
FIGURE 1.
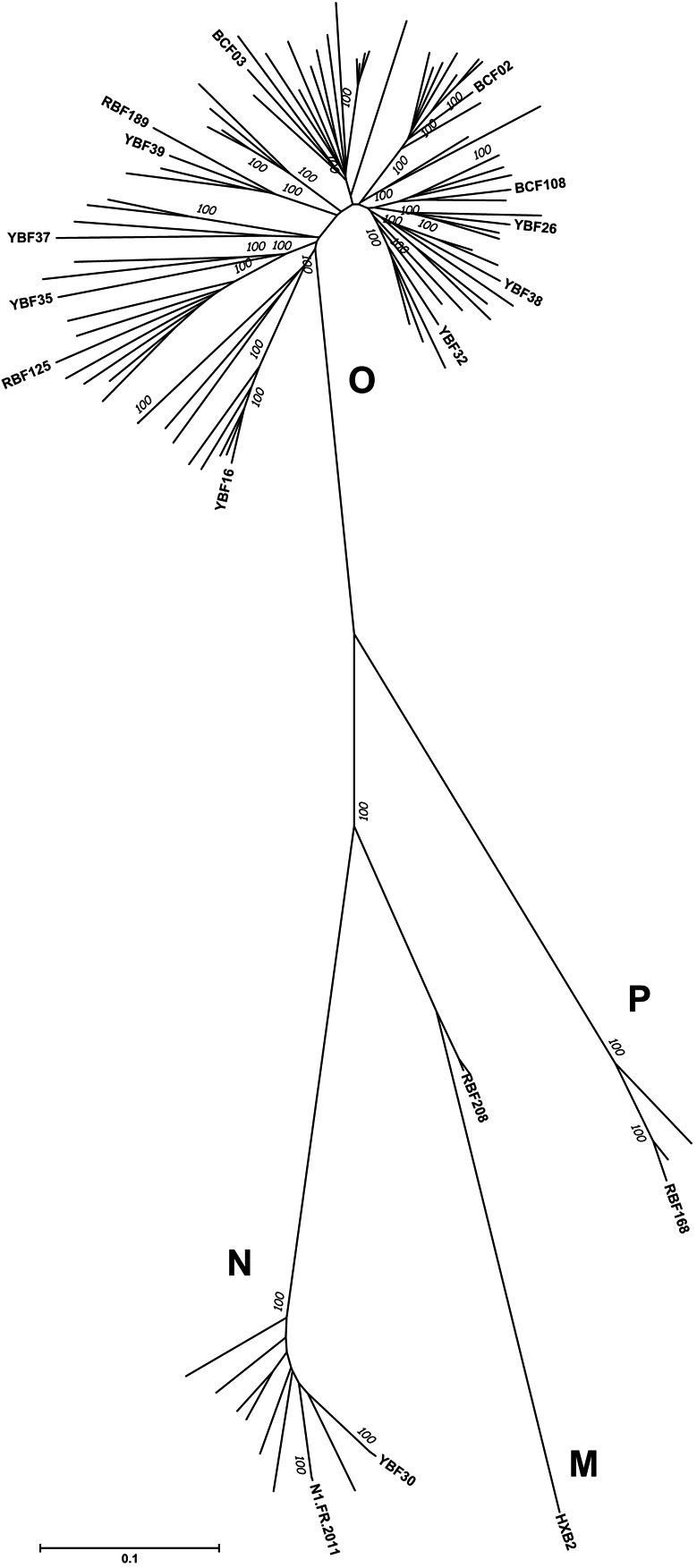
Phylogenetic analysis of env sequences. The 16 env sequences of the PIs included in the study were aligned with 72 env sequences from non-M variants that were available in the Los Alamos database. The evolutionary history was inferred using the Neighbor-Joining method.34 The optimal tree with the sum of branch length = 752,171,464 is shown. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches.35 The tree is drawn to scale with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Tamura-Nei method36 and are in the units of the number of base substitutions per site. The rate variation among sites was modeled with a gamma distribution (shape parameter = 1). All ambiguous positions were removed for each sequence pair. There were a total of 3165 positions in the final data set. Evolutionary analyses were conducted in MEGA6.37