
Figure 8. Effects of GSK3β signal pathway on β-catenin and PTMA expression.
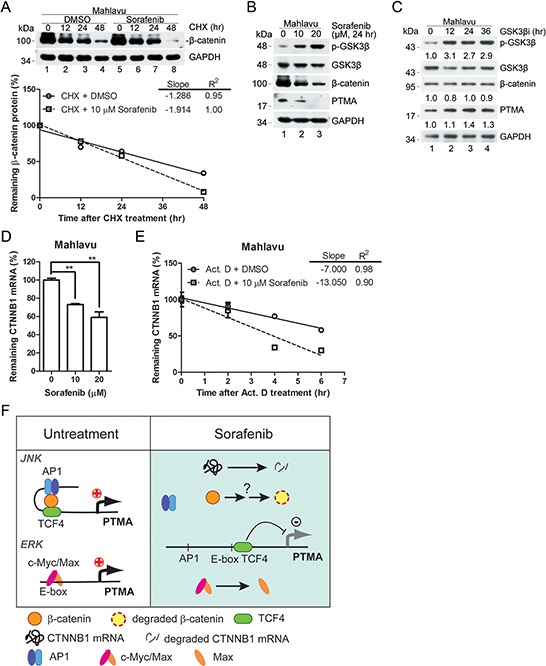
A. Degradation rate of β-catenin protein in sorafenib-treated Mahlavu cells. Cells were pre-incubated with cycloheximide (CHX), an inhibitor of protein synthesis, followed by the indicated incubation. Linear regressions of the kinetic pattern of PTMA protein level are shown at bottom. B. Decreased GSK3β signaling in sorafenib-treated Mahlavu cells. GSK3β inactivation is shown based on the phosphorylated form of the kinase by sorafenib in a dose-dependent manner. Simultaneous decrease of PTMA protein level was observed in the same treatment cells. C. Lack of reduction of β-catenin and PTMA protein level following treatment with GSK3β inhibitor (GSK3βi) for the indicated time. Cells were treated with 50 nM of GSK3β inhibitor for the time indicated. Fold change, relative to untreated control, is indicated. D. Reduction of CTNNB1 mRNA level by sorafenib in Mahlavu cells. E. Increased degradation rate of the CTNNB1 mRNA in sorafenib-treated Mahlavu cells. Cells were pre-incubated with actinomycin D (Act. D), an inhibitor of RNA synthesis, followed by the indicated incubation. Results are shown as linear regression of the kinetic pattern of CTNNB1 mRNA level. F. Working model for the involvement of the β-catenin/JNK/PTMA and ERK/c-Myc/Max/PTMA axes in HCC sensitivity to sorafenib. In the absence of sorafenib (left panel, untreatment), β-catenin acts in concert with TCF4 and the AP-1/JNK pathway to stimulate PTMA transcription. In the presence of sorafenib (right panel), β-catenin was degraded and the components AP-1/TCF4 were dissociated, leading to downregulation of PTMA transcription. The ERK/c-Myc/Max axis in regulation of PTMA previously identified [6] is also included.