

Abstract
Ribavirin remains an important component of hepatitis C treatment in certain clinical scenarios, but it causes hemolytic anemia. A quantitative understanding of the ribavirin exposure‐anemia relationship is important in dose individualization/optimization. We developed a model relating ribavirin triphosphate (RTP) exposure in red blood cells (RBCs), RBC lifespan, feedback regulation of RBC production when anemia occurs, and the resulting hemoglobin decline. Inosine triphosphatase (ITPA) and interleukin 28B (IL28B) genetics were found to be significant covariates. Clinical trial simulations predicted that anemia is least severe in IL28B non‐CC (rs12979860, CT or TT), ITPA variant subjects, followed by IL28B non‐CC, ITPA wild‐type, IL28B CC, ITPA variant, and IL28B CC, ITPA wild‐type subjects (most severe). Reducing the ribavirin dose from 1,200/1,000 mg to 800/600 mg could reduce the proportions of grade 2 anemia by about half. The resulting model framework will aid the development of dosing strategies that minimize the incidence of anemia in treatment regimens that include ribavirin.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC? ☑ Accumulation of RBV triphosphate in RBC has been shown to induce anemia through ATP depletion that causes membrane oxidative damage. Indirect response models have been used to characterize hemoglobin decline during RBV treatment. However, the two previously published models use dose rate (mg/kg/day), or empirical accumulation equation for ribavirin pharmacokinetics and a fixed feedback effect on RBC production rate, which limits the utility of these models to explore different dosing regimens or dose adjustment algorithms. • WHAT QUESTION DOES THIS STUDY ADDRESS? ☑ Instead of using dose rate or empirical accumulation equation for RBV PK, this study incorporated the systemic and intracellular phosphorylation kinetic component. We established the relationship between RBV triphosphate concentration in RBC on decreasing RBC lifespan, the feedback autoregulation to increase RBC (and therefore hemoglobin) production rate, and how ITPA and IL28B genetics influence these processes. • WHAT THIS STUDY ADDS TO OUR KNOWLEDGE ☑ The current model is a population model accounting for the interindividual variabilities in the kinetics of RTP concentration in RBC, the effect of RTP on RBC lifespan, and feedback effect. We also identified ITPA wild‐type and IL28B CC as significant factors increasing patient susceptibility to RBV‐induced anemia. • HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY AND THERAPEUTICS ☑ The PK/PD model allows for clinical trial simulations to evaluate alternative covariate‐adjusted RBV initial dose and/or dose adjustment algorithms that will render the hemoglobin reduction more tolerable.
Ribavirin has been an important component of hepatitis C virus (HCV) treatment for decades. Although several new direct‐acting antiviral agents have received regulatory approval in recent years, ribavirin remains part of the HCV treatment regimen for many patient populations, including individuals with HCV genotypes 2 or 3, those with decompensated liver disease, and in combination with ritonavir‐boosted paritaprevir, ombitasvir, and dasabuvir in individuals with HCV genotype 1a.1 Ribavirin, however, is associated with a dose‐limiting toxicity, hemolytic anemia, which requires monitoring of hemoglobin during therapy. The ribavirin dose is often reduced when hemoglobin decreases to <10 g/dL or declines by >3 g/dL, and discontinued if hemoglobin becomes <8.5 g/dL. Historically, ribavirin dose reduction/discontinuation because of anemia occurred in 9–31% of patients treated with pegylated interferon (peg‐IFN) and ribavirin.2, 3, 4 Moreover, the addition of telaprevir was associated with more anemia (37% vs. 19% in the ADVANCE trial).5 In general, rates of anemia are lower in interferon (IFN)‐free regimens with direct‐acting antiviral and ribavirin (5–11%) compared with the rates of anemia when given with peg‐IFN.6, 7, 8, 9 In the peg‐IFN era, ribavirin dose reductions compromised the likelihood of achieving viral cure (also known as a sustained virologic response),10, 11 but it is unknown if this is the case with newer agents.
The mechanism of ribavirin‐induced anemia has been studied previously. Ribavirin, a nucleoside analog, undergoes intracellular phosphorylation to form ribavirin monophosphate (RMP), ribavirin diphosphate (RDP), and ribavirin triphosphate (RTP). Because of lack of dephosphorylating enzymes in red blood cells (RBCs), accumulation of RTP in RBCs is significantly higher than other cell types. RTP not only lowers adenosine triphosphate (ATP) levels,12, 13 but it also impairs ATP‐dependent transport systems, resulting in membrane oxidative damage.14 In addition, ribavirin induces a morphological change in the RBCs favoring an echinocytic form and increases phosphatidylserine exposure on the RBC membrane.15 The combination of these events leads to premature RBC senescence and accelerated phagocytic removal by the reticuloendothelial system.14, 15, 16 It has also been suggested that ribavirin inhibits RBC release from the bone marrow by delaying erythroid differentiation.17, 18 The result of these ribavirin‐induced processes is a clinically observable decrease in the lifespan of RBCs16, 19, 20 as well as reduced hemoglobin levels.
Mathematical models of ribavirin‐induced anemia have been reported previously by Tod et al.21 and Krishnan and Dixit.22, 23 The Tod model uses a maximum effect model of ribavirin dosage administration rate stimulating the elimination of hemoglobin, and a static proportional term describing the feedback that increases hemoglobin production under ribavirin treatment. The Krishnan model uses an empirical accumulation equation for ribavirin plasma concentration, a one‐compartment model linking the plasma concentration to the concentration of total phosphorylated ribavirin in erythrocyte, a linear term describing the stimulatory effect of ribavirin on the death rate of erythrocytes, as well as a feedback term that increases the RBC production rate as a function of RBC population density. The first version of the Krishnan and Dixit22 model uses partial differential equations, whereas the second version (ref. 23) simplifies the model to ordinary differential equations with very similar parameter estimates. Rather than using the dosage administration rate or empirical accumulation equation, our approach links the previously published model of plasma and intracellular ribavirin phosphorylation kinetics24 to the effect of RTP accumulation on RBC homeostasis.
Another purpose of this study was to investigate and model the contribution of inosine triphosphatase (ITPA) and IL28B genetics to the RBC lifespan and likelihood of anemia. Single nucleotide polymorphisms in ITPA (located on chromosome 20) and three‐bases upstream of IL28B (located on chromosome 19) have been associated with clinical outcomes in HCV therapy. A missense variant in exon 2 (rs1127354, P32T) and a splice‐altering single nucleotide polymorphism in intron 2 (rs7270101) of the ITPA gene have been significantly associated with protection against declines in hemoglobin (hemoglobin decline >3 g/dL) at week four of HCV treatment,25, 26 and a lower rate of ribavirin dose reduction.25 The IL28B single nucleotide polymorphism (rs12979860) has been strongly associated with sustained virologic response27 and spontaneous viral clearance,28, 29 but it is unknown if there is any influence of IL28B genotype on ribavirin‐induced anemia. Identification of factors that significantly affect the mechanisms of ribavirin‐induced anemia, together with the development of a population model, can serve as a basis for investigating alternative dosing strategies. For example, a lower ribavirin dose may be recommended for any subpopulation with higher risk of developing severe anemia. The resulting model framework may therefore aid the development of dosing strategies that minimize the incidence of anemia in treatment regimens that include ribavirin.
MATERIALS AND METHODS
Details of the clinical trial design, ITPA and IL28B genotyping methods, and systemic and intracellular kinetic model have been described previously.24 Briefly, 36 treatment naive patients with genotype 1 chronic HCV were administered with either dual therapy (peg‐IFN α‐2a at 180 μg weekly and ribavirin at 1,000 or 1,200 mg daily) or triple therapy (peg‐IFN α‐2a at 180 μg weekly, ribavirin at 1,000 or 1,200 mg daily and the HCV protease inhibitor, telaprevir at 750 mg three times daily for the first 12 weeks and then continued treatment with peg‐IFN α‐2a and ribavirin for an additional 12 or 24 weeks). Response‐guided therapy was used and individual treatment durations ranged from 12 to 48 weeks depending on clinical factors and antiviral response at week four of treatment. Plasma ribavirin concentration were measured at predose and 1, 2, 3, 4, 6, 8, 10, and 12 hours postdose during the 12‐hour intensive‐pharmacokinetic (PK) visits (day 1 and the steady state at weeks 9–13) and at various times postdose (i.e., convenience sampling) at the other visits (weeks 1, 2, 4, 16, 24, and 48). RMP, RDP, and RTP concentration in peripheral blood mononuclear cells and RBCs were measured at predose and 2 and 6 hours postdose during the 12‐hour intensive‐PK visits (day 1 and weeks 9–13) and at various times postdose at the other visits (weeks 1, 2, 4, 16, 24, and 48). Hemoglobin was measured per protocol at screening, predose at first dose (day 1), and weeks 1, 2, 4, steady‐state (weeks 9–13), weeks 16, 24, and 48 as part of the complete hematology profile and analyzed using an automated cell counter (several subjects had additional hemoglobin measurements obtained for clinical care, but because these samples were obtained off‐protocol they were not included in the analysis). The total number of hemoglobin concentrations included in the analysis was 251. Two subjects discontinued HCV treatment because of the side effects after less than one week on treatment and thus had only baseline hemoglobin measures. Eight subjects had a ribavirin dose reduction at least once during the study because of anemia. In addition, three subjects discontinued treatment because of anemia—their final hemoglobin values were 8, 8, and 8.3 g/dL, respectively.
Anemia model
The population analysis of hemoglobin data was performed sequentially using the individual plasma and RBC phosphorylation parameter estimates obtained previously.24 Briefly, the plasma PK component is a two‐compartment model with first‐order absorption. The transport of ribavirin into the cells and its initial phosphorylation to RMP (kmp) is driven by the plasma ribavirin concentration, whereas the disappearance of RMP follows a first‐order process (kmpout). The parameter kmpout represents several mechanisms, including metabolism (dephosphorylation) of phosphorylated anabolites, degradation of peripheral blood mononuclear cells or RBCs, and potential efflux of RMP by nucleoside transporters. Both RDP and RTP rapidly reached equilibrium with RMP (Rdpmp = RDP/RMP and Rtpdp = RTP/RDP). The shrinkage values in these models were: 2.5%, 4.3%, 8.5%, 5.0%, and 11.4% for the parameters CLt, Vc, ka, CLd, and Vp, respectively; 5.0%, 3.0%, 12.1%, and 10.4% for the parameters kmp, kmpout, Rdpmp, and Rtpdp, respectively. These low shrinkage values showed that model parameters were well‐informed by the available data,30 and therefore the use of the sequential approach is reasonable.
The anemia model component describes the hemoglobin reduction under ribavirin treatment. A one‐compartment model is used to simplify the turnover of the RBC population. Because hemoglobin and RBC counts in a given subject are proportionally related through the mean corpuscular hemoglobin, the differential equation for hemoglobin has the same first‐order elimination rate constant as that of RBC (kout = 1/RBC lifespan). RTP in the RBC accelerates removal of RBCs, as described above and is incorporated in the model through a term (kRTP) relating RTP concentration to a linear increase in the RBC elimination rate constant kout, resulting in a shortened RBC lifespan. Other models that included saturable effect or exponential relationship were also examined but were rejected because of higher standard errors and Akaike information criterion. A feedback term (Hb0/Hb)γ was used to incorporate the physiological feedback mechanisms that upregulate the production rate of RBC in response to anemia through the hormone erythropoietin. Figure 1 shows the ribavirin‐induced anemia model. The initial condition for the hemoglobin compartment Hb0 is estimated. The initial lifespan is set to normal average RBC lifespan of 120 days (kout = 1/120 day−1).31 Because we assume pretreatment homeostasis, the initial hemoglobin production rate Rin0 can be derived as Hb0·kout.
Figure 1.
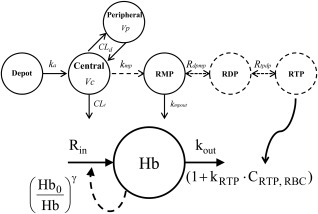
Composite model describing the plasma PK of ribavirin, its intracellular phosphorylation kinetics, and effect of RTP on RBC turnover. CLt, total clearance. Vc, central compartment volume of distribution. CLd, distributional clearance. Vp, peripheral compartment volume of distribution. ka: absorption rate constant. kmp, transport/formation rate constant for ribavirin monophosphate (RMP); kmpout, rate constant for the disappearance of phosphorylated ribavirin. Rdpmp, rapid equilibrium partition coefficient between ribavirin diphosphate (RDP) and RMP; Rtpdp, rapid equilibrium partition coefficient between ribavirin triphosphate (RTP) and RDP; Rin, zero‐order hemoglobin production rate; kout, rate constant for RBC elimination (inverse of RBC lifespan); kRTP, effect of RTP concentration in RBC on RBC lifespan; γ, feedback term characterizing RBC production rate increase in response to anemia.
The differential equations that described the final model were as follows:
Population analyses of hemoglobin data were performed using the maximum likelihood expectation maximization algorithm in the ADAPT, version 5, software.32 Model parameters were assumed to follow a multivariate lognormal distribution. Various models of residual variability (defined as the difference between observed and predicted hemoglobin concentration) were tested including additive, proportional, and combined additive/proportional error models. The final residual error model was selected based on model estimate precision and the Akaike information criterion.
The following covariates were examined for their influence on the anemia model: age, sex, race, degree of fibrosis, baseline HCV RNA level, telaprevir coadministration, ITPA, and IL28B genetics. Covariate‐parameter relationships were identified on the basis of exploratory graphics, scientific interest, and mechanistic plausibility. Categorical covariates were incorporated as proportional to the typical value of the parameters, whereas continuous covariates were included through power equations and centered on their median values. The final covariate model was selected based on the conventional approach, as presented in Wahlby et al.33 using forward addition (P < 0.05) followed by elimination (P < 0.01) of covariates. As a model qualification method, a visual predictive check based on 500 simulation replicates was performed.
Model simulation
A population simulation with 500 subjects receiving standard weight‐based ribavirin dose was performed using Nonmem, version 7.2, software (ICON Development Solutions, Ellicott City, MD) with an Intel Fortran compiler in order to simulate RBC lifespan and incidence of anemia with different ribavirin doses. The 500 subjects’ baseline weight, creatinine clearance, and sex were sampled from the study dataset to preserve the correlation among these covariates. The simulation was replicated for four cases depending on IL28B and ITPA genetics: IL28B non‐CC (CT or TT), ITPA wild‐type; IL28B non‐CC, ITPA variant; IL28B CC, ITPA wild‐type; and IL28B CC, ITPA variant.
The mean hemoglobin time profiles and distribution of hemoglobin reduction at week 12 were visually compared among the four cases. Furthermore, the nadir hemoglobin, inferred nadir RBC lifespan, and the proportion of subjects with hemoglobin reduction >3 g/dL or with hemoglobin <10 g/dL were summarized.
RESULTS
Exploratory data analyses revealed that telaprevir use was associated with greater hemoglobin reductions. After telaprevir was discontinued at week 12, the hemoglobin level slowly increased to a similar steady‐state level as in subjects who did not receive telaprevir. Female subjects had lower baseline hemoglobin, lower nadir hemoglobin, but smaller hemoglobin declines. Furthermore, subjects with ITPA variants had a slower slope of hemoglobin decline compared to wild‐type subjects. At week 4, the mean ± SD hemoglobin reductions were −3.11 ± 1.17 g/dL in the ITPA wild‐type group vs. −1.73 ± 1.41 g/dL in the ITPA variant group (P = 0.016). Finally, subjects with IL28B CC genotype had the same slope of hemoglobin decline as IL28B non‐CC subjects, but they had lower hemoglobin nadir as well as greater hemoglobin reduction. At week 12, the mean ± SD hemoglobin reductions were −3.32 ± 1.45 g/dL in the IL28B non‐CC group vs. −4.35 ± 1.33 g/dL in the IL28B CC group (P = 0.078).
The model shown in Figure 1 was able to describe the hemoglobin data well. Model parameters for the fixed effects were estimated with relative standard error <25% (Table 1). The interindividual random effects were estimated with relative standard error of 26–90%. Age, race, degree of fibrosis, baseline HCV RNA level, and telaprevir coadministration were not statistically significant covariates. The final model incorporated three covariate effects: Female sex was associated with 0.910 (P = 0.00196) times lower baseline hemoglobin, ITPA variant phenotype was associated with 0.431 (P = 0.000586) times lower kRTP, and IL28B CC genotype was associated with 0.629 (P = 0.00254) times lower feedback γ. Upon inclusion of the covariates, the interindividual variability of Hb0, kRTP, and γ decreased from 8.2 to 6.4%, 88 to 45%, and 55 to 30%, respectively. The goodness of fit plots (observed vs. population and individual prediction, and residuals vs. prediction and time) in Figure 2 showed that the model described the observed hemoglobin concentration range with no systematic prediction biases (individual subject plots are provided in the Supplementary Figure S1). Additionally, Figure 3 shows the average individual predicted hemoglobin time profiles (top panels), RBC lifespan (middle panels), and hemoglobin production rate (bottom panels), stratified by ITPA (left panels) or IL28B genotype (right panels).
Table 1.
Population estimates (relative standard error) for the parameters of the ribavirin‐induced anemia model without and with covariates
| Parameter (unit) | Without covariates | With covariates |
|---|---|---|
| AIC | 741.477 | 710.705 |
| kout (h−1) | 0.000347 (fixed) | 0.000347 (fixed) |
| Hb0 (g/dL) | 14.9 (1.72) | 15.5 (2.47) |
| kRTP (106 cells/pmol) | 0.0184 (29.0) | 0.0244 (24.7) |
| γ | 3.57 (16.1) | 4.46 (15.6) |
| Proportional error | 0.0546 (7.49) | 0.0551 (8.18) |
| IIV Hb0 | 8.20 (24.4) | 6.44 (26.0) |
| IIV kRTP | 88.2 (50.6) | 45.4 (52.3) |
| IIV γ | 54.7 (58.8) | 30.1 (89.7) |
| Female on Hb0 | 0.910 (3.55) | |
| ITPA variant on kRTP | 0.431 (23.4) | |
| IL28B CC on γ | 0.629 (17.7) |
AIC, Akaike information criterion; Hb0, baseline hemoglobin; IL, interleukin; ITPA, inosine triphosphatase; kout, rate constant for red blood cell (RBC) elimination (inverse of RBC lifespan); kRTP, effect of ribavirin triphosphate (RTP) concentration in RBC on RBC lifespan; γ, feedback term characterizing RBC production rate increase in response to anemia.
Figure 2.
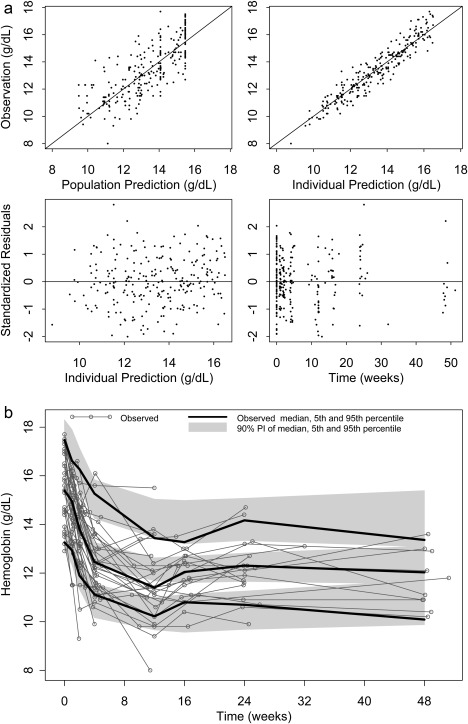
(a) Goodness‐of‐fit plots of the ribavirin‐induced anemia model. Top left: observed hemoglobin vs. population prediction. Top right: observed hemoglobin vs. individual prediction. Bottom left: standardized residuals vs. individual prediction. Bottom right: standardized residuals vs. time. (b) Visual predictive check overlaying hemoglobin individual profiles (gray thin lines) with simulation. Black thick lines: median, 5th, and 95th percentiles of the observed data. Gray shaded areas: 90% prediction intervals of the median, 5th, and 95th percentiles of the simulated profiles.
Figure 3.
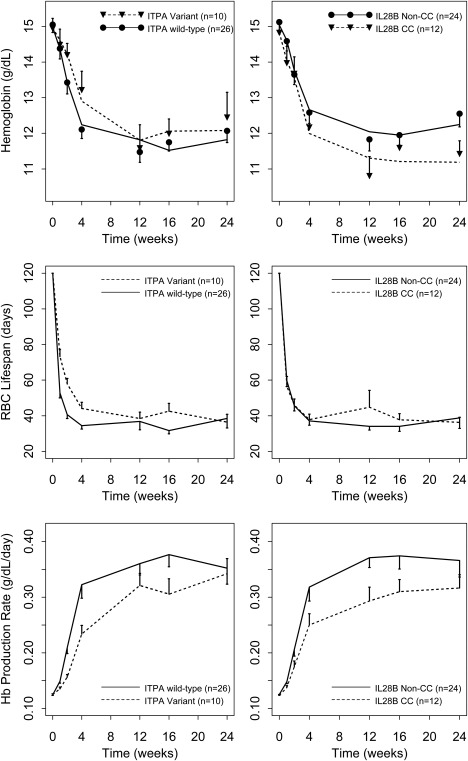
Top left: Means and standard errors of hemoglobin time profiles stratified by ITPA. Black circles: ITPA wild‐type. Black triangles: ITPA variant. Solid lines: mean individual predictions for ITPA wild‐type. Dashed lines: mean individual predictions for ITPA variant. Top right: Mean and standard errors of hemoglobin time profiles stratified by IL28B. Black circles: IL28B Non‐CC. Black triangles: IL28B CC. Solid lines: mean individual predictions for IL28B Non‐CC. Dashed lines: mean individual predictions for IL28B CC. Middle left: Mean and standard errors of model‐predicted RBC lifespan time profiles stratified by ITPA. Middle right: Mean and standard errors of model‐predicted RBC lifespan time profiles stratified by IL28B. Bottom left: Mean and standard errors of model‐predicted hemoglobin production rate time profiles stratified by ITPA. Bottom right: Mean and standard errors of model‐predicted hemoglobin production rate time profiles stratified by IL28B.
The population simulation results (Figure 4 and Table 2) revealed that the general order of anemia severity from the least severe to the most severe was: IL28B non‐CC, ITPA variant; IL28B non‐CC, ITPA wild‐type; IL28B CC, ITPA variant; and IL28B CC, ITPA wild‐type. Greater anemia severity differences were noted between IL28B non‐CC (black lines) vs. CC (green lines), compared to between ITPA variant (dashed lines) vs. wild‐type (solid lines). Reducing the ribavirin dose from 1,200/1,000 mg daily dosing to 800/600 mg increases the steady‐state hemoglobin by 0.72 (90% PI: −2.34 to 3.64) g/dL and reduces the proportion of subjects with hemoglobin <10 g/dL (grade 2 anemia) by about half (from 17.6 to 7.2% for IL2B non‐CC, ITPA wild‐type, from 13.4 to 6.4% for IL28B non‐CC, ITPA variant, from 45.8 to 22.2% for IL28B CC, ITPA wild‐type, and from 38.6 to 18.0% for IL28B CC, ITPA variant).
Figure 4.
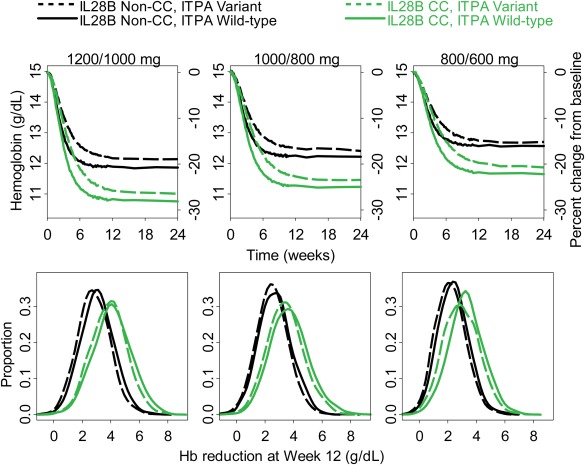
Simulation of hemoglobin profiles (top), distribution of hemoglobin level reduction from baseline at week 12 (bottom), for standard 1,200/1,000 mg daily dosing (left) 1,000/800 mg (middle), and 800/600 mg (right). Solid black lines: IL28B Non‐CC, ITPA wild‐type. Dashed black lines: IL28B Non‐CC, ITPA variant. Solid green lines: IL28B CC, ITPA wild‐type. Dashed green lines: IL28B CC, ITPA variant.
Table 2.
Summary of hemoglobin outcome for the three dosing regimens stratified by IL28B and ITPA genetics (mean baseline hemoglobin is 14.9 g/dL)
| 1,200/1,000 | 1,000/800 | 800/600 | |
| Steady‐state Hb, g/dL: median (5th–95th percentiles) | |||
| IL28B non‐CC, ITPA variant | 11.5 (9.4, 13.9) | 11.8 (9.6, 14.2) | 12.1 (9.9, 14.2) |
| IL28B non‐CC, ITPA wild‐type | 11.3 (8.8, 13.7) | 11.7 (9.7, 13.8) | 11.9 (9.8, 14.5) |
| IL28B CC, ITPA variant | 10.5 (8.2, 12.7) | 10.8 (8.5, 13.4) | 11.3 (8.9, 13.8) |
| IL28B CC, ITPA wild‐type | 10.2 (7.7, 13.3) | 10.7 (8.2, 13.0) | 11.1 (8.8, 13.4) |
| Steady‐state RBC lifespan, days: median (5th–95th percentiles) | |||
| IL28B non‐CC, ITPA variant | 38.8 (17.1, 68.2) | 43.9 (20.5, 72.7) | 49.8 (25.2, 80.1) |
| IL28B non‐CC, ITPA wild‐type | 33.7 (16.4, 61.8) | 41.4 (20.3, 69.0) | 45.6 (22.6, 77.0) |
| IL28B CC, ITPA variant | 36.5 (19.3, 67.2) | 42.9 (19.4, 70.3) | 50.1 (26.1, 79.5) |
| IL28B CC, ITPA wild‐type | 34.4 (15.4, 63.8) | 40.9 (18.6, 68.7) | 46.3 (23.0, 76.4) |
| W4 Hb decline, g/dL: median (5th–95th percentiles) | |||
| IL28B non‐CC, ITPA variant | −1.81 (−3.44, −0.17) | −1.66 (−3.39, −0.12) | −1.30 (−2.91, 0.06) |
| IL28B non‐CC, ITPA wild‐type | −2.43 (−4.33, −0.79) | −2.09 (−3.85, −0.42) | −1.79 (−3.32, −0.18) |
| IL28B CC, ITPA variant | −2.30 (−3.95, −0.44) | −1.99 (−4.08, −0.28) | −1.51 (−3.19, −0.05) |
| IL28B CC, ITPA wild‐type | −3.02 (−5.38, −1.06) | −2.55 (−4.63, −0.51) | −2.14 (−4.14, −0.49) |
| W12 Hb decline >3 g/dL (% subjects) | |||
| IL28B non‐CC, ITPA variant | 39.2 | 31.2 | 21.6 |
| IL28B non‐CC, ITPA wild‐type | 52.8 | 37.6 | 28.8 |
| IL28B CC, ITPA variant | 75.8 | 63.6 | 45.2 |
| IL28B CC, ITPA wild‐type | 81.2 | 66.6 | 61.2 |
| W12 Hb <10 g/dL (% subjects) | |||
| IL28B non‐CC, ITPA variant | 13.4 | 9.8 | 6.4 |
| IL28B non‐CC, ITPA wild‐type | 17.6 | 9.0 | 7.2 |
| IL28B CC, ITPA variant | 38.6 | 29.0 | 18.0 |
| IL28B CC, ITPA wild‐type | 45.8 | 31.2 | 22.2 |
Hb, hemoglobin; IL, interleukin; ITPA, inosine triphosphatase; RBC, red blood cell.
DISCUSSION
The ability to quantitatively characterize and predict hemoglobin decline in patients with HCV may provide insights for current clinical practice, which recommends monitoring of hemoglobin and dose reduction or discontinuation depending on the severity of anemia. The ribavirin‐induced anemia model described the hemoglobin data in our study reasonably well. The model also accounted for the mechanism of how RTP accumulation in RBCs increases the turnover of RBCs (or shortens the RBC lifespan) and the autoregulation or feedback mechanism for RBC homeostasis in anemia. To provide further insight into these processes, the secondary parameters RBC lifespan (1/(kout·(1+kRTP·RTP))) and hemoglobin production rate (Rin·(Hb0/Hb)γ) were plotted along with hemoglobin profiles in Figure 3. On average, RBC lifespan decreased from 120 days to 38.5 ± 22.4 days, and the hemoglobin/RBC production rate increased 2.72 ± 0.93‐fold. Our predicted nadir RBC lifespan is consistent with the estimates of RBC lifespan of 39 ± 13 days based on carbon monoxide measurements in 12 patients receiving IFN‐α and ribavirin (RBV).20 The inferred increase in the RBC production rate is also similar to the expected two to threefold RBC production increase for anemia of short duration.34
The model presented in the current work accounts for ribavirin's systemic PK, in contrast to previously reported models,21, 22, 23 allowing different dosing regimens to be simulated. Furthermore, covariate effects of sex and ITPA and IL28B genetics were incorporated into the anemia model, in addition to the previously reported effects of weight, creatinine clearance, sex, telaprevir use, and ITPA phenotype on the plasma PK, and the effects of ITPA phenotype on RBC phosphorylation.24 As mentioned above, we did not find telaprevir to be a significant covariate for the ribavirin‐induced anemia model. Although the hemoglobin decline in subjects receiving telaprevir was greater in the triple therapy group compared to that in the dual therapy group, this was not the result of a direct effect of telaprevir on RBC turnover, but an indirect effect caused by a telaprevir‐induced elevated ribavirin exposure.24, 35
Krishnan and Dixit23 previously reported a model‐based exploration of the effect of ITPA polymorphism on ribavirin‐induced anemia, by examining three possible mechanisms of action: lower sensitivity of RBC lifespan on total phosphorylated ribavirin concentration (higher C50); reduced rate of phosphorylated ribavirin (lower kp); and both C50 and kp as adjustable parameters. However, their results were inconclusive and they could not distinguish among these three mechanisms. In a previous publication, we reported that ITPA variant subjects had more than twofold higher RTP in RBCs compared to ITPA wild‐type subjects.24 If RTP had the same effect in ITPA wild‐type and variant patients, anemia would be expected to be more severe in ITPA variant patients. It is observed clinically, however, that the ITPA variant confers a protective effect against RBV‐induced anemia. The results reported herein suggest that ITPA variant subjects are less sensitive to RTP because of a smaller value of kRTP in the model, which in turn leads to less reduction in hemoglobin compared with ITPA wild‐type subjects, despite the elevated RTP concentrations in these subjects.
Previous reports have suggested several hypotheses to explain the protective effect of ITPA deficiency against RBV‐induced anemia. Thompson et al.25 suggested that higher levels of inosine triphosphate (ITP) may lead to a decrease in the available phosphate Pi, thereby restricting the conversion and accumulation of RTP in RBC. Domingo et al.36 suggested that ITP may complex with RTP and confer protection against hemolysis. Fellay et al.26 suggested that ITPA activity and/or ITPA levels directly or indirectly influence RBV PK. The current model, in contrast, supports the hypothesis that ITPA deficiency reduces the effect of RBV through restoration of ATP levels. The role of ITPA enzyme is to recycle ITP to inosine monophosphate, in which inosine monophosphate is then converted to ATP via adenosine monophosphate through or to guanosine‐5′‐triphosphate via guanosine monophosphate.37 Hitomi et al.38 further suggested that accumulation of ITP in subjects with ITPA variants compared to wild‐type subjects allowed more ATP biosynthesis because: (a) RBV inhibits inosine monophosphate dehydrogenase, which is the first reaction in the pathway converting inosine monophosphate to guanosine‐5′‐triphosphate; and (b) ITP can replace guanosine‐5′‐triphosphate as an energy source for the reaction of adenylosuccinate synthase, which is the first reaction in the pathway converting inosine monophosphate to ATP. Therefore, the lower kRTP parameter for ITPA variant subjects in our model suggests that the membrane oxidative damage and other effects of ATP deficiency resulting from RTP accumulation were attenuated in subjects with deficient ITPA enzyme activity, which leads to a slower hemoglobin decline.
Our model also found an effect of IL28B genotype on RBC feedback term γ, which governs the bone marrow responsiveness to anemia in increasing the RBC production rate. Because IL28B CC genotype is also associated with higher antiviral responsiveness to IFN‐α,39, 40, 41 it is possible that these subjects are also more susceptible to more severe IFN‐induced suppression of hematopoietic progenitor cells.42, 43 As suggested by our data and model, subjects with IL28B CC genotype, therefore, had significantly lower nadir hemoglobin and larger hemoglobin reduction compared with subjects with IL28B non‐CC genotype. Further studies are needed to explore the mechanism underlying this association.
There were some limitations to this work that limit generalizability. Because peg‐IFN concentrations were not measured, any possible effect of peg‐IFN on kRTP and γ could not be explored. In this study, subjects were taking peg‐IFN plus ribavirin with or without telaprevir, which is no longer the standard of care for HCV. However, ribavirin remains an important component of HCV treatment and anemia remains problematic. Additionally, our dosing simulations assumed mean baseline hemoglobin of 15 g/dL and did not model the impact of dose adjustments on the likelihood of sustained virologic response. Although dose reductions may mitigate the anemia associated with ribavirin treatment, studies are needed to reassure that high rates of sustained virologic response are maintained.
In summary, a robust model, including a feedback mechanism, described well the anemia effect induced by RBV treatment. The link between plasma PK, RBC phosphorylation kinetic, and the effect of RTP on RBC turnover was established. Furthermore, our analyses shed light on the significance of both ITPA and IL28B genetics in the development of anemia. The model framework can serve as a tool for conducting future clinical trial simulations to evaluate alternative covariate‐adjusted RBV initial dose regimens and/or dosing adjustment algorithms that will render the hemoglobin reduction more tolerable.
Supporting information
Supporting Information
Author Contributions
D.Z.D., L.S.W., and J.J.K. wrote the manuscript. D.Z.D., L.S.W., and J.J.K. designed the research. D.Z.D., L.S.W., L.C.J., C.E.M., and J.J.K. performed the research. L.S.W. and L.C.J. analyzed the data.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
We gratefully acknowledge the patients who participated in the study, Genentech and Vertex for supplying the study medications, and our funding support from the National Institutes of Health (K23 DK 082621 [J.K.]; R03 DK 09612 [J.K.]; P41 EB 001978 [D.Z.D.]), and the National Institutes of Health/National Center for Advancing Translational Sciences Colorado CTSI (UL1 TR001082).
References
- 1. AASLD/IDSA/IAS‐USA . Hepatitis C treatment guidelines. ≪www.hcvguidelines.org≫.
- 2. Fried, M.W. et al Peginterferon alfa‐2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 347, 975–982 (2002). [DOI] [PubMed] [Google Scholar]
- 3. Manns, M.P. et al Peginterferon alfa‐2b plus ribavirin compared with interferon alfa‐2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 358, 958–965 (2001). [DOI] [PubMed] [Google Scholar]
- 4. McHutchison, J.G. et al Peginterferon alfa‐2b or alfa‐2a with ribavirin for treatment of hepatitis C infection. N. Engl. J. Med. 361, 580–593 (2009). [DOI] [PubMed] [Google Scholar]
- 5. Jacobson, I.M. et al Telaprevir for previously untreated chronic hepatitis C virus infection. N. Engl. J. Med. 364, 2405–2416 (2011). [DOI] [PubMed] [Google Scholar]
- 6. Poordad, F. et al ABT‐450/r‐ombitasvir and dasabuvir with ribavirin for hepatitis C with cirrhosis. N. Engl. J. Med. 370, 1973–1982 (2014). [DOI] [PubMed] [Google Scholar]
- 7. Zeuzem, S. et al Retreatment of HCV with ABT‐450/r‐ombitasvir and dasabuvir with ribavirin. N. Engl. J. Med. 370, 1604–1614 (2014). [DOI] [PubMed] [Google Scholar]
- 8. Feld, J.J. et al Treatment of HCV with ABT‐450/r‐ombitasvir and dasabuvir with ribavirin. N. Engl. J. Med. 370, 1594–1603 (2014). [DOI] [PubMed] [Google Scholar]
- 9. Sovaldi [package insert]. Foster City, CA: Gilead Sciences, 2015. [Google Scholar]
- 10. Hadziyannis, S.J. et al Peginterferon‐alpha2a and ribavirin combination therapy in chronic hepatitis C: a randomized study of treatment duration and ribavirin dose. Ann. Intern. Med. 140, 346–355 (2004). [DOI] [PubMed] [Google Scholar]
- 11. Reddy, K.R. et al Impact of ribavirin dose reductions in hepatitis C virus genotype 1 patients completing peginterferon alfa‐2a/ribavirin treatment. Clin. Gastroenterol. Hepatol. 5, 124–129 (2007). [DOI] [PubMed] [Google Scholar]
- 12. Page, T. & Connor, J.D. The metabolism of ribavirin in erythrocytes and nucleated cells. Int. J. Biochem. 22, 379–383 (1990). [DOI] [PubMed] [Google Scholar]
- 13. Jimmerson, L.C. et al Effects of ribavirin on red blood cell concentrations of endogenous purines in patients with hepatitis C virus undergoing ribavirin‐based treatment. 65th Annual Meeting of the American Association for the Study of Liver Diseases: the Liver Meeting, Boston, MA, 7–11 November 2014.
- 14. De Franceschi, L. et al Hemolytic anemia induced by ribavirin therapy in patients with chronic hepatitis C virus infection: role of membrane oxidative damage. Hepatology 31, 997–1004 (2000). [DOI] [PubMed] [Google Scholar]
- 15. Homma, M. , Hosono, H. , Hasegawa, Y. & Kohda Y. Morphological transformation and phosphatidylserine exposure in erythrocytes treated with ribavirin. Biol. Pharm. Bull. 32, 1940–1942 (2009). [DOI] [PubMed] [Google Scholar]
- 16. Canonico, P.G. , Kastello, M.D. , Spears, C.T. , Brown, J.R. , Jackson, E.A. & Jenkins, D.E. Effects of ribavirin on red blood cells. Toxicol. Appl. Pharmacol. 74, 155–162 (1984). [DOI] [PubMed] [Google Scholar]
- 17. Canonico, P.G. et al Hematological and bone marrow effects of ribavirin in rhesus monkeys. Toxicol. Appl. Pharmacol. 74, 163–172 (1984). [DOI] [PubMed] [Google Scholar]
- 18. Ronzoni, L. et al Ribavirin suppresses erythroid differentiation and proliferation in chronic hepatitis C patients. J. Viral Hepat. 21, 416–423 (2014). [DOI] [PubMed] [Google Scholar]
- 19. Virtue, M.A. , Furne, J.K. , Ho, S.B. & Levitt, M.D. Use of alveolar carbon monoxide to measure the effect of ribavirin on red blood cell survival. Am. J. Hematol. 76, 107–113 (2004). [DOI] [PubMed] [Google Scholar]
- 20. Krishnan, S.M. & Dixit, N.M. Estimation of red blood cell lifespan from alveolar carbon monoxide measurements. Transl. Res. 154, 15–17 (2009). [DOI] [PubMed] [Google Scholar]
- 21. Tod, M. et al Pharmacokinetic/pharmacodynamic and time‐to‐event models of ribavirin‐induced anaemia in chronic hepatitis C. Clin. Pharmacokinet. 44, 417–428 (2005). [DOI] [PubMed] [Google Scholar]
- 22. Krishnan, S.M. & Dixit, N.M. Ribavirin‐induced anemia in hepatitis C virus patients undergoing combination therapy. PLoS Comput. Biol. 7, e1001072 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Krishnan, S.M. & Dixit, N.M. A formula to estimate the optimal dosage of ribavirin for the treatment of chronic hepatitis C: influence of ITPA polymorphisms. Antivir. Ther. 17, 1581–1592 (2012). [DOI] [PubMed] [Google Scholar]
- 24. Wu, L.S. et al Population pharmacokinetic modeling of plasma and intracellular ribavirin concentrations in patients with chronic hepatitis C virus infection. Antimicrob. Agents Chemother. 59, 2179–2188 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thompson, A.J. et al Variants in the ITPA gene protect against ribavirin‐induced hemolytic anemia and decrease the need for ribavirin dose reduction. Gastroenterology 139, 1181–1189 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fellay, J. et al ITPA gene variants protect against anaemia in patients treated for chronic hepatitis C. Nature 464, 405–408 (2010). [DOI] [PubMed] [Google Scholar]
- 27. Ge, D. et al Genetic variation in IL28B predicts hepatitis C treatment‐induced viral clearance. Nature 461, 399–401 (2009). [DOI] [PubMed] [Google Scholar]
- 28. Thomas, D.L. et al Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 461, 798–801 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Spada, E. et al Role of IL28B gene polymorphism and cell‐mediated immunity in spontaneous resolution of acute hepatitis C. Clin. Infect. Dis. 57, 803–811 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Savic, R.M. & Karlsson, M.O. Importance of shrinkage in empirical bayes estimates for diagnostics: problems and solutions. AAPS J. 11, 558–569 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Berlin, N.I. , Waldmann, T.A. & Weissman, S.M. Life span of red blood cell. Physiol. Rev. 39, 577–616 (1959). [DOI] [PubMed] [Google Scholar]
- 32. D'Argenio, D.Z. , Schumitzky, A. & Wang, X. ADAPT 5 User's Guide: Pharmacokinetic/Pharmacodynamic Systems Analysis Software. (Biomedical Simulations Resource, Los Angeles, CA, 2009). [Google Scholar]
- 33. Wahlby, U. , Jonsson, E.N. & Karlsson, M.O. Comparison of stepwise covariate model building strategies in population pharmacokinetic‐pharmacodynamic analysis. AAPS PharmSci. 4, E27 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Giblett, E.R. , Coleman, D.H. , Pirziobiroli, G. , Donohue, D.M. , Motulsky, A.G. & Finch, C.A. Erythrokinetics: quantitative measurements of red cell production and destruction in normal subjects and patients with anemia. Blood 11, 291–309 (1956). [PubMed] [Google Scholar]
- 35. De Nicolò, A. et al Telaprevir‐S isomer enhances ribavirin exposure and the ribavirin‐related haemolytic anaemia in a concentration‐dependent manner. Antiviral Res. 109, 7–14 (2014). [DOI] [PubMed] [Google Scholar]
- 36. Domingo, P. et al Association of ITPA gene polymorphisms and the risk of ribavirin‐induced anemia in HIV/hepatitis C virus (HCV)‐coinfected patients receiving HCV combination therapy. Antimicrob. Agents Chemother. 56, 2987–2993 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sumi, S. et al Genetic basis of inosine triphosphate pyrophosphohydrolase deficiency. Hum. Genet. 111, 360–367 (2002). [DOI] [PubMed] [Google Scholar]
- 38. Hitomi, Y. et al Inosine triphosphate protects against ribavirin‐induced adenosine triphosphate loss by adenylosuccinate synthase function. Gastroenterology 140, 1314–1321 (2011). [DOI] [PubMed] [Google Scholar]
- 39. Li, M. , Liu, X. , Zhou, Y. & Su, S.B. Interferon‐lambdas: the modulators of antivirus, antitumor, and immune responses. J. Leukoc. Biol. 86, 23–32 (2009). [DOI] [PubMed] [Google Scholar]
- 40. Zhang, L. et al IL28B inhibits hepatitis C virus replication through the JAK‐STAT pathway. J. Hepatol. 55, 289–298 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marcello, T. et al Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 131, 1887–1898 (2006). [DOI] [PubMed] [Google Scholar]
- 42. Ganser, A. , Carlo–Stella, C. , Greher, J. , Völkers, B. & Hoelzer, D. Effect of recombinant interferons alpha and gamma on human bone marrow‐derived megakaryocytic progenitor cells. Blood 70, 1173–1179 (1987). [PubMed] [Google Scholar]
- 43. Peck–Radosavljevic, M. et al Rapid suppression of hematopoiesis by standard or pegylated interferon‐alpha. Gastroenterology 123, 141–151 (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information