

Abstract
Diacylglycerol kinase (DGK) consists of 10 isozymes. The α-isozyme enhances the proliferation of cancer cells. However, DGKα facilitates the nonresponsive state of immunity known as T-cell anergy; therefore, DGKα enhances malignant traits and suppresses immune surveillance. The aim of this study was to identify a novel small molecule that selectively and potently inhibits DGKα activity. We screened a library containing 9,600 chemical compounds using a newly established high-throughput DGK assay. As a result, we have obtained a promising compound, 5-[(2E)-3-(2-furyl)prop-2-enylidene]-3-[(phenylsulfonyl)amino]2-thioxo-1,3-thiazolidin-4-one) (CU-3), which selectively inhibited DGKα with an IC50 value of 0.6 μM. CU-3 targeted the catalytic region, but not the regulatory region, of DGKα. CU-3 competitively reduced the affinity of DGKα for ATP, but not diacylglycerol or phosphatidylserine. Moreover, this compound induced apoptosis in HepG2 hepatocellular carcinoma and HeLa cervical cancer cells while simultaneously enhancing the interleukin-2 production of Jurkat T cells. Taken together, these results indicate that CU-3 is a selective and potent inhibitor for DGKα and can be an ideal anticancer drug candidate that attenuates cancer cell proliferation and simultaneously enhances immune responses including anticancer immunity.
Keywords: cancer, apoptosis, phosphatidic acid, lipid kinases, anergy, T cell, interleukin-2, high-throughput screening, anticancer drug
Diacylglycerol kinase (DGK) phosphorylates diacylglycerol (DG) to generate phosphatidic acid (1–6). To date, 10 mammalian DGK isozymes (α, β, γ, δ, ε, ζ, η, θ, ι, and κ) have been identified. These DGK isozymes are divided into five groups (type I–V) according to their structural features (1–6). Type I DGK isozymes (DGKs α, β, and γ) commonly contain tandem repeats of two EF-hand motif domains and are classified as members of the EF-hand family of Ca2+ binding proteins. In addition to the Ca2+ binding EF-hand motifs, all type I DGK isozymes contain an N-terminal recoverin homology domain, two cysteine-rich C1 domains, and the C-terminal catalytic region (1–6).
DGKα (7, 8) is highly expressed in hepatocellular carcinoma and melanoma cells (9, 10). DGKα expression is involved in hepatocellular carcinoma progression and is a positive regulator of the proliferative activity of hepatocellular carcinoma through the Ras/Raf/MEK (mitogen-activated protein kinase/ERK kinase)/ERK pathway (9). In melanoma cells, DGKα positively regulates the tumor necrosis factor-α-dependent nuclear factor-κB (p65) activation via the protein kinase C ζ-mediated Ser311 phosphorylation of p65 (11). Therefore, the suppression of DGKα activity is expected to inhibit the progression of these cancers. On the other hand, DGKα is abundantly expressed in T lymphocytes where it facilitates the nonresponsive state known as anergy (12, 13). Anergy induction in T cells represents the main mechanism for advanced tumors to avoid immune action.
As described above, DGKα has antiapoptosis and proproliferation activities in cancer cells (9, 10) and also induces T-cell anergy (12, 13). Therefore, if a DGKα-selective inhibitor is identified and developed, it would reversely attenuate cancer cell proliferation and simultaneously activate T-cell function. There are two commercially available DGK inhibitors, 6-(2-{4-[(4-fluorophenyl)phenylmethylene]1-piperidinyl}ethyl)-7-methyl-5H-thiazolo-(3,2-a)pyrimidin-5-one (R59022) (14, 15) and 3-(2-{4-[bis-(4-fluorophenyl)methylene]1-piperidinyl}ethyl)-2,3-dihydro-2-thioxo-4(1H)quinazolinone (R59949) (16, 17); however, we found that R59022 and R59949 only semiselectively inhibited type I, III, and V DGKs α, ε, and θ, and type I and II DGKs α, γ, δ, and κ, respectively (18). Moreover, the IC50 values of R59022 and R59949 were ∼25 μM and 18 μM, respectively (18). Therefore, R59022 and R59949 are low selective and low effective inhibitors of DGKα.
To develop highly effective and DGKα-selective inhibitors, a system for high-throughput screening (HTS) is required; however, the conventional DGK assay is quite laborious and requires technical skill. For example, the conventional assay requires the use of a radioisotope ([γ-32P]ATP) and the manipulation of thin-layer chromatography with multiple extraction steps. We recently established a simple DGK assay (18) that is useful for constructing an HTS system for detecting DGK inhibitors from chemical compound libraries.
In this study, we screened a library containing 9,600 chemical compounds to find DGKα-specific inhibitors using the newly established DGK assay for HTS (18). As a result, we have obtained a promising compound, 5-[(2E)-3-(2-furyl)prop-2-enylidene]-3-[(phenylsulfonyl)amino]2-thioxo-1,3-thiazolidin-4-one (CU-3), which selectively inhibited DGKα with an IC50 value of 0.6 μM. Moreover, CU-3 simultaneously enhanced the apoptosis of cancer cells and the activation of T cells.
MATERIALS AND METHODS
Cell culture and transfection
COS-7 and HeLa cells were grown in Dulbecco’s modified Eagle’s medium (Wako Pure Chemical Industries, Tokyo, Japan) supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin (Wako Pure Chemical Industries). HepG2 cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS, 1% nonessential amino acids (Wako Pure Chemical Industries), 100 U/ml penicillin, and 100 μg/ml streptomycin. Jurkat T cells were maintained in 75 cm2 flasks in RPMI-1640 medium (Wako Pure Chemicals Industries) containing 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. The cells were maintained at 37°C in an atmosphere containing 5% CO2.
COS-7 cells were seeded in 60 mm dishes at a density of 2.5 × 105 cells/dish. cDNA was transfected into COS-7 cells by electroporation with a Gene Pulser XcellTM Electropolation System (Bio-Rad Laboratories) according to the manufacturer’s instructions.
cDNA constructs
The expression plasmids, p3xFLAG-CMV-pig DGKα (19), -rat DGKβ (19), -human DGKγ (19), -human DGKδ1-ΔSAM (20, 21), -human DGKη1 (22), -human DGKε (23), -human DGKζ (24), -human DGKι (25), -human DGKθ (26) and -human DGKκ (27) were generated as described (18). cDNAs of DGKα-Δ1–196 and DGKα-Δ1–332 (28) were amplified by PCR and inserted into p3xFLAG-CMV vector.
Western blot analysis
COS-7 cell lysates expressing the 3xFLAG tagged proteins were separated on SDS-polyacrylamide gel electrophoresis. The separated proteins were transferred to a polyvinylidene difluoride (PVDF) membrane (Pall Corporation, Tokyo, Japan) and blocked with Block Ace (Dainippon Pharmaceutical, Tokyo, Japan). The membrane was incubated with an anti-FLAG antibody (Sigma-Aldrich, St. Louis, MO) in 10% Block Ace for 1 h. The immunoreactive bands were then visualized using a peroxidase-conjugated anti-mouse IgG antibody (Jackson ImmunoResearch Laboratories) and the Enhanced Chemiluminescence Western Blotting Detection System (GE Healthcare, Tokyo, Japan).
HTS
For HTS, 384-well plates were predispensed with 60 nl (final concentration: 30 or 50 μM) of each compound. Glutathione S-transferase-fused pig DGKα was expressed in Sf9 insect cells and purified with a glutathione-Sepharose column (GE Healthcare). The DGK assay was performed using the ADP-Glo™ Kinase Assay Kit (Promega, Tokyo, Japan) at 30°C for 2 h as described previously (18). The chemiluminescence generated in this assay correlates to the amount of ADP generated, which is equivalent to the phosphatidic acid (PA) produced, in the kinase assay, indicating kinase activity. The chemiluminescence was measured using a PHERAstar microplate reader (BMG LABTECH, Offenburg, Germany). The assay performance was consistent across all plates, which was evident due to the robust Z’ factor.
Chemical compounds
CU-1, -2, -3, and -4 were obtained from Drug Discovery Initiative, University of Tokyo. For further characterization of CU-3 [inhibition mechanisms of CU-3, inhibition effects of CU-3 in cells, induction of apoptosis in cancer cells by CU-3, and enhancement of the interleukin-2 (IL-2) production by CU-3 (see Results)], highly pure CU-3 was resynthesized and supplied by Namiki Shoji (Tokyo, Japan).
Determination of DGK activity in vitro
COS-7 cells (60 mm dish) expressing the various 3xFLAG-tagged DGK isozymes were harvested in lysis buffer (0.5 ml/60 mm dish) containing 50 mM HEPES (pH 7.2), 150 mM NaCl, 5 mM MgCl2, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, and the Complete Protease Inhibitor mixture (Roche Molecular Biochemicals, Tokyo, Japan). After centrifugation at 400 g for 5 min, the resultant supernatant was used for the DGK activity assays.
The octyl glucoside mixed micellar DGK activity assay (29) was modified and performed in a 96-well microplate. The assay mixture (25 μl) contained 50 mM MOPS (pH 7.4), 50 mM n-octyl-β-d-glucoside (Dojindo Laboratories, Kumamoto, Japan), 1 mM dithiothreitol, 100 mM NaCl, 20 mM NaF, 10 mM MgCl2, 1 μM CaCl2, 10 mM (27 mol%) phosphatidylserine (PS; Sigma-Aldrich), 2 mM (5.4 mol%) 1,2-dioleoyl-sn-glycerol (Sigma-Aldrich), 0.2 mM ATP, and 1.25 μg of purified glutathione S-transferase-fused pig DGKα or 2.5 μg of the COS-7 cell lysates expressing the 3xFLAG-tagged DGKα or other isozymes. We confirmed that the assays were linear with respect to protein concentration and time.
Determination of the DGK activity in cells by LC/MS
Determination of the DGK activity in cells by LC/MS was carried out as described previously (30, 31). COS-7 cells expressing DGKα-Δ1–196 (a constitutively active mutant) were harvested in phosphate-buffered saline. Total lipids were extracted from the cells according to the method of Bligh and Dyer (32). The extracted cellular lipids (10 μl) containing 65 pmol of the 28:0-PA internal standard (Sigma-Aldrich) were separated on the LC system (Accela LC Systems, Thermo Fisher Scientific, Tokyo, Japan) using a UK-Silica column (3 μm, 150 × 2.0 mm inner diameter; Imtakt, Kyoto, Japan) (30, 31). Mobile phase A consisted of chloroform-methanol-ammonia (89:10:1), and mobile phase B consisted of chloroform-methanol-ammonia-water (55:39:1:5). The gradient elution program was as follows: 30% B for 5 min, 30–60% B over 25 min, 60–70% over 10 min, followed by 70% B for 5 min. The flow rate was 0.3 ml/min, and the chromatography was performed at 25°C.
The LC system described above was coupled online to an Exactive Orbitrap MS (Thermo Fisher Scientific) equipped with an ESI source. The ion spray voltage was set to –5 kV and 5 kV in the negative and positive ion mode, respectively. The capillary temperature was set to 300°C. The other parameters were set according to the manufacturer’s recommendations. Individual phospholipids were measured by scanning from m/z 450 to 1,100 in the negative or positive ion modes using an Orbitrap Fourier Transform MS with a resolution of 50,000. The MS peaks were identified based on their m/z value and were presented in the form of X:Y, where X is the total number of carbon atoms and Y is the total number of double bonds in both acyl chains of the phospholipid.
Apoptosis analysis
HepG2, HeLa, and COS-7 cells were incubated in a 96-well plate in the presence or absence of CU-3 (5 μM) for 24 h. The caspase-3/7 assay (Caspase-Glo® 3/7; Promega) was conducted according to the manufacturer’s description. After a 1 h incubation at 25°C, each sample was measured in a microplate reader (GloMax®-Multi+ Detection System; Promega).
Assay for IL-2 mRNA expression in Jurkat T cells
The assay for IL-2 mRNA expression in Jurkat T cells was carried out as previously reported (33). Jurkat cells were preincubated in 35 mm culture dishes filled with 2 ml of RPMI in the presence or absence of CU-3 (5 μM) for 5 min. Concanavalin A (Con A) was then added to the media, and the cells were further incubated for 3 h, collected by centrifugation (400 g, 5 min), and lysed with 1 ml of ISOGEN (Wako Pure Chemical Industries). Total RNA was prepared, and 1 μg of total RNA was reverse transcribed into cDNA according to the manufacturer’s instructions (for ISOGEN). PCR amplification (34 cycles) was performed using the following primers: 5′-ATGTACAGGATGCAACTCCTGTCTT-3′ and 5′-GTT AGTGTTGAGATGATGCTTTGAC-3′ for IL-2 and 5′-ACCACAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′ for glyceraldehyde-3-phosphate dehydrogenase. PCR products were then separated by 1% agarose gel electrophoresis and visualized with ethidium bromide. The visualized bands were digitized and quantified using Adobe Photoshop and National Institutes of Health (NIH) Image software.
RNA interference of DGKα
DGKα Stealth select RNAi (catalog number HSS102626; Invitrogen, Tokyo, Japan) was used. Jurkat T cells suspended in unsupplemented, prewarmed RPMI, with 500 nM of human si-DGKα or nontargeting control siRNA (Invitrogen) in 4 mm cuvettes (Nepa Gene) were electroporated at 210 V and 950 microfarads using the GenePulsar Xcell Electroporation System (Bio-Rad Laboratories). After electroporation, cells were incubated for 48 h in culture medium.
Western blot analysis
Lysates of HepG2, HeLa, Jurkat, or COS-7 cells were separated on SDS-polyacrylamide gel electrophoresis. The separated proteins were transferred to a PVDF membrane (Bio-Rad Laboratories) and blocked with Block Ace (Dainippon Pharmaceutical). The membrane was incubated with anti-DGKα (34) antibody in 10% Block Ace for 1 h. The immunoreactive bands were then visualized using a peroxidase-conjugated anti-rabbit IgG antibody (Jackson ImmunoResearch Laboratories) and the Enhanced Chemiluminescence Western Blotting Detection System (GE Healthcare).
RESULTS
Core library screen to identify compounds that inhibit DGKα activity
To identify specific inhibitors for DGKα activity, we screened the Core Library (Drug Discovery Initiative, University of Tokyo) that consists of 9,600 compounds using a recently established, simple DGK assay (18). Purified DGKα was incubated with these compounds at a concentration of 30 μM. We set the threshold for hit compounds at >20% inhibition for all compounds and identified 103 hit compounds. These hit compounds were then subjected to the second screen at a concentration of 50 μM to assess the reproducibility.
We selected four compounds, CU-1, -2, -3, and -4 (Table 1), that reproducibly and strongly inhibited DGKα activity. Their IC50 values were ∼8, 27, 0.6, and 6 μM, respectively (Fig. 1A–D). Doses of 10 μM and 30 μM of CU-3 and CU-4, respectively, almost completely inhibited DGKα activity (Fig. 1C, D).
TABLE 1.
Molecular structures of CU-1, CU-2, CU-3, CU-3-1, CU-3-2, CU-3-3, and CU-4
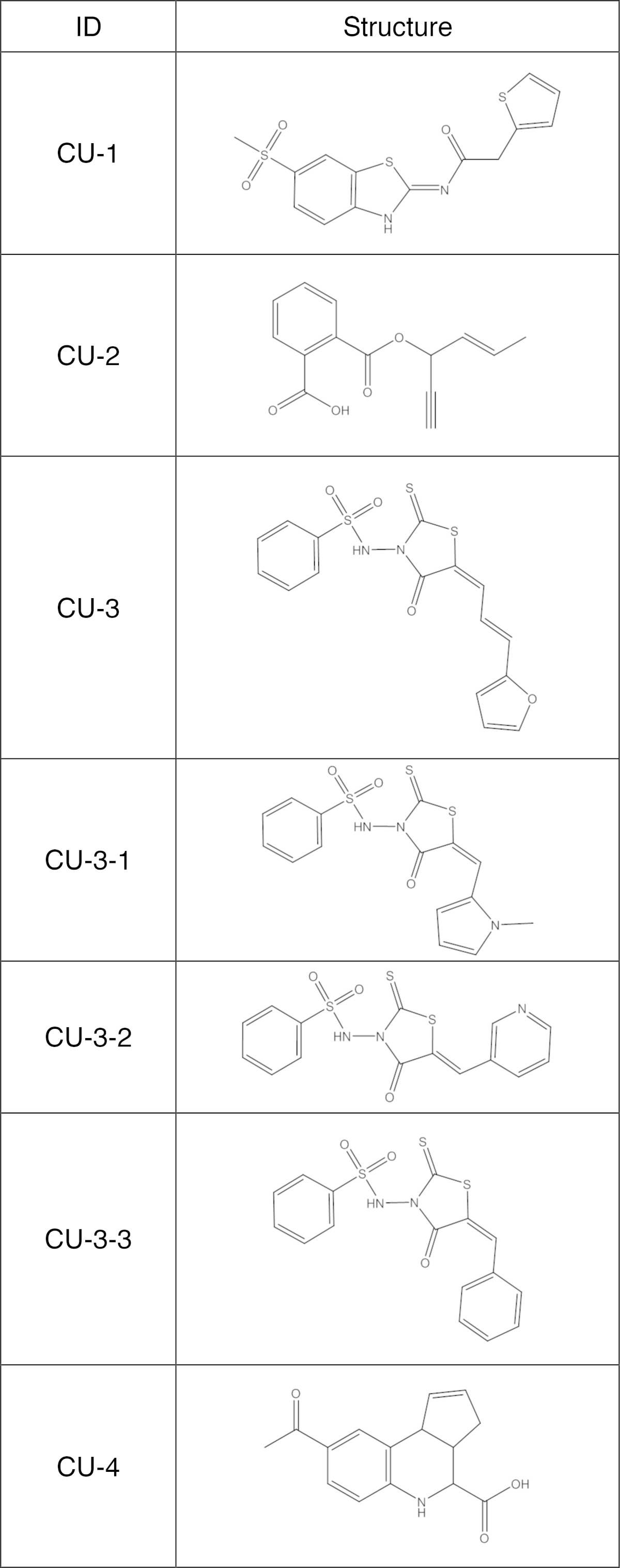
Fig. 1.

Effects of CU-1, CU-2, CU-3, and CU-4 on DGKα activity. Purified DGKα (1.25 μg) was incubated with various concentrations of CU-1 (A), CU-2 (B), CU-3 (C), or CU-4 (D) as indicated for 5 min. Values are the average of duplicate determinations, which were within 5% of the mean. Similar results were obtained in two repeated experiments (experiments 1 and 2).
Comparison of the inhibitory activities of CU-1, -2, -3, and -4 against the 10 DGK isozymes
We have already demonstrated that R59022 and R59949 only semiselectively inhibited the 10 DGK isozymes (18). Using the same system, we next compared the inhibitory activities of CU-1, -2, -3, and -4 against all 10 DGK isozymes. As previously reported (18), the expression levels of the DGK isozymes were comparable to each other. The concentrations of these compounds at their approximate IC50 values (10, 30, 1, and 5 μM, respectively) against DGKα (see Fig. 1) were used. When DGKα activity was inhibited to <50% by CU-1, -2, and -3, at least >75, 80, and 75% activities of the other nine isozymes remained, respectively (Fig. 2A–C); however, in addition to DGKα, CU-4 strongly inhibited the δ-, ε-, and ι-isozymes as well (Fig. 2D). These results indicate that CU-1, -2, and -3, but not CU-4, selectively inhibit DGKα.
Fig. 2.

Effects of CU-1, CU-2, CU-3, and CU-4 on DGK isozyme activities. The 12,000 g supernatant (5 μg) of the extracts from COS-7 cells expressing DGKα, β, γ, δ, η, κ, ε, ζ, ι, or θ) was incubated with CU-1 (10 μM) (A), CU-2 (30 μM) (B), CU-3 (1 μM) (C), or CU-4 (5 μM) (D) as indicated for 5 min. The values in the absence of CU-1, CU-2, CU-3, and CU-4 (supplementary Fig. 1) were set to 100%. * P < 0.05, ** P < 0.01.
Among CU-1, -2, and -3, the IC50 value of CU-2 (27 μM) is relatively high (Fig. 1). Therefore, we focused on CU-1 and -3 and further determined their selectivity for DGKα. The IC50 values of CU-1 and -3 against all 10 DGK isozymes were compared (Table 2). The apparent IC50 values of CU-1 and -3 against the β- to θ-isozymes were 3- to 6-fold and 12- to 60-fold higher than those of DGKα, respectively (Table 2). Therefore, compared with CU-1, CU-3 showed clearly higher selectivity for DGKα. Hence, we selected CU-3 for further analyses.
TABLE 2.
Apparent IC50 values of CU-1 and CU-3 against 10 DGK isozymes
| DGK isozyme | IC50 of CU-1 (μM) | IC50 of CU-3 (μM) |
| α (type I) | 8 | 0.6 |
| β (type I) | 43 | 36 |
| γ (type I) | 37 | 28 |
| δ (type II) | 33 | 36 |
| η (type II) | 32 | 7 |
| κ (type II) | 46 | 32 |
| ε (type III) | 24 | 8 |
| ζ (type IV) | 47 | 30 |
| ι (type IV) | 27 | 11 |
| θ (type V) | 30 | 15 |
The effects of the three derivatives (CU-3-1, CU-3-2, and CU-3-3) of CU-3 (Table 1) were examined. Furan in CU-3 is substituted with N-methylpyrrole, pyridine, and benzene for CU-3-1, CU-3-2, and CU-3-3, respectively (Table 1). Moreover, although CU-3 has three carbons between 2-thioxo-1,3-thiazolidin-4-one and furan, CU-3-1, CU-3-2, and CU-3-3 have only one carbon between 2-thioxo-1,3-thiazolidin-4-one and N-methylpyrrole, pyridine, and benzene (Table 1). As shown in Fig. 3, the inhibitory effect of CU-3 on DGKα activity was stronger than that of CU-3-1, CU-3-2, and CU-3-3, indicating that CU-3 is the strongest DGKα inhibitor among these derivatives.
Fig. 3.

Effects of CU-3 and its derivatives (CU-3-1, CU-3-2, and CU-3-3) on DGKα activity. The 12,000 g supernatant (5 μg) of the extracts from COS-7 cells expressing DGKα was incubated for 5 min in the presence or absence (DMSO alone) of CU-3 (A) and its derivatives: CU-3-1 (B), CU-3-2 (C), or CU-3-3 (D). The values in the absence of CU-3, CU-3-1, CU-3-2, and CU-3-3 (supplementary Fig. 2) were set to 100%.
Inhibition mechanisms of CU-3
We next attempted to reveal the inhibition mechanisms of CU-3. We first examined which region of DGKα was targeted by CU-3. We prepared truncation mutants lacking the recoverin homology domain–the EF-hand motifs (DGKα-Δ1–196) and the recoverin homology domain–the C1 domains (DGKα-Δ1–332) (Fig. 4A). CU-3 inhibited the DGK activities of the wild-type enzyme and these mutants to a similar extent (Fig. 4B). These results indicate that CU-3 targets the catalytic domain, not the regulatory region, of DGKα. Although DGKα is activated by Ca2+ (7, 35), these mutants commonly lack the Ca2+ binding EF-hand motifs and show strong Ca2+-independent activity (28, 36). Therefore, it is likely that Ca2+ is not involved in the inhibition mechanism of CU-3.
Fig. 4.

Effects of CU-3 on the activities of the DGKα truncation mutants. A: Schematic presentation of DGKα truncation mutants. RVH, recoverin homology. B: The 12,000 g supernatant (5 μg) of the extracts from COS-7 cells expressing wild-type DGKα, DGKα-Δ1–196, or DGKα-Δ1–332 was incubated for 5 min in the presence (1 μM) or absence (DMSO alone) of CU-3. The activities of wild-type DGKα, DGKα-Δ1–196, and DGKα-Δ1–332 in the absence of CU-3 are defined as 100%.
It was reported that in addition to wild-type DGKα (35), DGKα-Δ1–332 (catalytic domain alone) was also activated by PS (28). Therefore, we next determined the EC50 values of PS for DGKα activity in the presence and absence of CU-3. PS strongly activated DGKα, with activation reaching a maximum at ∼23.0 mol% in the absence of CU-3 (Fig. 5). The apparent EC50 of the isozyme for PS was 12.0 mol% ± 0.0 (n = 3) in the absence of CU-3 (Fig. 5 and Table 3). The addition of CU-3 did not markedly affect the EC50 value of PS (13.0 ± 0.6 mol%, n = 3) (Fig. 5 and Table 3).
Fig. 5.
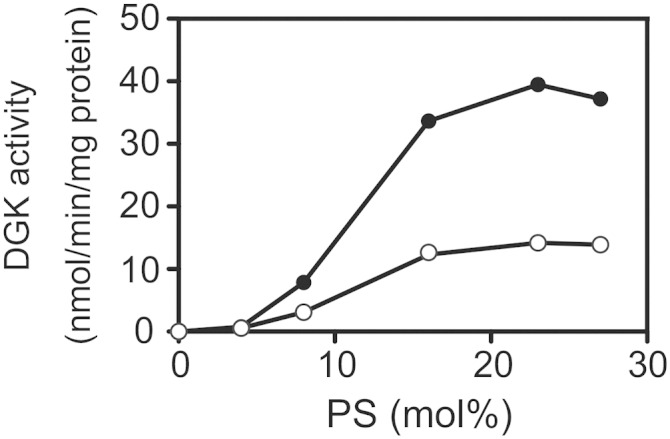
Effect of CU-3 on the PS dependency of DGKα. The 12,000 g supernatant (5 μg) of the extracts from COS-7 cells expressing DGKα was incubated with various concentrations of PS as indicated for 5 min in the presence (1 μM, open circle) or absence (DMSO alone, closed circle) of CU-3. The values are the averages of duplicate determinations. The data shown are representative of three separate experiments.
TABLE 3.
Apparent EC50 values of DGKα for PS in the presence or absence of CU-3
| EC50 (mol%) | P | |
| DMSO | 12.0 ± 0.0 | — |
| CU-3 | 13.0 ± 0.6 | NS |
The values are presented as the mean ± SD (n = 3).
We next measured the kinetic parameter for DG. The activity of DGKα increased in a DG dose-dependent manner (Fig. 6A). A double reciprocal plot provided the Km value for DG in the absence of CU-3 (3.4 ± 1.0 mol%, n = 3; Fig. 6B and Table 4). As shown in Fig. 6B and Table 4, CU-3 did not significantly affect the apparent Km value for DG (2.9 ± 0.5 mol%, n = 3).
Fig. 6.

Effect of CU-3 on the affinity of DGKα for DG. A: The 12,000 g supernatant (5 μg) of the extracts from COS-7 cells expressing DGKα was incubated with various concentrations of DG as indicated for 5 min in the presence (1 μM, open circle) or absence (DMSO alone, closed circle) of CU-3. B: Lineweaver-Burk plots of the data. The values are the averages of duplicate determinations. The data shown are representative of three separate experiments.
TABLE 4.
Apparent Km values of DGKα for DG in the presence or absence of CU-3
| Km (mol%) | P | |
| DMSO | 3.4 ± 1.0 | — |
| CU-3 | 2.9 ± 0.5 | NS |
The values are presented as the mean ± SD (n = 3).
We next determined the kinetic parameter for ATP in the presence and absence of CU-3. The activity of DGKα was increased in an ATP dose-dependent manner (Fig. 7A). A double reciprocal plot provided the apparent Km value for ATP in the absence of CU-3 (0.25 ± 0.07 mM, n = 3; Fig. 7B and Table 5). However, the Km value for ATP in the presence of CU-3 was significantly increased to 0.48 ± 0.07 mM, n = 3 (Fig. 6B and Table 5). The Vmax value was not markedly changed in the presence of CU-3 (Fig. 7B). These results strongly suggest that CU-3 competitively inhibited the affinity of DGKα for ATP.
Fig. 7.

Effect of CU-3 on the affinity of DGKα for ATP. A: The 12,000 g supernatant (5 μg) of the extracts from COS-7 cells expressing DGKα was incubated with various concentrations of ATP as indicated for 5 min in the presence (1 μM, open circle) or absence (DMSO alone, closed circle) of CU-3. B: Lineweaver-Burk plots of the data. The values are the averages of duplicate determinations. The data shown are representative of three separate experiments.
TABLE 5.
Apparent Km values of DGKα for DG in the presence or absence of CU-3
| Km (mM) | P | |
| DMSO | 0.25 ± 0.07 | — |
| CU-3 | 0.48 ± 0.07 | 0.023 |
The values are presented as the mean ± SD (n = 3).
Inhibition effects of CU-3 in cells
We showed that CU-3 intensely inhibited DGKα activity in vitro (Figs. 1–7). Next, the inhibition effects of CU-3 on DGKα in cell were determined using our newly established LC/MS method (30, 31). The overexpression of DGKα-Δ1–196 (Fig. 8A), a constitutively active mutant, clearly increased the total amount of PA (approximately a 15% increase) in COS-7 cells (Fig. 8B). Moreover, the addition of CU-3 significantly reduced the total PA amount that was increased by the overexpression of DGKα-Δ1–196 (Fig. 8B).
Fig. 8.

Effect of CU-3 on the DGKα activity in cells. COS-7 cells were transfected with p3xFLAG-DGKα-Δ1–196 or p3xFLAG vector alone (Vector). After a 24 h incubation, 2 μM of CU-3 or DMSO alone was added and further incubated for 24 h. A: Expression of endogenous DGKα and DGKα-Δ1–196 in COS-7 cells. COS-7 cells transfected with p3xFLAG-CMV vector alone or p3xFLAG-CMV-DGKα-Δ1–196 were harvested, and the cell lysates (15 μg of protein) were analyzed by Western blotting using anti-DGKα antibody. B–D: The amounts of the total PAs (B) and major PA molecular species (C) in COS-7 cells were quantified using the LC/MS method. The values are presented as the mean ± SD (n = 3). B: * P < 0.05, *** P < 0.005. C: * P < 0.05, ** P < 0.01 (p3xFLAG-vector-transfected cells in the absence of CU-3 vs. p3xFLAG-DGKα-Δ1–196-transfected cells in the absence of CU-3); # P < 0.05, ## P < 0.01, ### P < 0.005 (p3xFLAG-DGKα-Δ1–196-transfected cells in the absence of CU-3 vs. p3xFLAG-DGKα-Δ1–196-transfected cells in the presence of CU-3). p3xFLAG-vector-transfected cells in the absence of CU-3, white bars; p3xFLAG-vector-transfected cells in the presence of CU-3, dark gray bars; p3xFLAG-DGKα-Δ1–196-transfected cells in the absence of CU-3, light gray bars; p3xFLAG-DGKα-Δ1–196-transfected cells in the presence of CU-3, black bars. (D) The results are presented as the relative value of major PA molecular species of p3xFLAG-DGKα-Δ1–196-transfected cells versus p3xFLAG vector-transfected cells in the presence (black bars) or absence (white bars) of CU-3. # P < 0.05, ## P < 0.01, ### P < 0.005.
In addition to the total amount of PA (Fig. 8B), the amounts of each PA molecular species were also determined (Fig. 8C). To facilitate comparison, relative changes (p3xFLAG-DGKα-Δ1–196-transfected cells vs. p3xFLAG vector-transfected cells in the presence or absence of CU-3) were also shown (Fig. 8D). The amounts of broad PA molecular species such as 34:1-, 34:0-, 36:4-, 36:3-, 38:5-, and 38:4-PA were clearly increased by the overexpression of DGKα-Δ1–196 in COS-7 cells (Fig. 8C, D). Particularly, the amount of 38:4-PA was increased to ∼200%. As shown in Fig. 8C, D, CU-3 (2 μM) significantly inhibited the amounts of the PA molecular species that were increased by the overexpression of DGKα-Δ1–196, indicating that the inhibitor negatively affected the activity of DGKα in the cells.
Induction of apoptosis in cancer cells by CU-3
We next examined whether CU-3 induces apoptosis in a human hepatocellular carcinoma cell line HepG2 because DGKα was highly expressed in the cell lines (Fig. 9A) and significantly enhanced their cell growth (9). To assess this possibility, caspase-3/7 activity was determined. As shown in Fig. 9B, CU-3 markedly enhanced the caspase-3/7 activity (an ∼50% increase) of HepG2 cells. Moreover, we utilized HeLa cells (a human cervical carcinoma cell line) because DGKα was highly expressed in the cells (Fig. 9A) and a DGK inhibitor R59022 induced their cell death (37). As shown in Fig. 9C, essentially the same results (an ∼24% increase of caspase-3/7 activity) were obtained with HeLa cells. However, the caspase-3/7 activity of COS-7 epithelial cells (monkey kidney-derived, noncancerous cell line) was not augmented by CU-3 (Fig. 9D), suggesting that CU-3 is not nonspecifically toxic to cells.
Fig. 9.

Effect of CU-3 on the apoptosis of HepG2, HeLa, and COS-7 cells. A: Expression of DGKα [80 kDa and 70 kDa (10)] in HepG2, HeLa, and COS-7 cells. HepG2, HeLa, and COS-7 cells were harvested, and the cell lysates (15 μg of protein) were analyzed by Western blotting using anti-DGKα antibody. B–D: After the addition of 5 μM of CU-3 or DMSO alone, HepG2 (B), HeLa (C), and COS-7 (D) cells were incubated for 24 h. The caspase-3/7 assay was conducted according to the manufacturer’s description. After a 1 h incubation at 25°C, each sample was measured in a microplate reader. The values are presented as the mean ± SD (n = 6). *** P < 0.005.
Enhancement of the IL-2 production by CU-3
We next tested whether CU-3 enhanced the function of T cells. To assess this possibility, we determined the Con A-induced IL-2 mRNA production activity of Jurkat cells (a human T-cell line). As shown in Fig. 10, IL-2 mRNA in Jurkat T cells was not detectable in the absence of Con A, and Con A markedly induced the IL-2 mRNA production of the cells. CU-3 further enhanced IL-2 mRNA production activity (an ∼50% increase) of Jurkat T cells (Fig. 10). These results indicate that CU-3 enhanced the function of T cells.
Fig. 10.

Effect of CU-3 on the IL-2 production of Jurkat T cells. A: Jurkat T cells were preincubated in 35 mm culture dishes filled with 2 ml RPMI in the presence or absence of CU-3 (5 μM) for 5 min. Con A was then added to the media, and the cells were further incubated for 3 h. Total RNA was reverse transcribed into cDNA, and PCR amplification (34 cycles) was performed using primers for IL-2 or GAPDH. The PCR products were then separated by agarose gel electrophoresis and visualized with ethidium bromide. The visualized bands were digitized and quantified using Adobe Photoshop and NIH Image software. B: The value in the presence of Con A and in the absence of CU-3 was set to 100%. The values are presented as the mean ± SD (n = 5). ** P < 0.01.
To demonstrate that CU-3 causes IL-2 production through the inhibition of DGKα, we compared the effects of CU-3 and DGKα-siRNA side by side and examined whether the RNA interference further enhances the effects of the inhibitor. CU-3 and DGKα-siRNA increased IL-2 production to almost the same extents (Fig. 11). Moreover, DGKα-siRNA did not further enhance the effect of CU-3 (Fig. 11), indicating that CU-3 and DGKα-siRNA inhibit the same target, DGKα.
Fig. 11.
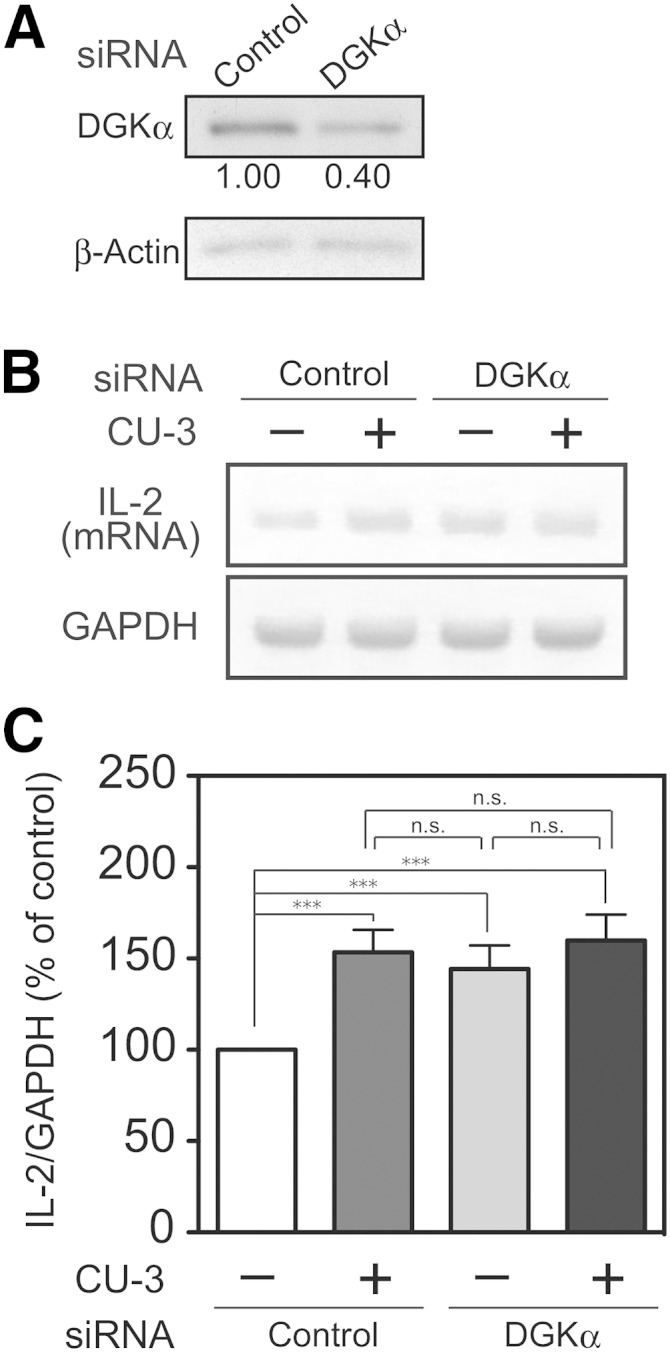
Effects of DGKα knockdown and CU-3 on IL-2 production of Jurkat T cells. Jurkat T cells were transfected with DGKα-siRNA or nontargeting siRNA (Invitrogen) as indicated. A: After transfection with siRNAs for 48 h, the cells were harvested, and cell lysates (15 μg of protein) were analyzed by Western blotting with anti-DGKα antibody (upper panel) or anti-β-actin antibody (lower panel). The relative intensity of each band is shown below upper panel. B, C: After transfection with siRNAs for 48 h, the cells were preincubated in the presence or absence of CU-3 (5 μM) for 5 min. Con A was then added to the media, and the cells were further incubated for 3 h. Total RNA was reverse transcribed into cDNA, and PCR amplification (34 cycles) was performed using primers for IL-2 or GAPDH. The PCR products were then separated by agarose gel electrophoresis and visualized with ethidium bromide. The visualized bands were digitized and quantified using Adobe Photoshop and NIH Image software. The value of control siRNA-treated samples without CU-3 was set to 100%. The values are presented as the mean ±SD (n = 3). *** P < 0.005; n.s., not significant.
DISCUSSION
We screened a library containing 9,600 compounds using a high-throughput chemiluminescence-based assay that was recently established (18). Among the compounds, CU-3 was identified as a potent and selective inhibitor against the α-isozyme of DGK (Figs. 1 and 2; Table 2). Compared with commercially available DGK inhibitors, R59022 and R59949 (18), CU-3 exhibited higher efficiency and selectivity against DGKα. The IC50 value of CU-3 (0.6 μM) (Fig. 1) was markedly lower than that of R59022 and R59949 (∼25 μM and 18 μM, respectively) (18). R59022 and R59949 only semiselectively inhibited type I, III, and V DGKs α, ε, and θ, and type I and II DGKs α, γ, δ, and κ, respectively (18). Moreover, the Km value of CU-3 for DGKα was at least ∼12 times lower than those for other DGK isozymes (Fig. 2 and Table 2). Stemphone, a fungal metabolite, has also been reported to be a DGK inhibitor (38). However, its DGK-isozyme selectivity is not known. Therefore, this study is the first report of a highly α-isozyme selective inhibitor.
DGKα-Δ1–332 (catalytic domain alone) (Fig. 4A) lacks the regulatory region including the Ca2+ binding EF-hand motifs. CU-3 inhibited DGK activities of the full-length enzyme and the mutant to a similar extent (Fig. 4B). The results indicate that the target of CU-3 is the catalytic domain of DGKα. Therefore, it is likely that Ca2+ is not involved in the inhibition mechanism of CU-3.
CU-3 did not affect the dependence on an activator PS (Fig. 5 and Table 3). Moreover, CU-3 did not change the Km value of DGKα for DG (Fig. 6 and Table 4). These results indicate that the affinities for PS and DG failed to be affected by the inhibitor. However, CU-3 significantly decreased the Km value of DGKα for ATP (Fig. 7 and Table 5). Therefore, it is likely that the inhibitor competitively decreased the affinity for ATP.
CU-3 is not strikingly similar to R59022 (14), R59949 (16), or stemphone (39). CU-3 contains 2-thioxo-1,3-thiazolidin-4-one (Table 1), which is partly similar to ATP. Therefore, it is reasonable to speculate that the inhibitor may partly mimic ATP and eventually decrease the affinity for the substrate of DGK, ATP, as observed in Fig. 7. The primary structures of the catalytic domains of DGK isozymes are highly similar to each other (1–6). Therefore, it is unknown how the isozyme selectivity occurs. To evaluate this issue, a determination of the 3D structure is needed.
In addition to in vitro, CU-3 inhibited cellular DGKα activity in COS-7 cells (Fig. 8). It was noted that a concentration of 2 μM, which is only ∼3-fold higher than the IC50 value (0.6 μM) (Fig. 1 and Table 2), strongly inhibited PA production by overexpressed DGKα (Fig. 8). This result suggests that CU-3 has high cell permeability. Therefore, CU-3 is expected to effectively inhibit DGKα in vivo as well.
We demonstrated that CU-3 induced apoptosis in HepG2 hepatocellular carcinoma and HeLa cervical cancer cells (Fig. 9B, C). We also reported that DGKα-specific siRNA attenuated the proliferation of hepatocellular carcinoma (9) and induced apoptosis in melanoma cells (10). Supporting our results, Torres-Ayuso et al. (40) also demonstrated that the growth of colon and breast cancer cell lines was significantly inhibited by DGKα-siRNA and R59949, which attenuates DGKα activity (18). In addition, Dominguez et al. (37) reported that R59022, which most strongly inhibits DGKα (18), negatively affected the proliferation of glioblastoma, melanoma, breast cancer, and cervical cancer cells. It is interesting to investigate the effect of CU-3 on the cell growth of a variety of cancer cells. They also observed that in marked contrast to cancer cells, R59022 did not weaken the growth of noncancerous astrocytes and fibroblasts (37). In this study, we also observed that although CU-3 enhanced the caspase-3/7 activities of HepG2 hepatocellular carcinoma and HeLa cervical cancer cells (Fig. 9B, C), the compound did not increase the caspase-3/7 activity of the noncancer-derived COS-7 cells (Fig. 9D). Therefore, we reproduced their results (37). These findings suggest that cancer-derived cells and noncancer-derived cells may utilize different pathways to induce apoptosis. DGKα seems to be particularly relevant for cancer cells. For example, we have revealed that the expression of DGKα was not detectable in normal hepatocytes, whereas this isozyme was expressed in several hepatocellular carcinoma cell lines (9). Moreover, although normal melanocytes did not express DGKα, several melanoma cell lines did express this isozyme (10). It has been shown that DGKα positively regulated angiogenesis signaling (41). Therefore, it is possible that CU-3 also attenuates the angiogenesis of cancer cells in addition to their cell death. It is notable that DGKα-knockout mice are generally healthy, although the mice have a defect in T-cell anergy (12, 13, 42). These results allow us to speculate that DGKα-specific inhibitors would not have severe side effects.
In addition to the induction on cancer cell apoptosis (Fig. 9), the inhibitor promotes IL-2 production (Fig. 10), which is one of the indicators of T-cell activation. Olenchock et al. (12) and Zha et al. (13) previously reported that DGKα is abundantly expressed in T cells, where it facilitates the nonresponsive state, anergy. Thus, the inhibitor of DGKα was expected to enhance T-cell activity. In this study, we found that CU-3 indeed enhanced the IL-2 production of T cells (Fig. 10). Although it has already been reported that the DGKα inhibitor inhibited cancer cell proliferation (37), this study provides a new aspect of the DGKα inhibitor, an enhancer of the immune system. Inactivation (anergy induction) of T cells is the main mechanism for advanced tumors to avoid immune action. Therefore, it is expected that CU-3 is able to activate cancer immunity.
General anticancer drugs attenuate the proliferation and function of both cancer and bone marrow cells (43, 44). Therefore, general anticancer drugs induce not only the attenuation of cancer cell proliferation but also bone marrow suppression/myelosuppression, which is one of the most commonly observed side effects of chemotherapy. Bone marrow suppression results in a decreased number of T cells and, consequently, causes severe side effects, such as recurrent infectious diseases. However, there is no drug that has both protumoral and anti-immunogenic effects. Interestingly, because DGKα has both protumoral and anti-immunogenic properties (4), the DGKα-selective inhibitor CU-3 would simultaneously have antitumoral and proimmunogenic effects. Therefore, CU-3 can be a lead compound to develop an ideal anticancer drug without infectious side effects. Moreover, in addition to the direct effects on apoptosis induction in cancer cells, CU-3 can indirectly induce the death of cancer cells through activation of the immune system. In addition to the development of an anticancer drug, CU-3 will be a great tool for biological science concerning cancer and immunity.
In this study, we were able to obtain DGK isozyme-selective inhibitors, although the 10 DGK isozymes are highly similar to each other. Therefore, our current results encourage us to identify and develop specific inhibitors/activators against every DGK isozyme that can be effective regulators and drugs against a wide variety of physiological events and diseases (4). For example, DGKα: cancer cell growth (9, 10, 37, 40), angiogenesis (41), T-cell proliferation (45), and T-cell anergy (12, 13); DGKβ: bipolar disorder (46, 47); DGKγ: actin reorganization (19), allergy (48), and insulin secretion (49); DGKδ: epidermal growth factor receptor signaling (50) and type 2 diabetes (51, 52); DGKη: epidermal growth factor receptor signaling (53), lung cancer (54), bipolar disorder (55, 56), unipolar depression (56), and attention-deficit/hyperactivity disorder (56); DGKκ: hypospadias (57); DGKε: seizure susceptibility/long-term potentiation (58) and Huntington’s disease (59); DGKζ: cell growth (60), T-cell receptor response (61), and brain ischemia (62); DGKι: Ras signaling (63); and DGKθ: Parkinson’s disease (64) and bile acid signaling (65).
Supplementary Material
Acknowledgments
The authors thank Drs. Isabel Merida (Consejo Superior de Investigaciones Científica, Spain) and Andrea Graziani (Università del Piemonte Orientale, Italy) for valuable discussions.
Footnotes
Abbreviations:
- Con A
- concanavalin A
- CU-3
- 5-[(2E)-3-(2-furyl)prop-2-enylidene]-3-[(phenylsulfonyl)amino]2-thioxo-1,3-thiazolidin-4-one
- DG
- diacylglycerol
- DGK
- diacylglycerol kinase
- HTS
- high-throughput screening
- IL-2
- interleukin-2
- PA
- phosphatidic acid
- PS
- phosphatidylserine
- R59022
- 6-(2-{4-[(4-fluorophenyl)phenylmethylene]1-piperidinyl}ethyl)-7-methyl-5H-thiazolo-(3,2-a)pyrimidin-5-one
- R59949
- 3-(2-{4-[bis-(4-fluorophenyl)methylene]1-piperidinyl}ethyl)-2,3-dihydro-2-thioxo-4(1H)quinazolinone
This work was supported by Platform for Drug Discovery, Informatics, and Structural Life Science from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. This work was also supported by MEXT/JSPS KAKENHI Grant Numbers 22370047 [Grant-in-Aid for Scientific Research (B)], 23116505 (Grant-in-Aid for Scientific Research on Innovative Areas), 25116704 (Grant-in-Aid for Scientific Research on Innovative Areas), 26291017 [Grant-in-Aid for Scientific Research (B)], and 15K14470 (Grant-in-Aid for Challenging Exploratory Research); the Japan Science and Technology Agency (AS221Z00794F, AS231Z00139G, AS251Z01788Q, and AS2621643Q), the Naito Foundation, the Hamaguchi Foundation for the Advancement of Biochemistry, the Daiichi-Sankyo Foundation of Life Science, the Terumo Life Science Foundation, the Futaba Electronic Memorial Foundation, the Daiwa Securities Health Foundation, the Ono Medical Research Foundation, the Japan Foundation for Applied Enzymology, the Food Science Institute Foundation, the Skylark Food Science Institute, and the Venture Business Laboratory of Chiba University (F.S.). The authors declare that they have no conflicts of interest to disclose.
The online version of this article (available at http://www.jlr.org) contains a supplement.
REFERENCES
- 1.Goto K., Hozumi Y., and Kondo H.. 2006. Diacylglycerol, phosphatidic acid, and the converting enzyme, diacylglycerol kinase, in the nucleus. Biochim. Biophys. Acta. 1761: 535–541. [DOI] [PubMed] [Google Scholar]
- 2.Mérida I., Avila-Flores A., and Merino E.. 2008. Diacylglycerol kinases: at the hub of cell signalling. Biochem. J. 409: 1–18. [DOI] [PubMed] [Google Scholar]
- 3.Sakane F., Imai S., Kai M., Yasuda S., and Kanoh H.. 2007. Diacylglycerol kinases: why so many of them? Biochim. Biophys. Acta. 1771: 793–806. [DOI] [PubMed] [Google Scholar]
- 4.Sakane F., Imai S., Kai M., Yasuda S., and Kanoh H.. 2008. Diacylglycerol kinases as emerging potential drug targets for a variety of diseases. Curr. Drug Targets. 9: 626–640. [DOI] [PubMed] [Google Scholar]
- 5.Topham M. K. 2006. Signaling roles of diacylglycerol kinases. J. Cell. Biochem. 97: 474–484. [DOI] [PubMed] [Google Scholar]
- 6.van Blitterswijk W. J., and Houssa B.. 2000. Properties and functions of diacylglycerol kinases. Cell. Signal. 12: 595–605. [DOI] [PubMed] [Google Scholar]
- 7.Sakane F., Yamada K., Kanoh H., Yokoyama C., and Tanabe T.. 1990. Porcine diacylglycerol kinase sequence has zinc finger and E-F hand motifs. Nature. 344: 345–348. [DOI] [PubMed] [Google Scholar]
- 8.Schaap D., de Widt J., van der Wal J., Vandekerckhove J., van Damme J., Gussow D., Ploegh H. L., van Blitterswijk W. J., and van der Bend R. L.. 1990. Purification, cDNA-cloning and expression of human diacylglycerol kinase. FEBS Lett. 275: 151–158. [DOI] [PubMed] [Google Scholar]
- 9.Takeishi K., Taketomi A., Shirabe K., Toshima T., Motomura T., Ikegami T., Yoshizumi T., Sakane F., and Maehara Y.. 2012. Diacylglycerol kinase alpha enhances hepatocellular carcinoma progression by activation of Ras-Raf-MEK-ERK pathway. J. Hepatol. 57: 77–83. [DOI] [PubMed] [Google Scholar]
- 10.Yanagisawa K., Yasuda S., Kai M., Imai S., Yamada K., Yamashita T., Jimbow K., Kanoh H., and Sakane F.. 2007. Diacylglycerol kinase α suppresses tumor necrosis factor-α-induced apoptosis of human melanoma cells through NF-κB activation. Biochim. Biophys. Acta. 1771: 462–474. [DOI] [PubMed] [Google Scholar]
- 11.Kai M., Yasuda S., Imai S., Toyota M., Kanoh H., and Sakane F.. 2009. Diacylglycerol kinase α enhances protein kinase Cζ-dependent phosphorylation at Ser311 of p65/RelA subunit of nuclear factor-κB. FEBS Lett. 583: 3265–3268. [DOI] [PubMed] [Google Scholar]
- 12.Olenchock B. A., Guo R., Carpenter J. H., Jordan M., Topham M. K., Koretzky G. A., and Zhong X. P.. 2006. Disruption of diacylglycerol metabolism impairs the induction of T cell anergy. Nat. Immunol. 7: 1174–1181. [DOI] [PubMed] [Google Scholar]
- 13.Zha Y., Marks R., Ho A. W., Peterson A. C., Janardhan S., Brown I., Praveen K., Stang S., Stone J. C., and Gajewski T. F.. 2006. T cell anergy is reversed by active Ras and is regulated by diacylglycerol kinase-α. Nat. Immunol. 7: 1166–1173. [DOI] [PubMed] [Google Scholar]
- 14.de Chaffoy de Courcelles D. C., Roevens P., and Van Belle H.. 1985. R 59 022, a diacylglycerol kinase inhibitor. Its effect on diacylglycerol and thrombin-induced C kinase activation in the intact platelet. J. Biol. Chem. 260: 15762–15770. [PubMed] [Google Scholar]
- 15.Sakane F., Yamada K., and Kanoh H.. 1989. Different effects of sphingosine, R59022 and anionic amphiphiles on two diacylglycerol kinase isozymes purified from porcine thymus cytosol. FEBS Lett. 255: 409–413. [DOI] [PubMed] [Google Scholar]
- 16.de Chaffoy de Courcelles D., Roevens P., Van Belle H., Kennis L., Somers Y., and De Clerck F.. 1989. The role of endogenously formed diacylglycerol in the propagation and termination of platelet activation. A biochemical and functional analysis using the novel diacylglycerol kinase inhibitor, R 59 949. J. Biol. Chem. 264: 3274–3285. [PubMed] [Google Scholar]
- 17.Jiang Y., Sakane F., Kanoh H., and Walsh J. P.. 2000. Selectivity of the diacylglycerol kinase inhibitor 3-{2-(4-[bis-(4-fluorophenyl)methylene]-1-piperidinyl)ethyl}-2, 3-dihydro-2-thioxo-4(1H)quinazolinone (R59949) among diacylglycerol kinase subtypes. Biochem. Pharmacol. 59: 763–772. [DOI] [PubMed] [Google Scholar]
- 18.Sato M., Liu K., Sasaki S., Kunii N., Sakai H., Mizuno H., Saga H., and Sakane F.. 2013. Evaluations of the selectivities of the diacylglycerol kinase inhibitors R59022 and R59949 among diacylglycerol kinase isozymes using a new non-radioactive assay method. Pharmacology. 92: 99–107. [DOI] [PubMed] [Google Scholar]
- 19.Tsushima S., Kai M., Yamada K., Imai S., Houkin K., Kanoh H., and Sakane F.. 2004. Diacylglycerol kinase γ serves as an upstream suppressor of Rac1 and lamellipodium formation. J. Biol. Chem. 279: 28603–28613. [DOI] [PubMed] [Google Scholar]
- 20.Imai S., Sakane F., and Kanoh H.. 2002. Phorbol ester-regulated oligomerization of diacylglycerol kinase δ linked to its phosphorylation and translocation. J. Biol. Chem. 277: 35323–35332. [DOI] [PubMed] [Google Scholar]
- 21.Sakane F., Imai S., Yamada K., Murakami T., Tsushima S., and Kanoh H.. 2002. Alternative splicing of the human diacylglycerol kinase δ gene generates two isoforms differing in their expression patterns and in regulatory functions. J. Biol. Chem. 277: 43519–43526. [DOI] [PubMed] [Google Scholar]
- 22.Murakami T., Sakane F., Imai S., Houkin K., and Kanoh H.. 2003. Identification and characterization of two splice variants of human diacylglycerol kinase η. J. Biol. Chem. 278: 34364–34372. [DOI] [PubMed] [Google Scholar]
- 23.Tang W., Bunting M., Zimmerman G. A., McIntyre T. M., and Prescott S. M.. 1996. Molecular cloning of a novel human diacylglycerol kinase highly selective for arachidonate-containing substrates. J. Biol. Chem. 271: 10237–10241. [PubMed] [Google Scholar]
- 24.Bunting M., Tang W., Zimmerman G. A., McIntyre T. M., and Prescott S. M.. 1996. Molecular cloning and characterization of a novel human diacylglycerol kinase ζ. J. Biol. Chem. 271: 10230–10236. [PubMed] [Google Scholar]
- 25.Ding L., Traer E., McIntyre T. M., Zimmerman G. A., and Prescott S. M.. 1998. The cloning and characterization of a novel human diacylglycerol kinase, DGKι. J. Biol. Chem. 273: 32746–32752. [DOI] [PubMed] [Google Scholar]
- 26.Houssa B., Schaap D., van der Val J., Goto K., Kondo H., Yamakawa A., Shibata M., Takenawa T., and van Blitterswijk W. J.. 1997. Cloning of a novel human diacylglycerol kinase (DGKθ) containing three cysteine-rich domains, a proline-rich Region, and a pleckstrin homology domain with an overlapping Ras-associating domain. J. Biol. Chem. 272: 10422–10428. [DOI] [PubMed] [Google Scholar]
- 27.Imai S., Kai M., Yasuda S., Kanoh H., and Sakane F.. 2005. Identification and characterization of a novel human type II diacylglycerol kinase, DGKκ. J. Biol. Chem. 280: 39870–39881. [DOI] [PubMed] [Google Scholar]
- 28.Sakane F., Kai M., Wada I., Imai S., and Kanoh H.. 1996. The C-terminal part of diacylglycerol kinase α lacking zinc fingers serves as a catalytic domain. Biochem. J. 318: 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sakane F., Imai S., Kai M., Wada I., and Kanoh H.. 1996. Molecular cloning of a novel diacylglycerol kinase isozyme with a pleckstrin homology domain and a C-terminal tail similar to those of the EPH family of protein tyrosine kinase. J. Biol. Chem. 271: 8394–8401. [DOI] [PubMed] [Google Scholar]
- 30.Mizuno S., Sakai H., Saito M., Kado S., and Sakane F.. 2012. Diacylglycerol kinase-dependent formation of phosphatidic acid molecular species during interleukin-2 activation in CTLL-2 T-lymphocytes. FEBS Open Bio. 2: 267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sakai H., Kado S., Taketomi A., and Sakane F.. 2014. Diacylglycerol kinase δ phosphorylates phosphatidylcholine-specific phospholipase C-dependent, palmitic acid-containing diacylglycerol species in response to high glucose levels. J. Biol. Chem. 289: 26607–26617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bligh E. G., and Dyer W. J.. 1959. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37: 911–917. [DOI] [PubMed] [Google Scholar]
- 33.Tanaka S., Akaishi E., Hosaka K., Okamura S., and Kubohara Y.. 2005. Zinc ions suppress mitogen-activated interleukin-2 production in Jurkat cells. Biochem. Biophys. Res. Commun. 335: 162–167. [DOI] [PubMed] [Google Scholar]
- 34.Yamada K., Sakane F., and Kanoh H.. 1989. Immunoquantitation of 80 kDa diacylglycerol kinase in pig and human lymphocytes and several other cells. FEBS Lett. 244: 402–406. [DOI] [PubMed] [Google Scholar]
- 35.Sakane F., Yamada K., Imai S., and Kanoh H.. 1991. Porcine 80-kDa diacylglycerol kinase is a calcium-binding and calcium/phospholipid-dependent enzyme and undergoes calcium-dependent translocation. J. Biol. Chem. 266: 7096–7100. [PubMed] [Google Scholar]
- 36.Sakane F., Imai S., Yamada K., and Kanoh H.. 1991. The regulatory role of EF-hand motifs of pig 80K diacylglycerol kinase as assessed using truncation and deletion mutants. Biochem. Biophys. Res. Commun. 181: 1015–1021. [DOI] [PubMed] [Google Scholar]
- 37.Dominguez C. L., Floyd D. H., Xiao A., Mullins G. R., Kefas B. A., Xin W., Yacur M. N., Abounader R., Lee J. K., Wilson G. M., et al. . 2013. Diacylglycerol kinase α is a critical signaling node and novel therapeutic target in glioblastoma and other cancers. Cancer Discov. 3: 782–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nobe K., Miyatake M., Nobe H., Sakai Y., Takashima J., and Momose K.. 2004. Novel diacylglycerol kinase inhibitor selectively suppressed an U46619-induced enhancement of mouse portal vein contraction under high glucose conditions. Br. J. Pharmacol. 143: 166–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huber C. S. 1975. The structure of stemphone, a yellow fungal metabolite. Acta Crystallogr. B. 31: 108–113. [Google Scholar]
- 40.Torres-Ayuso P., Daza-Martin M., Martin-Perez J., Avila-Flores A., and Merida I.. 2014. Diacylglycerol kinase α promotes 3D cancer cell growth and limits drug sensitivity through functional interaction with Src. Oncotarget. 5: 9710–9726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baldanzi G., Mitola S., Cutrupi S., Filigheddu N., van Blitterswijk W. J., Sinigaglia F., Bussolino F., and Graziani A.. 2004. Activation of diacylglycerol kinase α is required for VEGF-induced angiogenic signaling in vitro. Oncogene. 23: 4828–4838. [DOI] [PubMed] [Google Scholar]
- 42.Guo R., Wan C. K., Carpenter J. H., Mousallem T., Boustany R. M., Kuan C. T., Burks A. W., and Zhong X. P.. 2008. Synergistic control of T cell development and tumor suppression by diacylglycerol kinase α and ζ. Proc. Natl. Acad. Sci. USA. 105: 11909–11914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chabner B. A., and Roberts T. G. Jr. 2005. Timeline: chemotherapy and the war on cancer. Nat. Rev. Cancer. 5: 65–72. [DOI] [PubMed] [Google Scholar]
- 44.Pérez-Herrero E., and Fernández-Medarde A.. 2015. Advanced targeted therapies in cancer: drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 93: 52–79. [DOI] [PubMed] [Google Scholar]
- 45.Jones D. R., Flores I., Diaz E., Martinez-A C., and Merida I.. 1998. Interleukin-2 stimulates a late increase in phosphatidic acid production in the absence of phospholipase D activation. FEBS Lett. 433: 23–27. [DOI] [PubMed] [Google Scholar]
- 46.Kakefuda K., Oyagi A., Ishisaka M., Tsuruma K., Shimazawa M., Yokota K., Shirai Y., Horie K., Saito N., Takeda J., et al. . 2010. Diacylglycerol kinase β knockout mice exhibit lithium-sensitive behavioral abnormalities. PLoS One. 5: e13447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shirai Y., Kouzuki T., Kakefuda K., Moriguchi S., Oyagi A., Horie K., Morita S. Y., Shimazawa M., Fukunaga K., Takeda J., et al. . 2010. Essential role of neuron-enriched diacylglycerol kinase (DGK), DGKβ in neurite spine formation, contributing to cognitive function. PLoS One. 5: e11602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sakuma M., Shirai Y., Ueyama T., and Saito N.. 2014. Diacylglycerol kinase gamma regulates antigen-induced mast cell degranulation by mediating Ca(2+) influxes. Biochem. Biophys. Res. Commun. 445: 340–345. [DOI] [PubMed] [Google Scholar]
- 49.Kurohane Kaneko Y., Kobayashi Y., Motoki K., Nakata K., Miyagawa S., Yamamoto M., Hayashi D., Shirai Y., Sakane F., and Ishikawa T.. 2013. Depression of type I diacylglycerol kinases in pancreatic β-cells from male mice results in impaired insulin secretion. Endocrinology. 154: 4089–4098. [DOI] [PubMed] [Google Scholar]
- 50.Crotty T., Cai J., Sakane F., Taketomi A., Prescott S. M., and Topham M. K.. 2006. Diacylglycerol kinase δ regulates protein kinase C and epidermal growth factor receptor signaling. Proc. Natl. Acad. Sci. USA. 103: 15485–15490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chibalin A. V., Leng Y., Vieira E., Krook A., Bjornholm M., Long Y. C., Kotova O., Zhong Z., Sakane F., Steiler T., et al. . 2008. Downregulation of diacylglycerol kinase delta contributes to hyperglycemia-induced insulin resistance. Cell. 132: 375–386. [DOI] [PubMed] [Google Scholar]
- 52.Miele C., Paturzo F., Teperino R., Sakane F., Fiory F., Oriente F., Ungaro P., Valentino R., Beguinot F., and Formisano P.. 2007. Glucose regulates diacylglycerol intracellular levels and protein kinase C activity by modulating diacylglycerol-kinase subcellular localization. J. Biol. Chem. 282: 31835–31843. [DOI] [PubMed] [Google Scholar]
- 53.Yasuda S., Kai M., Imai S., Takeishi K., Taketomi A., Toyota M., Kanoh H., and Sakane F.. 2009. Diacylglycerol kinase η augments C-Raf activity and B-Raf/C-Raf heterodimerization. J. Biol. Chem. 284: 29559–29570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakano T., Iravani A., Kim M., Hozumi Y., Lohse M., Reichert E., Crotty T. M., Stafforini D. M., and Topham M. K.. 2014. Diacylglycerol kinase eta modulates oncogenic properties of lung cancer cells. Clin. Transl. Oncol. 16: 29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baum A. E., Akula N., Cabanero M., Cardona I., Corona W., Klemens B., Schulze T. G., Cichon S., Rietschel M., Nothen M. M., et al. . 2008. A genome-wide association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Mol. Psychiatry. 13: 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weber H., Kittel-Schneider S., Gessner A., Domschke K., Neuner M., Jacob C. P., Buttenschon H. N., Boreatti-Hummer A., Volkert J., Herterich S., et al. . 2011. Cross-disorder analysis of bipolar risk genes: further evidence of DGKH as a risk gene for bipolar disorder, but also unipolar depression and adult ADHD. Neuropsychopharmacology. 36: 2076–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van der Zanden L. F., van Rooij I. A., Feitz W. F., Knight J., Donders A. R., Renkema K. Y., Bongers E. M., Vermeulen S. H., Kiemeney L. A., Veltman J. A., et al. . 2011. Common variants in DGKK are strongly associated with risk of hypospadias. Nat. Genet. 43: 48–50. [DOI] [PubMed] [Google Scholar]
- 58.Rodriguez de Turco E. B., Tang W., Topham M. K., Sakane F., Marcheselli V. L., Chen C., Taketomi A., Prescott S. M., and Bazan N. G.. 2001. Diacylglycerol kinase ε regulates seizure susceptibility and long-term potentiation through arachidonoyl- inositol lipid signaling. Proc. Natl. Acad. Sci. USA. 98: 4740–4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang N., Li B., Al-Ramahi I., Cong X., Held J. M., Kim E., Botas J., Gibson B. W., and Ellerby L. M.. 2012. Inhibition of lipid signaling enzyme diacylglycerol kinase ε attenuates mutant huntingtin toxicity. J. Biol. Chem. 287: 21204–21213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Topham M. K., Bunting M., Zimmerman G. A., McIntyre T. M., Blackshear P. J., and Prescott S. M.. 1998. Protein kinase C regulates the nuclear localization of diacylglycerol kinase-zeta. Nature. 394: 697–700. [DOI] [PubMed] [Google Scholar]
- 61.Zhong X. P., Hainey E. A., Olenchock B. A., Jordan M. S., Maltzman J. S., Nichols K. E., Shen H., and Koretzky G. A.. 2003. Enhanced T cell responses due to diacylglycerol kinase ζ deficiency. Nat. Immunol. 4: 882–890. [DOI] [PubMed] [Google Scholar]
- 62.Nakano T., Hozumi Y., Ali H., Saino-Saito S., Kamii H., Sato S., Kayama T., Watanabe M., Kondo H., and Goto K.. 2006. Diacylglycerol kinase zeta is involved in the process of cerebral infarction. Eur. J. Neurosci. 23: 1427–1435. [DOI] [PubMed] [Google Scholar]
- 63.Regier D. S., Higbee J., Lund K. M., Sakane F., Prescott S. M., and Topham M. K.. 2005. Diacylglycerol kinase ι regulates Ras guanyl-releasing protein 3 and inhibits Rap1 signaling. Proc. Natl. Acad. Sci. USA. 102: 7595–7600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pankratz N., Wilk J. B., Latourelle J. C., DeStefano A. L., Halter C., Pugh E. W., Doheny K. F., Gusella J. F., Nichols W. C., Foroud T., et al. . 2009. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum. Genet. 124: 593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cai K., and Sewer M. B.. 2013. Diacylglycerol kinase θ couples farnesoid X receptor-dependent bile acid signalling to Akt activation and glucose homoeostasis in hepatocytes. Biochem. J. 454: 267–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.