
Figure 3. Cellular maps of areas with increased β-actin translation.
(A) Super-resolution PALM density map composed of all 63,940 β-actin mRNA localizations from 6,235 trajectories. The PALM density plot was generated with VISP (El Beheiry and Dahan, 2013) from a simultaneous two-color movie with 500 consecutive frames. (B) Corresponding super-resolution PALM density map of all 19,420 ribosome localizations from 2,811 trajectories. (C) A map of the local apparent diffusion coefficients from all 6,235 β-actin mRNA trajectories. (D) A map of the local apparent diffusion coefficients from the 300 trajectories that were observed to be co-moving. The red arrows mark areas of enriched ribosome association with slowly diffusing β-actin mRNA. (E) Tracking of ribosome trajectories in cells where β-actin mRNA is tethered to focal adhesions. Super-resolution PALM density map composed of all detected localizations (1827) from 215 trajectories from 500 consecutive frames of ribosome imaging (see Video 7). (F) In shades of pink, overlay of the 33 trajectories (15.4% ) that co-localize with the focal-adhesion marker with tethered β-actin mRNA. (G) A map of the apparent diffusion coefficients of co-localized ribosome trajectories exhibits predominately slow diffusion characteristics when mRNA is tethered to focal adhesions. The red arrows mark the hotspots where the tethered mRNA is being actively translated.

Figure 3—figure supplement 1. Tracking of individual ribosome molecules with β-actin mRNA tethered to focal adhesions.
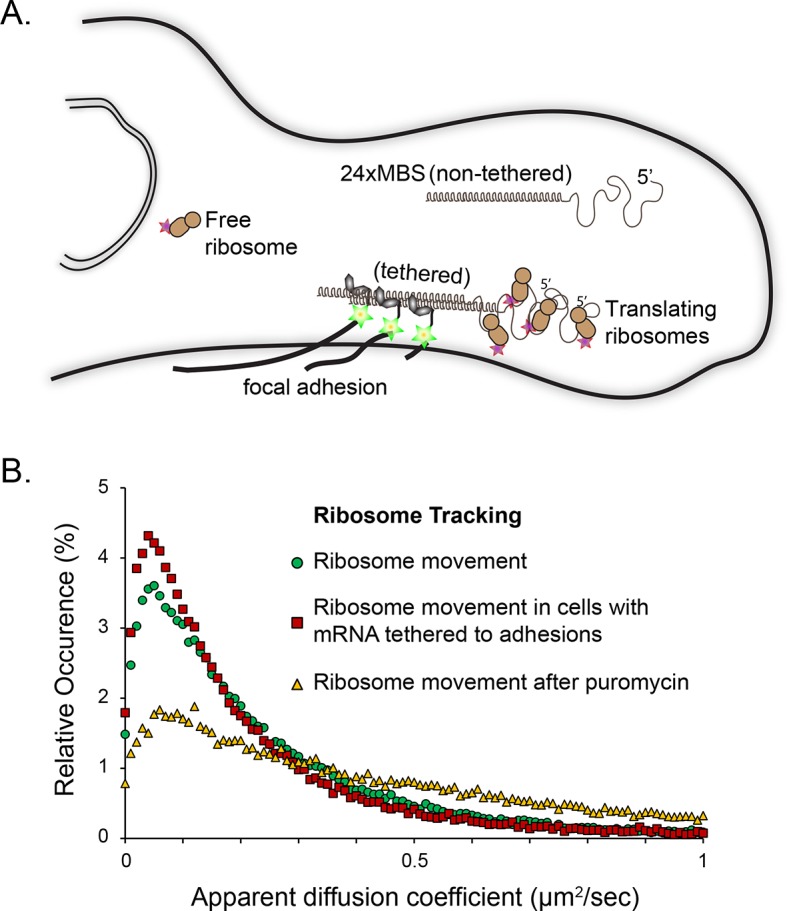
Figure 3—figure supplement 2. Diffusion characteristics of β-actin mRNA co-moving with ribosomes and without ribosomes.

Figure 3—figure supplement 3. mRNA/ribosome colocalization statistics in the absence and presence of puromycin.
