwise, the BASDAI was set to missing. For other end points, only observed data were used in the analyses, and no missing data were imputed.
An “objective signs of inflammation” (OSI) population consisting of patients with baseline evidence of sacroiliitis on MRI and/or a screening CRP level greater than the ULN was also analyzed for the primary and key secondary efficacy end points and overall AEs. Patients in the OSI population used for efficacy and safety end point analyses were derived from the full analysis set and “all patients as treated” (APaT) populations, respectively. The APaT population consisted of all patients who received at least 1 dose of study medication. The APaT population was used for the analysis of safety data. AEs were analyzed according to the method described by Miettinen and Nurminen for between‐treatment differences in the percentage of patients with events 30.
RESULTS
Patient disposition
Of the 393 patients screened for inclusion in the study, 198 were randomized (98 were assigned to the golimumab group and 100 were assigned to the placebo group) (Figure 1B). Of these patients, 190 (96%) completed the study through week 16. One patient in the golimumab group was not treated (due to positive results of a pregnancy test), and 7 patients discontinued participation in the study (4 in the golimumab group and 3 in the placebo group) (Figure 1B).
Baseline characteristics
The 2 treatment groups were similar with respect to baseline demographics and disease characteristics, except sex (Table 1). The majority of the patients were male (57.1%), with a greater percentage of male patients in the golimumab group compared with the placebo group (62.2% versus 52.0%). The mean age of the patients was 31 years, and all patients were white. Disease duration was similar between the 2 treatment groups, and the majority of the patients (66.7%) had a disease duration of <1 year since diagnosis. Most patients (82.3%) were HLA–B27 positive, the mean BASDAI score was 6.5, and the mean ASDAS was 3.5. The baseline characteristics of the OSI population were consistent with those of the overall study population.
Table 1.
Baseline characteristics of the 198 patients randomized to receive golimumab or placeboa
| Characteristic | Golimumab (n = 98) | Placebo (n = 100) |
|---|---|---|
| Male sex | 61 (62.2) | 52 (52.0) |
| Age, mean ± SD years | 30.7 ± 7.1 | 31.7 ± 7.2 |
| White race | 98 (100.0) | 100 (100.0) |
| Geographic region | ||
| Eastern Europe | 52 (53.1) | 53 (53.0) |
| Western Europe and US | 46 (46.9) | 47 (47.0) |
| BMI, mean ± SD kg/m2 | 25.6 ± 4.7 | 25.1 ± 4.9 |
| Disease duration since diagnosis | ||
| 1 year | 67 (68.4) | 65 (65.0) |
| 1–2 years | 20 (20.4) | 19 (19.0) |
| 3–5 years | 11 (11.2) | 16 (16.0) |
| BASDAI, mean ± SD (10‐cm VAS) | 6.6 ± 1.6 | 6.4 ± 1.5 |
| BASFI, mean ± SD (10‐cm VAS) | 5.3 ± 2.4 | 4.8 ± 2.5 |
| SPARCC SI MRI score, mean ± SD (range 0–72)b | 9.9 ± 12.3 | 12.7 ± 15.4 |
| MRI‐positive for sacroiliitisc | 66 (67.3) | 66 (66.0) |
| ASDAS, mean ± SD | 3.6 ± 0.9 | 3.5 ± 0.8 |
| CRP concentration, mean ± SD mg/dl | 1.5 ± 2.9 | 1.3 ± 2.0 |
| CRP > upper limit of normal | 40 (40.8) | 41 (41.0) |
| HLA–B27 positive | 81 (82.7) | 82 (82.0) |
Except where indicated otherwise, values are the number (%) of patients. BMI = body mass index; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; VAS = visual analog scale; BASFI = Bath Ankylosing Spondylitis Functional Index; ASDAS = Ankylosing Spondylitis Disease Activity Score; CRP = C‐reactive protein.
The Spondyloarthritis Research Consortium of Canada magnetic resonance imaging index for sacroiliac joint inflammation (SPARCC SI MRI) score was determined in 91 patients in the golimumab group and 96 patients in the placebo group.
A central reader reported the presence or absence of active inflammation of SI joints.
Exposure and compliance
Most patients received the planned 16 weeks of treatment with golimumab or placebo (190 [96%] of 198 patients). Mean compliance (number of doses taken divided by the number of doses expected) was 99.5%.
Efficacy results
Clinical end points
The percentage of patients achieving an ASAS20 response at 16 weeks was significantly higher in the golimumab group compared with the placebo group (71.1% versus 40.0%; change in response 31.2% [95% confidence interval (95% CI) 17.5%, 43.6%]; P < 0.0001) (Figure 2A). Analysis of the ASAS20 response in the OSI patient population (n = 158 [78 in the golimumab group and 80 in the placebo group]) demonstrated a greater difference in response between golimumab‐ and placebo‐treated patients (76.9% versus 37.5%; change in response 39.6% [95% CI 24.6%, 52.6%]; P < 0.0001), whereas patients with negative MRI results and a normal CRP level showed no difference in the ASAS20 response (golimumab 47.4% versus placebo 50.0%; change in response −2.6% [95% CI −32.7%, 27.9%]; P = 0.8711) (Figure 2A). The results of the sensitivity analysis substantiated the results of the primary analysis (data not shown), thus confirming the robustness of the data.
Figure 2.
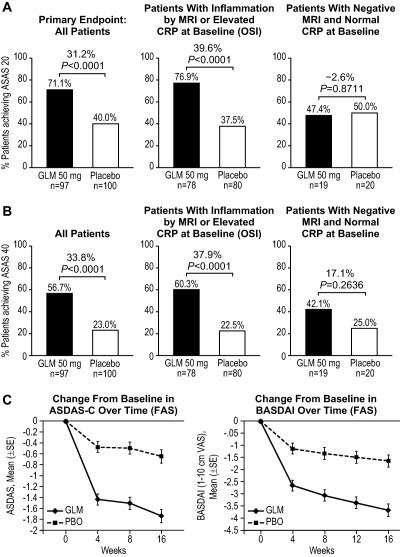
A and B, Percentages of patients in the golimumab (GLM) and placebo (PBO) groups achieving 20% improvement according to the Assessment of SpondyloArthritis international Society criteria (ASAS20) (A) and ASAS40 criteria (B) at week 16, in the full analysis set (FAS) and according to magnetic resonance imaging (MRI) status (positive or negative for the presence of sacroiliitis) and C‐reactive protein (CRP) status (objective signs of inflammation [OSI] population). Differences were derived using the stratified method described by Miettinen and Nurminen (30). C, Change from baseline in the Ankylosing Spondylitis Disease Activity Score using the CRP level (ASDAS‐C) and change from baseline in the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) response over time.
Assessment of the ASAS20 response according to subgroup based on prespecified demographic and baseline factors demonstrated responses favoring golimumab over placebo in most cases; the exception was the subgroup of patients with the combination of negative MRI findings and a CRP level within normal limits at baseline, in whom no differences in the ASAS20 response rate were observed between those receiving golimumab and those receiving placebo (Figure 3). Among the subgroup of patients achieving an ASAS20 response at week 16 who had baseline evidence of sacroiliitis on MRI, the response was greater in those receiving golimumab compared with those receiving placebo (73.8% versus 37.9%; change in response 36.0% [95% CI 19.3%, 50.7%]; P < 0.0001). Among the subgroup of patients with an elevated CRP level at baseline, the response was also greater in those receiving golimumab than in those receiving placebo (87.2% versus 36.6%; change in response 50.6% [95% CI 30.5%, 66.6%]; P < 0.0001).
Figure 3.
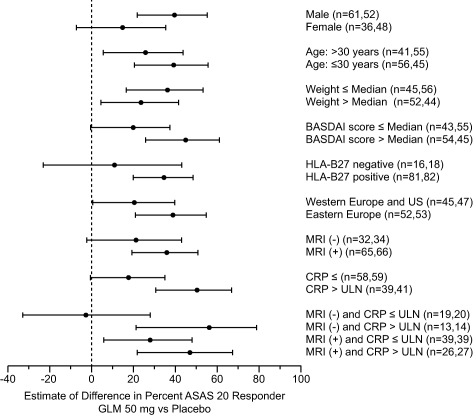
Differences in the percentage of patients achieving an ASAS20 response at week 16 between the golimumab group and the placebo group, according to subgroups at baseline. Values are the point estimates and 95% confidence intervals. ULN = upper limit of normal (see Figure 2 for other definitions).
For the key secondary end point, an ASAS40 response, the treatment group difference was similar to that observed for the primary end point (56.7% of patients in the golimumab group versus 23.0% of those in the placebo group; change in response 33.8% [95% CI 20.4%, 46.1%]; P < 0.0001) (Figure 2B). Findings similar to those for the primary end point were also observed in the ASAS40 subgroup analyses based on baseline evidence of MRI‐defined sacroiliitis and/or CRP level elevation (Figure 2B). Analysis of other key secondary end points, i.e., the BASDAI 50, ASAS partial remission, and mean change in the SPARCC MRI SI joint score, revealed significant improvement in patients treated with golimumab (P < 0.0001, P = 0.0136, and P < 0.0001, respectively) (Table 2). Among patients with baseline evidence of MRI‐defined sacroiliitis and/or an elevated CRP level, significant improvement was also observed in the golimumab group compared with the placebo group for these secondary end points (data not shown). No significant improvements were observed in patients without evidence of sacroiliitis by MRI and a normal CRP level at baseline (data not shown).
Table 2.
Efficacy assessments at 16 weeks in the full analysis seta
| Golimumab 50 mg | Placebo | Difference, golimumab vs. placebo | ||||||
|---|---|---|---|---|---|---|---|---|
| n | Baseline | Week 16 | n | Baseline | Week 16 | % (95% CI) | P | |
| Responders, no. (%) | ||||||||
| BASDAI 50 | 97 | – | 57 (57.7) | 100 | – | 30 (30.0) | 28.0 (14.4, 40.6) | <0.0001b |
| ASAS partial remission | 97 | – | 32 (33.0) | 100 | – | 18 (18.0) | 15.2 (3.2, 27.1) | 0.0136b |
| SPARCC MRI SI score | 74 | 9.9 ± 11.82 | 4.6 ± 7.92 | 87 | 12.7 ± 15.62 | 11.71 ± 14.79 | −4.3 | <0.0001c |
| CRP, mg/dl | 88 | 1.51 ± 2.94 | 0.43 ± 0.87 | 91 | 1.36 ± 2.08 | 1.06 ± 1.64 | −0.64 (−0.98, −0.30) | 0.0003d |
| BASDAI, 10‐cm VAS | 93 | 6.62 ± 1.57 | 2.93 ± 2.51 | 96 | 6.29 ± 1.45 | 4.68 ± 2.75 | −2.00 (−2.68, −1.35) | <0.0001d |
| BASFI | 93 | 5.26 ± 2.34 | 2.50 ± 2.53 | 97 | 4.70 ± 2.53 | 3.87 ± 2.83 | −1.73 (−2.33, −1.13) | <0.0001 |
| ASDAS | 88 | 3.59 ± 0.94 | 1.87 ± 1.02 | 90 | 3.40 ± 0.78 | 2.80 ± 1.22 | −1.05 (−1.37, −0.73) | <0.0001d |
| BASMI | 94 | 2.4 ± 1.30 | 1.93 ± 1.18 | 100 | 2.51 ± 1.32 | 2.42 ± 1.39 | −0.39 (−0.58, −0.20) | <0.0001d |
| MASES index score | 92 | 3.2 ± 3.36 | 1.7 ± 2.95 | 97 | 3.2 ± 3.35 | 2.5 ± 3.18 | −0.7 (−1.4, −0.1) | 0.0302d |
| SF‐36, physical summary scale | 91 | 32.85 ± 8.08 | 43.43 ± 10.21 | 96 | 34.97 ± 8.68 | 38.33 ± 9.65 | 6.56 (4.28, 8.83) | <0.0001d |
| SF‐36, mental summary scale | 91 | 41.10 ± 11.94 | 47.06 ± 11.08 | 96 | 41.55 ± 11.14 | 43.08 ± 11.84 | 4.24 (1.42, 7.07) | 0.0034d |
| EQ‐5D index score | 94 | 0.41 ± 0.32 | 0.68 ± 0.28 | 100 | 0.44 ± 0.33 | 0.54 ± 0.31 | 0.15 (0.08, 0.22) | <0.0001d |
| ASQoL questionnaire score | 94 | 11.1 ± 4.45 | 5.6 ± 5.16 | 100 | 10.2 ± 4.57 | 8.6 ± 5.09 | −3.5 (−4.7, −2.2) | <0.0001d |
| Total back pain, 10‐cm VAS | 93 | 6.98 ± 1.78 | 2.77 ± 2.78 | 97 | 6.61 ± 1.67 | 4.74 ± 3.17 | −2.13 (−2.94, −1.32) | <0.0001d |
Except where indicated otherwise, values are the mean ± SD. 95% CI = 95% confidence interval; BASDAI 50 = 50% improvement in the Bath Ankylosing Spondylitis Disease Activity Index; ASAS = Assessment of SpondyloArthritis international Society; SPARCC MRI SI = Spondyloarthritis Research Consortium of Canada magnetic resonance imaging index for sacroiliac joint inflammation; CRP = C‐reactive protein; VAS = visual analog scale; BASFI = Bath Ankylosing Spondylitis Functional Index; ASDAS = Ankylosing Spondylitis Disease Activity Score; BASMI = Bath Ankylosing Spondylitis Metrology Index; MASES = Maastricht Ankylosing Spondylitis Enthesitis Score; SF‐36 = Short Form 36; EQ‐5D = EuroQol 5‐domain; ASQoL = Ankylosing Spondylitis Quality of Life.
Differences derived using the stratified Miettinen and Nurminen method (30).
Based on Mann‐Whitney scores.
Derived using a constrained longitudinal data analysis.
Figure 2C shows the changes in the BASDAI score and ASDAS over 16 weeks of treatment. Marked improvements (P < 0.0001) were observed at week 4 after 1 injection of golimumab, and further improvements in disease activity continued through week 16. The degree of suppression of the CRP level at week 16 was greater in the golimumab group compared with the placebo group (P = 0.0003) (Table 2).
Functional, spinal mobility, and QoL end points
Results from functional, spinal mobility, and QoL assessments are summarized in Table 2. The mean changes from baseline in the BASFI, BASMI, and MASES at week 16 were greater in the golimumab group compared with the placebo group (P < 0.0001, P < 0.0001, and P = 0.03, respectively). Changes from baseline in the SJC and TJC were similar between the golimumab group and the placebo group (data not shown). Furthermore, the changes from baseline were greater in the golimumab group compared with the placebo group for the SF‐36 physical and mental component scores, ASQoL, EQ‐5D, and total back pain (Table 2).
Safety
Overall, golimumab was well tolerated; the incidence of serious AEs (SAEs) and other significant AEs was comparable between the patients treated with golimumab and those treated with placebo (Table 3). No new safety signals were identified during treatment of nonradiographic axial SpA in this study.
Table 3.
Adverse events (AEs) in the patients during 16 weeks of treatmenta
| Golimumab (n = 97) | Placebo (n = 100) | |
|---|---|---|
| Any AE | 40 (41.2) | 47 (47.0) |
| AE related to study medicationb | 13 (13.4) | 17 (17.0) |
| Serious AE | 1 (1.0) | 2 (2.0) |
| Female partner reported fetal death | 1 (1.0) | 0 |
| Cholelithiasis | 0 | 1 (1.0) |
| Back pain | 0 | 1 (1.0) |
| AE leading to early withdrawalc | 2 (2.1) | 1 (1.0) |
| Specific AEs of interest | ||
| Serious infections | 0 | 0 |
| Active tuberculosis | 0 | 0 |
| Malignancies | 0 | 0 |
| Serious systemic hypersensitivity | 0 | 0 |
| Deaths | 0 | 0 |
Values are the number (%).
As determined by the investigator.
Study medication withdrawn.
In the APaT population, AEs were reported for 87 (44.2%) of the 197 patients who received study medication. The incidence of AEs was lower in patients receiving golimumab compared with those receiving placebo (41.2% and 47.0%, respectively). No notable differences between the golimumab group and the placebo group were identified for AE categories, including drug‐related AEs (13 [13.4%] and 17 [17.0%], respectively).
For the most frequently reported clinical AEs, the incidence was generally lower in the golimumab group compared with the placebo group, with the exception of skin and subcutaneous tissue AEs (10.3% in the golimumab group versus 6% in the placebo group). A total of 3 SAEs in 3 patients were reported, with 2 events (back pain and cholelithiasis) in the placebo group and 1 event (fetal death; the female partner of the patient experienced spontaneous abortion at <20 weeks gestational age) in the golimumab group. None of the SAEs were considered by the investigator to be related to the study drug. No deaths were reported during this study. No events of special interest were reported (including serious infections, serious opportunistic infections, active TB, malignancies, or serious systemic hypersensitivity).
Antibodies to golimumab were present in 4 patients. All 4 of these patients achieved an ASAS20 response at week 16, suggesting that the immunogenicity response did not affect efficacy.
DISCUSSION
This phase III, randomized, double‐blind, placebo‐controlled, global study was conducted to evaluate the efficacy and safety of subcutaneous golimumab, a fully human anti‐TNF antibody, at a dose of 50 mg, in patients with active nonradiographic axial SpA. Treatment with golimumab every 4 weeks resulted in significant and sustained improvements in the signs and symptoms of nonradiographic axial SpA through week 16, with improvement occurring after the first injection of golimumab. Golimumab was safe and generally well tolerated in this study, and its safety profile was consistent with the known safety profile of golimumab when used for other indications.
In total, 71.1% of patients in the golimumab group achieved the primary end point, an ASAS20 response at week 16, compared with 40.0% of patients in the placebo group. Improvement in the ASDAS and BASDAI were already notable by the first postbaseline assessment (week 4) and were maintained and increased slightly between 4 weeks and 16 weeks. The treatment effects of golimumab were also significant for the key secondary efficacy measures, which included the ASAS40 response, the BASDAI 50 response, ASAS partial remission, and change in the SPARCC MRI SI joint score between baseline and week 16. Clinically meaningful improvements were observed in multiple other measures of disease activity, physical function, and QoL.
Patients with and those without objective signs of inflammation were included in this study. The OSI population, which was defined by the presence of sacroiliitis by MRI and/or an elevated CRP level, had a robust golimumab treatment effect, with 76.9% of these patients achieving an ASAS20 response at week 16 compared with 37.5% in the placebo group. In the 20% of patients without MRI‐confirmed sacroiliitis or an elevated CRP level, there were no differences in the efficacy end points, indicating that this subgroup of patients with nonradiographic axial SpA may not be candidates for treatment with golimumab, as was also recently shown for treatment of nonradiographic axial SpA with adalimumab, another anti‐TNF antibody 16. Consequently, current labeling in the EU for the treatment of nonradiographic axial SpA with TNF blockers such as adalimumab, certolizumab, or etanercept makes a normal CRP level and/or MRI positivity mandatory for the initiation of treatment.
Subgroup analyses to examine the impact of different baseline parameters on the ASAS20 response showed that golimumab treatment resulted in consistently better clinical responses (compared with placebo) in all but the subpopulation with no evidence of sacroiliitis by MRI and a normal CRP level at baseline. This is suggestive of a treatment benefit across a variety of demographics and baseline disease conditions. As reported previously 14, 32, 33, patients who were male, HLA–B27 positive, young, and who had MRI‐defined SI and/or an elevated CRP level had a slightly better response, although in general the number of patients in these subgroups was small. In this study, all patients underwent MRI of the SI joints at baseline, which was read centrally and used for study enrollment, according to the ASAS classification criteria 2, 7.
This is the first study of TNF blockers in patients with nonradiographic axial SpA that analyzed the treatment effect in the HLA–B27–positive/MRI‐negative group of patients who meet the so‐called clinical arm of the ASAS classification criteria. As suggested in Figure 3, patients from the clinical arm (MRI negative) who had a CRP level above the ULN responded to treatment slightly better compared with the group of MRI‐positive patients, and the response was in the same range as that in the overall group of patients with CRP levels above the ULN, while patients with CRP levels below the ULN from the clinical arm (equivalent to MRI negative and CRP levels less than or equal to the ULN) did not benefit from treatment with golimumab. Thus, in this study, patients with nonradiographic axial SpA who were HLA–B27–positive, MRI‐negative, and had a CRP level above the ULN seemed to respond at least as well as the overall population.
These data indicate that the treatment response to golimumab in patients with nonradiographic axial SpA is similar to the response observed in patients with AS treated with golimumab. For example, in a previous study of golimumab in patients with AS 34, an ASAS20 response was observed in 59.4% of the patients receiving golimumab at a dose of 50 mg compared with 21.8% of those receiving placebo (37.6% difference). In the current study, the differences were 31.2% in the overall population and 39.6% in the OSI population. Based on the ASAS20 and ASAS40 responses observed in this study, golimumab appears to be at least as efficacious as other anti‐TNF therapies that have been tested in a population of patients with nonradiographic axial SpA 15, 16, 35. Head‐to‐head trials would be needed in order to test for response differences between the drugs.
The percentage of patients in the placebo group who achieved an ASAS20 response was relatively high (40% at 16 weeks), and the potential reason for this is unclear. Differences in the participating centers and inclusion of patients in whom disease was less advanced (compared with cases that were rather advanced in the AS study) might play a role. A placebo response of similar magnitude was also observed in another trial of anti‐TNF therapy in patients with axial SpA, including patients with nonradiographic axial SpA and those with radiographic axial SpA (AS) 15, suggesting that the patient population investigated in current clinical trials differs from the AS population included in previous studies of TNF blockers. The subgroup analysis (see Figure 3) did not provide any further information to explain the relatively high number of responders in the placebo group observed in our study.
Golimumab was generally well tolerated in this young population. The incidence of AEs was slightly higher among patients in the placebo group compared with the golimumab group (47.0% versus 41.2%). No AEs of clinical interest and no deaths were observed in this study. A total of 3 SAEs were reported in 3 patients. None of the SAEs were considered by the investigators to be related to the study drug. The safety data observed in this study were consistent with the safety profile of golimumab in AS 34 and other rheumatic diseases 36, 37, as well as with that of other anti‐TNF therapies 38. No new safety signals were identified during the treatment of nonradiographic axial SpA.
Limitations of this study include the reporting of data up to only 16 weeks of treatment. Results from later time points will be needed in order to confirm a continued reduction in symptoms over time. Findings from later time points will be presented in a subsequent report.
In summary, this study demonstrated that golimumab, administered subcutaneously every 4 weeks, is efficacious and generally well tolerated in patients with nonradiographic axial SpA. Golimumab treatment led to a rapid reduction in the signs and symptoms of nonradiographic axial SpA, and this effect was sustained through week 16. Furthermore, golimumab provided substantial benefits to patients with nonradiographic axial SpA by also improving multiple disease features including inflammation identified by MRI, range of motion, physical function, and health‐related QoL. These results demonstrate a favorable risk/benefit profile for golimumab treatment in patients with nonradiographic axial SpA.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Sieper had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Sieper, van der Heijde, Dougados, Maksymowych, Boice, Curtis, Tzontcheva, Huyck, Weng.
Acquisition of data. Boice, Berd, Bergman.
Analysis and interpretation of data. Sieper, van der Heijde, Dougados, Maksymowych, Scott, Curtis, Tzontcheva, Huyck, Weng.
ROLE OF THE STUDY SPONSOR
Merck & Company sponsored the study. The study protocol was the responsibility of Merck & Company and was designed in collaboration with the scientific advisory committee comprised of the academic authors and approved by various health authorities. Data were collected by the investigators and were entered into a database that was maintained by Merck & Company. All analyses were conducted by statisticians and programmers at Merck & Company. Publication of this article was not contingent upon approval by Merck & Company.
ACKNOWLEDGMENT
We would like to thank Jennifer Pawlowski (Merck & Company Inc.) for her assistance with the submission of this manuscript.
ClinicalTrials.gov identifier: NCT01453725.
REFERENCES
- 1. Braun J, Sieper J. Ankylosing spondylitis. Lancet 2007;369:1379–90. [DOI] [PubMed] [Google Scholar]
- 2. Rudwaleit M, van der Heijde D, Landewe R, Listing J, Akkoc N, Brandt J, et al. The development of Assessment of SpondyloArthritis international Society classification criteria for axial spondyloarthritis (part II): validation and final selection. Ann Rheum Dis 2009;68:777–83. [DOI] [PubMed] [Google Scholar]
- 3. Sieper J, Rudwaleit M, Baraliakos X. The Assessement of Spondyloarthritis international Society (ASAS) handbook: a guide to assess spondylarthritis. Ann Rheum Dis 2009;68 Suppl 2:ii1–44. [DOI] [PubMed] [Google Scholar]
- 4. Van der Linden S, Valkenburg HA, Cats A. Evaluation of diagnostic criteria for ankylosing spondylitis: a proposal for modification of the New York criteria. Arthritis Rheum 1984;27:361–8. [DOI] [PubMed] [Google Scholar]
- 5. Freeston J, Barkham N, Hensor E, Emery P, Fraser A. Ankylosing spondylitis, HLA‐B27 positivity and the need for biologic therapies. Joint Bone Spine 2007;74:140–3. [DOI] [PubMed] [Google Scholar]
- 6. Rudwaleit M, van der Heijde D, Khan MA, Braun J, Sieper J. How to diagnose axial spondyloarthritis early. Ann Rheum Dis 2004;63:535–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rudwaleit M, Landewe R, van der Heijde D, Listing J, Brandt J, Braun J, et al. The development of Assessment of SpondyloArthritis international Society classification criteria for axial spondyloarthritis (part I): classification of paper patients by expert opinion including uncertainty appraisal. Ann Rheum Dis 2009;68:770–6. [DOI] [PubMed] [Google Scholar]
- 8. Feldtkeller E, Khan MA, van der Heijde D, van der Linden S, Braun J. Age at disease onset and diagnosis delay in HLA‐B27 negative vs. positive patients with ankylosing spondylitis. Rheumatol Int 2003;23:61–6. [DOI] [PubMed] [Google Scholar]
- 9. Barkhuizen A, Steinfeld S, Robbins J, West C, Coombs J, Zwillich S. Celecoxib is efficacious and well tolerated in treating signs and symptoms of ankylosing spondylitis. J Rheumatol 2006;33:1805–12. [PubMed] [Google Scholar]
- 10. Poddubnyy D, Rudwaleit M, Haibel H, Listing J, Marker‐Hermann E, Zeidler H, et al. Effect of non‐steroidal anti‐inflammatory drugs on radiographic spinal progression in patients with axial spondyloarthritis: results from the German Spondyloarthritis Inception Cohort. Ann Rheum Dis 2012;71:1616–22. [DOI] [PubMed] [Google Scholar]
- 11. Sieper J, Klopsch T, Richter M, Kapelle A, Rudwaleit M, Schwank S, et al. Comparison of two different dosages of celecoxib with diclofenac for the treatment of active ankylosing spondylitis: results of a 12‐week randomised, double‐blind, controlled study. Ann Rheum Dis 2008;67:323–9. [DOI] [PubMed] [Google Scholar]
- 12. Van der Heijde D, Baraf HS, Ramos‐Remus C, Calin A, Weaver AL, Schiff M, et al. Evaluation of the efficacy of etoricoxib in ankylosing spondylitis: results of a fifty‐two–week, randomized, controlled study. Arthritis Rheum 2005;52:1205–15. [DOI] [PubMed] [Google Scholar]
- 13. Wanders A, van der Heijde D, Landewe R, Behier JM, Calin A, Olivieri I, et al. Nonsteroidal antiinflammatory drugs reduce radiographic progression in patients with ankylosing spondylitis: a randomized clinical trial. Arthritis Rheum 2005;52:1756–65. [DOI] [PubMed] [Google Scholar]
- 14. Haibel H, Rudwaleit M, Listing J, Heldmann F, Wong RL, Kupper H, et al. Efficacy of adalimumab in the treatment of axial spondylarthritis without radiographically defined sacroiliitis: results of a twelve‐week randomized, double‐blind, placebo‐controlled trial followed by an open‐label extension up to week fifty‐two. Arthritis Rheum 2008;58:1981–91. [DOI] [PubMed] [Google Scholar]
- 15. Landewe R, Braun J, Deodhar A, Dougados M, Maksymowych WP, Mease PJ, et al. Efficacy of certolizumab pegol on signs and symptoms of axial spondyloarthritis including ankylosing spondylitis: 24‐week results of a double‐blind randomised placebo‐controlled phase 3 study. Ann Rheum Dis 2014;73:39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sieper J, van der Heijde D, Dougados M, Mease PJ, Maksymowych WP, Brown MA, et al. Efficacy and safety of adalimumab in patients with non‐radiographic axial spondyloarthritis: results of a randomised placebo‐controlled trial (ABILITY‐1). Ann Rheum Dis 2013;72:815–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sieper J, Landewe R, Rudwaleit M, van der Heijde D, Dougados M, Mease PJ, et al. Effect of certolizumab pegol over ninety‐six weeks in patients with axial spondyloarthritis: results from a phase III randomized trial. Arthritis Rheumatol 2015;67:668–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sieper J, Lenaerts J, Wollenhaupt J, Rudwaleit M, Mazurov VI, Myasoutova L, et al. on behalf of all INFAST Investigators. Efficacy and safety of infliximab plus naproxen versus naproxen alone in patients with early, active axial spondyloarthritis: results from the double‐blind, placebo‐controlled INFAST study, part 1. Ann Rheum Dis 2014;73:101–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Song IH, Hermann K, Haibel H, Althoff CE, Listing J, Burmester G, et al. Effects of etanercept versus sulfasalazine in early axial spondyloarthritis on active inflammatory lesions as detected by whole‐body MRI (ESTHER): a 48‐week randomised controlled trial. Ann Rheum Dis 2011;70:590–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anderson JJ, Baron G, van der Heijde D, Felson DT, Dougados M. Ankylosing Spondylitis Assessment Group preliminary definition of short‐term improvement in ankylosing spondylitis. Arthritis Rheum 2001;44:1876–86. [DOI] [PubMed] [Google Scholar]
- 21. Garrett S, Jenkinson T, Kennedy LG, Whitelock H, Gaisford P, Calin A. A new approach to defining disease status in ankylosing spondylitis: the Bath Ankylosing Spondylitis Disease Activity Index. J Rheumatol 1994;21:2286–91. [PubMed] [Google Scholar]
- 22. Maksymowych WP, Inman RD, Salonen D, Dhillon SS, Williams M, Stone M, et al. Spondyloarthritis Research Consortium of Canada Magnetic Resonance Imaging Index for assessment of sacroiliac joint inflammation in ankylosing spondylitis. Arthritis Rheum 2005;53:703–9. [DOI] [PubMed] [Google Scholar]
- 23. Van der Heijde D, Lie E, Kvien TK, Sieper J, van den Bosch F, Listing J, et al., for the Assessment of SpondyloArthritis international Society (ASAS) . ASDAS, a highly discriminatory ASAS‐endorsed disease activity score in patients with ankylosing spondylitis. Ann Rheum Dis 2009;68:1811–8. [DOI] [PubMed] [Google Scholar]
- 24. Calin A, Garrett S, Whitelock H, Kennedy LG, O'Hea J, Mallorie P, et al. A new approach to defining functional ability in ankylosing spondylitis: the development of the Bath Ankylosing Spondylitis Functional Index. J Rheumatol 1994;21:2281–5. [PubMed] [Google Scholar]
- 25. Jenkinson TR, Mallorie PA, Whitelock HC, Kennedy LG, Garrett SL, Calin A. Defining spinal mobility in ankylosing spondylitis (AS): the Bath AS Metrology Index. J Rheumatol 1994;21:1694–8. [PubMed] [Google Scholar]
- 26. Heuft-Dorenbosch L, Spoorenberg A, van Tubergen A, Landewe R, van ver Tempel H, Mielants H, et al. Assessment of enthesitis in ankylosing spondylitis. Ann Rheum Dis 2003;62:127–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ware JE Jr, Snow KK, Kosinski M, Gandek B. SF‐36 health survey: manual and interpretation guide. Boston: The Health Institute, New England Medical Center; 1993. [Google Scholar]
- 28. Helliwell PS, Doward L, Whalley D, Tennant A, McKenna S, Reynolds S, et al. Psychometric and scaline properties of a new quality of life instrument specific to ankylosing spondylitis [abstract]. Arthritis Rheum 1999;42 Suppl:S72. [Google Scholar]
- 29. Hurst NP, Kind P, Ruta D, Hunter M, Stubbings A. Measuring health‐related quality of life in rheumatoid arthritis: validity, responsiveness and reliability of EuroQol (EQ‐5D). Br J Rheumatol 1997;36:551–9. [DOI] [PubMed] [Google Scholar]
- 30. Miettinen O, Nurminen M. Comparative analysis of two rates. Stat Med 1985;4:213–26. [DOI] [PubMed] [Google Scholar]
- 31. Liang KY, Zeger SL. Longitudinal data analysis of continuous and discrete responses for pre‐post designs. Indian J Stat 2000;62:134–48. [Google Scholar]
- 32. Rudwaleit M, Listing J, Brandt J, Braun J, Sieper J. Prediction of a major clinical response (BASDAI 50) to tumour necrosis factor α blockers in ankylosing spondylitis. Ann Rheum Dis 2004;63:665–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rudwaleit M, Claudepierre P, Wordsworth P, Cortina EL, Sieper J, Kron M, et al. Effectiveness, safety, and predictors of good clinical response in 1250 patients treated with adalimumab for active ankylosing spondylitis. J Rheumatol 2009;36:801–8. [DOI] [PubMed] [Google Scholar]
- 34. Inman RD, Davis JC Jr, van der Heijde D, Diekman L, Sieper J, Kim SI, et al. Efficacy and safety of golimumab in patients with ankylosing spondylitis: results of a randomized, double‐blind, placebo‐controlled, phase III trial. Arthritis Rheum 2008;58:3402–12. [DOI] [PubMed] [Google Scholar]
- 35. Dougados M, van der Heijde D, Sieper J, Braun J, Maksymowych WP, Citera G, et al. Symptomatic efficacy of etanercept and its effects on objective signs of inflammation in early nonradiographic axial spondyloarthritis: a multicenter, randomized, double‐blind, placebo‐controlled trial. Arthritis Rheumatol 2014;66:2091–102. [DOI] [PubMed] [Google Scholar]
- 36. Kavanaugh A, McInnes I, Mease P, Krueger GG, Gladman D, Gomez‐Reino J, et al. Golimumab, a new human tumor necrosis factor α antibody, administered every four weeks as a subcutaneous injection in psoriatic arthritis: twenty‐four–week efficacy and safety results of a randomized, placebo‐controlled study [published erratum appears in Arthritis Rheum 2010;62:2555]. Arthritis Rheum 2009;60:976–86. [DOI] [PubMed] [Google Scholar]
- 37. Smolen JS, Kay J, Doyle MK, Landewe R, Matteson EL, Wollenhaupt J, et al. Golimumab in patients with active rheumatoid arthritis after treatment with tumour necrosis factor α inhibitors (GO‐AFTER study): a multicentre, randomised, double‐blind, placebo‐controlled, phase III trial. Lancet 2009;374:210–21. [DOI] [PubMed] [Google Scholar]
- 38. Selmi C, Ceribelli A, Naguwa SM, Cantarini L, Shoenfeld Y. Safety issues and concerns of new immunomodulators in rheumatology. Expert Opin Drug Saf 2015;14:389–99. [DOI] [PubMed] [Google Scholar]