

Abstract
The nano- and micropatterned biosilica cell walls of diatoms are remarkable examples of biological morphogenesis and possess highly interesting material properties. Only recently has it been demonstrated that biosilica-associated organic structures with specific nanopatterns (termed insoluble organic matrices) are general components of diatom biosilica. The model diatom Thalassiosira pseudonana contains three types of insoluble organic matrices: chitin meshworks, organic microrings, and organic microplates, the latter being described in the present study for the first time. To date, little is known about the molecular composition, intracellular assembly, and biological functions of organic matrices. Here we have performed structural and functional analyses of the organic microrings and organic microplates from T. pseudonana. Proteomics analysis yielded seven proteins of unknown function (termed SiMat proteins) together with five known silica biomineralization proteins (four cingulins and one silaffin). The location of SiMat1-GFP in the insoluble organic microrings and the similarity of tyrosine- and lysine-rich functional domains identifies this protein as a new member of the cingulin protein family. Mass spectrometric analysis indicates that most of the lysine residues of cingulins and the other insoluble organic matrix proteins are post-translationally modified by short polyamine groups, which are known to enhance the silica formation activity of proteins. Studies with recombinant cingulins (rCinY2 and rCinW2) demonstrate that acidic conditions (pH 5.5) trigger the assembly of mixed cingulin aggregates that have silica formation activity. Our results suggest an important role for cingulins in the biogenesis of organic microrings and support the hypothesis that this type of insoluble organic matrix functions in biosilica morphogenesis.
Keywords: algae, biomineralization, cell wall, intrinsically disordered protein, morphogenesis, post-translational modification (PTM), protein self-assembly, proteomics
Introduction
A hallmark of diatoms, a large group of single-celled eukaryotic algae, is the formation of porous, hierarchically nano- and micropoatterned, amorphous SiO2 (silica). The silica serves as the diatom's cell wall material and is believed to provide an ecological advantage over other phytoplankton microorganisms due to the energy efficiency of its production (1) and mechanical stability against plankton grazers (2). Furthermore, diatom biosilica formation is a well established model system for elucidating the molecular mechanisms of biomineral morphogenesis (3–7) and is of increasing interest as a material for applications in bio-/nanotechnology (8–16).
It has long been known that diatom biosilica is an organic-inorganic hybrid material (17, 18), as is the case with most other biominerals (19). The first biosilica-associated biomacromolecules were characterized about 20 years ago (20), leading to the identification of components that strongly influence the kinetics and morphogenesis of silica formation in vitro (21, 22). The biosilica-associated organic material can be separated into two different fractions: (i) organic molecules that become soluble after dissolving the biosilica with a mildly acidic solution of ammonium fluoride (soluble fraction) and (ii) organic material that remains insoluble after ammonium fluoride treatment (insoluble fraction). The soluble fraction is composed of long-chain polyamines and highly phosphorylated and often also glycosylated proteins (silaffins and silacidins) (23–27). The insoluble fraction contains organic material with well defined nano- and micropatterns that resemble the silica morphology of the respective diatom species (28–31). The biochemical composition of the insoluble organic material appears to be quite complex, containing both proteins and polysaccharide, but it has so far remained poorly characterized in any diatom species. Until now, the functions of the biosilica-associated organic matrices have remained speculative, and they may include (i) serving as an organic casing that protects the silica against dissolution (32), (ii) mechanical strengthening of the cell wall by connecting the different biosilica building blocks (girdle bands and valves) and through dissipating energy imposed on the cell wall by external and internal forces (e.g. impact from grazers or osmotic pressure) (33), or (iii) serving as templates for silica morphogenesis (29).
Gaining insight into the functions of biosilica-associated insoluble organic matrices requires an accurate characterization of its molecular composition. What are the chemical structures and properties of the biopolymers? How are they assembled into insoluble organic matrices with characteristic nano- and micropatterns?
We have chosen Thalassiosira pseudonana to investigate these questions because this diatom is a well established model organism (21). Previously, two types of biosilica-associated insoluble organic matrices have been identified in T. pseudonana (i.e. a chitin-based meshwork (28) and protein-based microrings (29)). It is not yet known whether the chitin-based meshwork is associated with all parts of the biosilica. There is evidence that proteins are attached to the chitin meshwork (28), but so far none has been identified. A diatom cell wall-associated protein (p150) containing a chitin binding domain was identified in T. pseudonana (34), but its association with the chitin meshwork has not yet been investigated. The organic microrings are specifically associated with the girdle band biosilica and have silica formation activity (29). They contain a family of six proteins termed cingulins, which were serendipitously identified through a bioinformatics screen for silaffin-like proteins in the T. pseudonana genome (29). Cingulins were mapped to the organic microrings through GFP tagging and fluorescence microscopy analysis (29). The present study aimed at obtaining comprehensive information on the biochemical composition of the organic microrings with a focus on the structural and functional analysis of its protein components.
Experimental Procedures
Chemicals and Reagents
Chemical and reagents were purchased from the following companies: oligonucleotides (Eurofins Genomics), isopropylthiogalactoside (Carl Roth), 6 n HCl sequencing grade (Thermo Scientific), phenyl isothiocyanate (Thermo Scientific), acetonitrile (VWR), phenol (Wako Chemicals), mass spectrometry solvents (Fisher), anhydrous hydrogen fluoride (GHC Gerling), ampicillin (Merck), nourseothricin (Jena Bioscience), tetramethoxysilane (Sigma-Aldrich), ammonium molybdate tetrahydrate (Merck), ammonium fluoride (Merck), chitinase from Streptomyces griseus (Sigma-Aldrich), EDTA (Merck), SDS (Merck), trypsin (mass spectrometry grade; Promega), chymotrypsin (sequencing grade; Roche Applied Science), endoproteinase Asp-N (sequencing grade; Roche Applied Science), endoproteinase Glu-C (sequencing grade; Roche Applied Science). Enzymes used for molecular genetics were purchased from Thermo Scientific. MilliQ-purified H2O (resistivity, 18.2 megaohms·cm) was used throughout this study.
Culture Conditions
T. pseudonana (Hustedt) Hasle and Heimdal clone CCMP1335 was grown in an enriched artificial seawater medium according to the Canadian Center for the Culture of Microorganisms at 18 °C under constant light at 5,000–10,000 lux.
Cloning, Expression, and Purification of Recombinant Proteins
DNA sequences encoding for the mature polypeptides (i.e. lacking the N-terminal signal peptide) of CinY2 (Uniprot ID F2YBR8, amino acids 16–248) and CinW2 (Uniprot ID F2YBS1, amino acids 16–383) were synthesized by DNA2.0 with their codons optimized for expression in Escherichia coli (see supplemental Fig. S1 for DNA sequences). The genes were incorporated into expression vector pJ404 (T5 promoter, ampicillin resistance, isopropyl 1-thio-β-d-galactopyranoside-inducible) by the vendor. DH5α cells were chemically transformed using the pJ404-rcinY2 and pJ404-rcinW2 expression vectors. For protein expression, 50 ml of LB medium containing 100 μg·ml−1 ampicillin were inoculated with a colony of DH5α carrying the pJ404-rcinY2 or pJ404-rcinW2 plasmid and grown in a shaker incubator at 37 °C and 180 rpm overnight. The following day, 1 liter of culture medium was inoculated with 50 ml of the overnight culture and grown at 37 °C and 180 rpm. At an A600 nm of ∼0.6, the culture was supplemented with 1 mm isopropyl 1-thio-β-d-galactopyranoside and further incubated for 3 h. Cells were harvested by centrifugation (6,000 × g, 30 min, 4 °C) and stored at −80 °C.
To achieve cell lysis, frozen cells were resuspended in 20 volumes of lysis buffer (50 mm Tris·HCl, pH 8.0, 1 m NaCl, 10 mm imidazole) supplemented with 10 mm PMSF and sonicated with a Bandelin (Germany) sonication tip (50% amplitude, 30-s intervals with 30-s breaks between). Cell lysis was inspected by bright field microscopy, and sonication continued until the majority of cells (∼80%) were lysed. The lysate was centrifuged (12,000 × g, 30 min, 4 °C), and the supernatant was subjected to immobilized metal affinity chromatography using HisPur nickel-nitrilotriacetic acid resin (Thermo Scientific). The resin was equilibrated in lysis buffer, subsequently mixed with the crude extract, and incubated overnight at 4 °C under constant mixing. The suspension was poured into a glass column that was equipped with a frit (Sigma Aldrich), the resin was washed with 16 column volumes of washing buffer (50 mm Tris·HCl, pH 8.0, 1 m NaCl, 25 mm imidazole), and elution of proteins was carried out with 12 column volumes elution buffer (50 mm Tris·HCl, pH 8.0, 1 m NaCl, 250 mm imidazole). rCinY2 and rCinW2 were then further purified by ion exchange chromatography. To drastically reduce the NaCl concentration in the IMAC eluates, they were diluted 20-fold with buffer A (50 mm Tris·HCl, pH 7.5, 50 mm NaCl) for rCinY2 and buffer B (50 mm Tris·HCl, pH 8.8, 50 mm NaCl, 2 m urea) for rCinW2. Note that in the case of rCinW2, 2 m urea had to be included in all buffers to prevent precipitation of the protein at low salt conditions and high protein concentration. The cingulin solutions were then adsorbed to ion exchange resins by incubation for 2 h at 4 °C under constant rotation. rCinY2 was adsorbed to High S (Bio-Rad) equilibrated with buffer A, and rCinW2 was adsorbed to High Q resin (Bio-Rad) equilibrated with buffer B. After extensive washing (15 column volumes) of the resin with buffer A and buffer B, respectively, cingulins were eluted by stepwise (5 column volumes for each step) increase of the NaCl concentration (50, 100, 200, 300, 500, 1,000 mm NaCl) in buffer A and buffer B, respectively. All eluate fractions were analyzed for the presence and purity of cingulins by SDS-PAGE and Coomassie Blue staining. Pure rCinY2 was present in the 300 and 500 mm NaCl eluates, and pure rCinW2 was present in the 500 and 1,000 mm NaCl eluates. The eluates containing pure proteins were combined (supplemental Fig. S3) and flash-frozen in liquid nitrogen and stored at −80 °C until use.
The recombinant silaffin, rSil3, was isolated as described previously (35). The pure protein was dialyzed against 20 mm ammonium acetate, flash-frozen in liquid nitrogen, and stored at −80 °C until use.
Dynamic Light Scattering
Solutions of pure rCinY2 and rCinW2 were dialyzed against 10 mm MOPS, pH 7.7, containing 150 mm or 1 m NaCl. The dialysates were centrifuged (5 min, 20,000 × g), and protein concentrations were determined by measuring UV absorbance (molar extinction coefficients at 280 nm were 28,435 m−1·cm−1 for rCinY2 and 123,870 m−1·cm−1 for rCinW2). The protein solutions were adjusted to the indicated protein concentration using 0.2-μm filtered dialysis buffer and adjusted to the indicated pH by the addition of acetic acid (note that during pH adjustment, the volume increased only by <10%). Following pH adjustment, the protein solutions were incubated for 1 h at room temperature prior to measuring dynamic light scattering with a Zetasizer Nano-ZS (Malvern Instruments). Samples were measured in a quartz cuvette (Hellma) with a 10-mm path length at 25 °C using the 173° backscatter option of the instrument with automatic determination of the measurement duration. Data processing was performed using the multiple narrow modes analysis model of the Zetasizer software. Data from three measurement runs were averaged.
Silica Formation Assay
Solutions of pure rCinY2 and rCinW2 were prepared as described under “Dynamic Light Scattering.” Silaffin rSil3 was prepared as described previously (35). Proteins were diluted to the desired concentration in sodium acetate pH 5.5 (final concentration, 50 mm), and the NaCl content was adjusted to a final concentration of 150 mm. Silicic acid was freshly prepared by hydrolysis of tetramethoxysilane in 1 mm HCl (15 min of shaking at room temperature). Silica deposition was initiated by adding silicic acid to the protein solutions at a final concentration of 100 mm. The reaction volume was 300 μl for solutions containing 1–3 μm protein and 100 μl for solutions with a protein concentration of 4–10 μm. After a 10-min incubation at room temperature, silica was pelleted by centrifugation at 16,000 × g for 5 min. The supernatant was removed, and the pellet was washed three times with 200 μl of H2O each by resuspension-centrifugation. The final pellet was resuspended in 40 μl of 2 m NaOH and dissolved by incubation at 95 °C for 1 h. The silica concentration was determined using the β-silicomolybdate method (36).
Electron Microscopy
For scanning electron microscopy analysis, silica precipitates produced by rCinY2, rCinW2, and rSil3 (see “Silica Formation Assays”) were resuspended in H2O and air-dried under ambient conditions on a platinum-coated polycarbonate membrane (0.1-μm pore size; Whatman).
Insoluble organic matrices were prepared for scanning electron microscopy as follows. Biosilica was isolated as described previously (29), resuspended in H2O, and then air-dried at ambient conditions on a gold- or platinum-coated polycarbonate membrane (0.1-μm pore size; Whatman). The biosilica was overlaid with a solution containing 10 m NH4F adjusted to pH 4.5 with HCl and incubated for 1 h at room temperature in a humid chamber. Subsequently, the samples were extensively washed with H2O. To enhance visibility of valve-derived organic matrices, the isolated biosilica was resuspended in 1 ml of H2O in a 1.5-ml tube and subjected to 5 s of sonication with an MS72 sonotrode tip (Bandelin), applying a total energy of 0.12 kJ. An aliquot of the sonicated suspension was dried on a gold- or platinum-coated polycarbonate membrane (0.1-μm pore size; Whatman); demineralized with 10 m NH4F, pH 4.5, for 1 h; and extensively washed with H2O. The insoluble organic matrices were imaged using a JSM 7500F emission scanning electron microscope (Jeol) at an acceleration voltage of 1 kV (gentle beam conditions) for imaging of organic matrices.
For transmission electron microscopy, biosilica was isolated as described previously (29), resuspended in H2O, and air-dried at ambient conditions on a Formvar-coated copper grid (EMS) that was strengthened with evaporated carbon. Imaging was performed with a Morgagni 268D (FEI) transmission electron microscope at an acceleration voltage of 80 kV.
For electron dispersive x-ray measurements, a Supra 40VP scanning electron microscope (Zeiss) equipped with a Quantax XFlash 6/100 detector (Bruker) was used at an acceleration voltage of 6 kV. The data obtained were analyzed with the Esprit 1.9 software (Bruker).
Expression of GFP-tagged Proteins in T. pseudonana
The native promoters and terminators of the cinY2 and cinW2 genes were cloned by replacing the nitrate reductase promoter and terminator regions in the vector pTpNR-GFP (37) in a stepwise procedure. First, the promoter and coding regions of cinY2 (Pciny2-cinY2) and cinW2 (Pcinw2-cinW2) were amplified from genomic DNA (isolated according to Ref. 36), assuming that the promoter is located within the region 1,000 bp upstream of the coding region comprising amino acids 1–237 in CinY2 (Uniprot ID F2YBR8) and amino acids 1–383 in CinW2 (Uniprot ID F2YBS1). The amplified region for cinY2 did not include the sequence coding for the last 11 amino acids at the C terminus. This C-terminal region was amplified together with the terminator region of cinY2 and thus is located downstream of the enhanced gfp gene in the final vector (see below).
The sense primer 5′-AAT GTC GGG CCC GAT GAA GTT GGG TCC-3′ and antisense primer 5′-AAC CTT CTGAT ATC ATT TCT CCT GAC GTA-3′ were used for amplification of Pciny2-cinY2, and primer pair 5′-TCC TCT TAC TCG AGC TCT CAG CTC TCC TC-3′ and 5′-TTG TGG TGA TAT CCC ATC CAC TGT ACC-3′ was used to amplify Pcinw2-cinW2 (ApaI site underlined, EcoRV site in boldface type, XhoI site in italic type). The resulting PCR products were then cloned into the ApaI/EcoRV (cinY2) or XhoI/EcoRV sites (cinW2) of the plasmid pTpNR-GFP, thereby replacing the nitrate reductase promoter of that plasmid and generating plasmids pPciny2-cinY2-GFP-Tnr and pPcinw2-cinW2-gfp-Tnr. Subsequently, the nitrate reductase terminator region in both of these plasmids was replaced against the terminator region of cinY2 (Tciny2) and cinW2 (Tcinw2). The terminator regions of the cingulins were amplified from genomic DNA using sense primer 5′-AAG GTT GGC GGC CGC AGA AGG TTG GGT GCT TC-3′ and antisense primer 5′-AGT TTT TAG GAT CCT CAA TAG GTT GAC TC-3′ for cinY2 (NotI site underlined, BamHI site in boldface type) and sense primer 5′-TGG TAC AGC GGC CGC TAA ATA ACC ACA ACT ATC-3′ and antisense primer 5′- TAT TGT CCC GGG TAT CAT CAT CTT GGC-3′ (NotI site underlined, SmaI site in italic type) for cinW2, assuming that the respective terminator regions of the cingulin genes are located within the first 900 bp downstream of the stop codon. Plasmids pPciny2-cinY2-gfp-Tnr and pPcinw2-cinW2-gfp-Tnr were treated with NotI/BamHI and NotI/SmaI, respectively, to excise the nitrate reductase terminator gene. Subsequently, the PCR products covering the terminator regions of cinY2 and cinW2 were inserted into the NotI/BamHI (cinY2) and NotI/SmaI (cinW2) sites of these two vectors, yielding plasmids pPciny2-cinY2-gfp-Tciny2 and pPcinw2-cinW2-gfp-Tcinw2.
Finally, the nourseothricin resistance gene (nat1) under control of the Tpfcp promoter and terminator was inserted downstream of the cingulin terminator regions of the plasmids. The pTpfcp/nat resistance cassette (nat1 gene, including the fcp promoter and terminator) was retrieved from plasmid pTpfcp/nat (37) by treatment with HindIII, blunting with Klenow polymerase, and then excising the cassette by digestion with XbaI. The Tpfcp/nat resistance cassette was inserted into BamHI(blunted)/XbaI-digested pPciny2-cinY2-gfp-Tciny2 and into SmaI/XbaI-digested pPcinw2-cinW2-gfp-Tcinw2, yielding the final plasmids pPciny2-cinY2-gfp-Tciny2-Tpfcp/nat and pPcinw2-cinW2-gfp-Tcinw2-Tpfcp/nat, respectively.
To generate an expression vector for GFP-tagged SiMat1 (Uniprot ID B8BT90), the vector pTpNR-GFP/fcpNat(-KpnI) (29) was modified to include an HpaI site at the 3′-end of the nitrate reductase terminator region and silencing of the NotI site within the fcpNAT cassette to generate pTpNR-GFPHpaI/fcpNat(-KpnI-NotI). The start and stop codons of SiMat1 were confirmed by 5′- and 3′-RACE PCR (supplemental Fig. S7). The SiMat1 promoter (1,068 bp) and coding region (covering amino acids 1–248) were amplified from genomic DNA using sense primer 5′-TAG TAG TGG GCC CTG TCG TCA TCG TCG TC-3′ and antisense primer 5′-ACA TCC GGT ACC CAC GGT AGG ACC AG-3′ (ApaI site underlined and KpnI site in italic type). The resulting PCR product was digested with ApaI and KpnI and cloned into the ApaI and KpnI sites of pTpNR-GFPHpaI/fcpNat(-KpnI-NotI), generating pPSiMat1-SiMat1-GFP-Tnr/fcpNAT(-KpnI-NotI). The terminator region (806 bp) of SiMat1 was amplified from genomic DNA using sense primer 5′-ACT GGT CGC GGC CGC GTA GAG AGG ATG TTG TAC-3′ and antisense primer 5′- ACT TGA CGT TAA CAG AAT GAG TGA GAG CG-3′ (NotI site underlined, HpaI site in italic type) and cloned into the NotI and HpaI sites of pPSiMat1-SiMat1-GFP-Tnr/fcpNAT(-KpnI-NotI), generating the final expression plasmid pPSiMat1-SiMat1-GFP-TSiMat1/fcpNAT. Transformation of T. pseudonana and selection of transformants on nourseothricin-containing agar plates was performed as described previously (37).
Fluorescence Microscopy
For confocal fluorescence microscopy, 10 μl of a cell suspension was transferred onto 22 × 50-mm coverslips and covered with a rectangular, roughly 0.5-cm2 slice of 1% (w/v) agarose prepared with enriched artificial seawater medium. Images were acquired using a Zeiss LSM780 inverted microscope equipped with a Zeiss Plan-Apochromat ×63 (1.4 numerical aperture) oil DIC M27 objective. GFP fluorescence and chlorophyll autofluorescence were detected in 1-track mode using an argon laser line (power set to 2%), an MBS 488 beam splitter, and a 32-channel GaAsP spectral detector. Two channels were acquired to separately monitor the GFP fluorescence (emission at 491–535 nm) and chloroplast fluorescence (emission at 654–693 nm). Images were analyzed using the ZEN2012 software (Zeiss).
Epifluorescence microscopy was carried out using a ×63 oil objective on a Zeiss Axiovert 200 inverted microscope equipped with a Piston filter (Chroma; excitation 450–490 nm, emission 500–530 nm) to image GFP.
Immunodetection of CinY2-GFP and CinW2-GFP in Biosilica and Organic Microrings
Biosilica was isolated by SDS/EDTA extraction as described previously (25) and fragmented by sonication with a MS72 sonotrode tip (Bandelin), applying a total of 1.612 kJ over 80 s. The insoluble organic matrix material was prepared by incubating the biosilica with 10 m NH4F (adjusted to pH 4.5 with HCl) for 1 h at room temperature, followed by washing twice with H2O through centrifugation (10 min, 10,000 × g) and resuspension.
The biosilica and the insoluble organic matrix were resuspended separately in blocking solution (Roti-ImmunoBlock; Carl Roth), 0.05% Tween 20 (Merck Millipore) and immobilized on poly-l-lysine-coated coverslips by incubation for 1 h at room temperature. Unbound material was removed from the coverslips by washing with blocking solution. The coverslips were overlaid with an anti-GFP (full-length) polyclonal rabbit antibody (15 μg×ml−1; Clontech, product number 632592) in blocking solution for 1 h. After washing for 4 × 5 min in TBS (50 mm Tris·HCl, pH 7.5, 150 mm NaCl) containing 0.05% Tween 20, the coverslips were overlaid with AlexaFluor647-conjugated goat anti-rabbit IgG antibodies (2 μg×ml−1; Thermo Fisher Scientific) in blocking buffer for 1 h. The coverslips were washed as described above, followed by two 5-min washes with TBS.
GFP- and AlexaFluor647-bearing samples were visualized using epi-illumination with a 488-nm laser and a 647-nm laser at 10 milliwatts and respective filter sets (laser bandpass (475/35, 628/40), dichroic longpass (H 488 LPXR, H 643 LPXR), and emission bandpass (525/45, 700/75). The recorded fluorescence intensities were adjusted to prevent saturation of the detector. GFP and AlexaFluor647 z-stage image series were subsequently acquired with the NIS-Elements software (Nikon) using an EM CCD camera (Ixon Ultra 897; Andor) mounted on an inverted fluorescence microscope (NSTORM, Nikon) equipped with a ×100 oil objective (CFI TIRF Apochromat, numerical aperture 1.49, WD 0.12 mm, Nikon) and an autofocus system (Nikon) at an exposure time of 300 ms (GFP) and 50 ms (AlexaFluor647) and 4 frames/μm. For the defined regions of interest, maximum projections displaying the highest value of each pixel in all frames of the z-stacks were created using the software NIS-Elements. Alexa647 fluorescence intensity values were normalized by dividing through the corresponding GFP fluorescence intensity values from the same region of interest, resulting in relative fluorescence intensities (RFIs).2 The degree of accessibility of organic microrings in the biosilica was calculated through dividing the RFI from biosilica by the RFI from microrings.
Isolation of Insoluble Organic Matrices for Biochemical Analyses
Biosilica was isolated as described previously (29). Chitin fibers were removed by incubation with 0.1 mg·ml−1 chitinase from S. griseus (∼0.2 units mg−1; Sigma-Aldrich) in chitinase buffer (50 mm potassium phosphate, pH 6.0, 0.05% (w/v) sodium azide, 1 mm PMSF; note that the chitinase solution was filtered through a polyethersulfone syringe filter with 0.2-μm pore size) at 37 °C in a shaker incubator for 4 days. The progress of chitin degradation was monitored by Calcofluor White staining as described previously (28). The chitinase-treated biosilica was washed once with 1% (w/v) SDS followed by 5× washing with H2O by repeated centrifugation-resuspension cycles. The final pellet (i.e. chitin-free biosilica) was resuspended in H2O and freeze-dried. The dry material was resuspended in 150 ml of 10 m NH4F and adjusted to pH 4.5 by the addition of HCl. The suspension was incubated at room temperature for 1 h and centrifuged at 3,200 × g for 30 min. The pellet was washed twice with H2O, twice with 20 mm ammonium acetate, and twice with H2O by resuspension-centrifugation (3,200 × g, 30 min). The chitin-based meshwork was removed by a 48-h chitinase treatment as described above, followed by washing once with 1% (w/v) SDS and washing five times with H2O. The resulting NH4F-insoluble organic matrix material was freeze-dried and stored at −20 °C until use.
Amino Acid Analysis
Analysis of standard amino acids in the insoluble organic matrix material was performed according to methods described previously (38). Briefly, the insoluble organic matrix material (∼0.5 mg) was hydrolyzed for 16 h at 110 °C using a solution of 6 m HCl containing 10% phenol. The amino acids in the hydrolysate were quantified as phenylthiocarbamyl derivatives using reverse phase high pressure liquid chromatography in combination with a UV detector at 254 nm. Tryptophan was quantified following alkaline hydrolysis of the insoluble organic matrix material in 2 m NaOH for 4 h at 110 °C and subsequent analysis by the Dionex AAA-Direct method (Thermo Scientific) high pressure anion exchange chromatography in combination with pulsed amperometric detection.
Phosphoamino acid analysis was performed following acid hydrolysis of the insoluble organic matrix material for 1 and 3 h at 110 °C using a solution of 6 m HCl containing 10% phenol. The hydrolysate was evaporated to dryness in a vacuum centrifuge at 40 °C and dissolved in H2O. Phosphoamino acids were detected using high pressure anion exchange chromatography-pulsed amperometric detection with an Aminopac PA-10 column (Thermo Scientific Dionex) according to a procedure published by the manufacturer (64).
Monosaccharide Analysis
The insoluble organic matrix material (∼0.3 mg) was hydrolyzed with either 2 m TFA or 4 m HCl for 1 or 4 h at 100 °C. The hydrolysate was evaporated to dryness in a vacuum centrifuge at 40 °C and dissolved in H2O, and monosaccharides were quantified by high pressure anion exchange chromatography-pulsed amperometric detection using a Carbopac PA-10 column (Thermo Scientific Dionex) according to a method described previously (38).
Phosphate Quantification
The total phosphate content of the organic matrix was determined as described (39). In a glass vial, a sample of dry insoluble organic matrix was mixed with 25 μl of 10% (w/v) MgNO3·6H2O in 95% ethanol. The sample was dried at 95 °C and subsequently heated in the flame until the evolution of brown gases ceased. The vial was cooled to room temperature, and 500 μl of 1.2 m HCl was added and incubated for 20 min at room temperature. An aliquot was removed from the vial and diluted with 1.2 m HCl to 600 μl and 200 μl of the assay reagent (1 volume 10% (w/v) (NH4)6Mo7O24·4H2O in 4 m HCl plus 3 volumes of 0.2% (w/v) malachite green in H2O). After incubation at room temperature for 10 min, A660 nm was determined. Potassium dihydrogen phosphate was used as a standard.
Extraction of Insoluble Organic Matrices with Anhydrous HF
Freeze-dried insoluble organic matrix material (3.5 mg) was mixed with freshly condensed anhydrous HF (1 ml) and incubated on ice for 1 h. The HF was evaporated using a gentle nitrogen stream followed by a final drying step in a Speedvac (25 °C, −0.8 bar, 1 h). The dry samples were immediately dissolved in an appropriate volume of 50 mm ammonium bicarbonate until the pH of the solution was neutral. The HF extract was analyzed by SDS-PAGE using 16% Tris-Tricine gels (40), which were stained using Coomassie G-250 dissolved in 50% methanol and 10% acetic acid.
Proteomics Analysis
HF extracts from the insoluble organic matrices were dissolved in 10 mm ammonium bicarbonate (pH 7.4), and in separate experiments, aliquots were digested with trypsin, chymotrypsin, Asp-N, or Glu-C endoproteases (final concentration of enzyme, ∼50 ng·μl−1) for 18 h at 37 °C and then dried in a vacuum centrifuge. For LC-MS/MS analyses, digests were dissolved in 50 μl of 5% aqueous formic acid, and 5 μl were injected into a Dionex Ultimate 3000 nano-HPLC system (Thermo Scientific). The system was equipped with a 300-μm inner diameter × 5-mm trap column and a 75 μm × 15-cm analytical column (Acclaim PepMap100 C18, 5 μm/100 Å and 3 μm/100 Å, respectively, both from Thermo Scientific). Solvents A and B were 2% acetonitrile and 60% acetonitrile in aqueous 0.1% formic acid, respectively. Samples were loaded on the trap column for 5 min with the flow of solvent A at 20 μl·min−1. Upon loading, the trap column was switched to the separation column, and the flow rate was set to 200 nl·min−1. Protein digests were analyzed using a 95-min elution program: 0% B for 15 min; linear gradient 0–60% of B in 65 min; maintained at 60% of B for 3 min; re-equilibration of the columns with 0% B for another 12 min. Spectra were acquired on an LTQ Orbitrap XL mass spectrometer (Thermo Scientific) in data-dependent acquisition mode. Fourier transform MS survey scans were acquired within the range of m/z = 300–1,700 with a target mass resolution of 60,000 (full width at half-maximum) at m/z = 400. The automated gain control target ion count was set to 5 × 105 for Fourier transform MS scans with maximal fill time of 500 ms. MS/MS spectra were acquired at the linear ion trap under normalized collision energy of 35%, dynamic exclusion time of 30 s, and precursor ion isolation width of 2 Da. Spectra were recorded in centroid mode, and target ion count was set to 1 × 104 with maximal fill time of 100 ms. A data-dependent acquisition cycle consisted of Fourier transform MS survey spectrum followed by “top 5” MS/MS spectra with a fragmentation threshold of 3,000 ion counts; singly charged precursor ions were excluded. Lock mass was set to the singly charged ion of dodecamethylcyclo-hexasiloxane (Si(CH3)2O))6 (m/z = 445.120025).
Proteins were identified by MASCOT version 2.2.04 software (Matrix Sciences Ltd.) by searching against a comprehensive (all species) NCBI protein sequences database (compiled in April 2015 and comprising 63,872,600 entries) or, where specified, against a T. pseudonana subset of the UniProt database supplemented with sequences of common protein contaminants (human and sheep keratins, enzymes used for proteolytic cleavage) containing 11,964 entries under the following settings: oxidation (methionine) and N-terminal acetylation as variable modifications; precursor mass tolerance of 5 ppm and MS/MS tolerance 0.6 Da; enzyme specificity “None”; up to 2 missed cleavages allowed; instrument profile “ESI-TRAP”; charge state inclusion “2+ and 3+”; search against decoy database “allowed.” Protein hits matching at least two peptides with a minimum score of 30 were reported. One-peptide hits were considered if the peptide ion score was above 45 and validated by manual inspection of MS/MS spectra and by independent de novo interpretation followed by MS BLAST (41). MS/MS spectra were subjected to batch de novo sequencing by the PepNovo program (42). Up to seven candidate peptide sequences for each interpreted tandem spectra were considered, and only candidates with a sequence quality score of 6 or above were used for subsequent MS BLAST searches. Candidate sequences were submitted to the MS BLAST server and searched against a non-redundant database using the LC-MS/MS presets option. To increase identification confidence, we only considered high scoring segment pairs with scores of 60 or above (43).
Mass Spectrometric Analysis of Modified Lysines
The acid hydrolysate of the insoluble organic matrices (see “Amino Acid Analysis”) was dried in a SpeedVac at 50 °C and dissolved in H2O. Samples were diluted in H2O/MeOH/HCO2H (49.9/50/0.1) and subsequently analyzed by electrospray ionization mass spectrometry using an AmaZon speed ETD mass spectrometer (Bruker). Samples were injected by direct infusion at a flow rate of 5 μl·min−1 using a standard electrospray ionization source. Measurement parameters were as follows: 4,500 V capillary voltage, 500 V end plate offset, 10 p.s.i. nebulizer, 4 liters·min−1 dry gas, 180 °C dry temperature, “Enhanced Resolution” scan mode, 50–500 m/z range, 5 averages, “Rolling Averaging” on, positive ion mode, 200,000 ICC target, 50-ms accumulation time, collision-induced fragmentation (cut-off was 27% of precursor mass, SmartFrag was enhanced, start amplitude was 80%, end amplitude was 120%, 50-ms fragmentation time), AutoMS(n) set to n = 3 in Xtreme scan mode, scan begin at 50 m/z, fragmentation amplitude of 60% (MS2) and 100% (MS3), number of precursor ions = 3, threshold absolute of 25,000 (MS2) and 2,500 (MS3), threshold relative of 5%, smart precursor selection on, and active exclusion on (after 1 spectrum, release after 0.2 min). MS data were averaged over a 5-min time window and charge-deconvoluted using the “Peptides/Small Molecule” settings in the data analysis software (Bruker). Only the peaks at m/z = 333.34 and m/z = 413.30 had doubly charged peaks at m/z = 167.17 and m/z = 207.16, respectively. Fragment ions were detected using the APEX peak finder and AutoMS(n) algorithms in the Data Analysis software (Bruker; intensity threshold was 100,000).
Results
Functional Characterization of Cingulins
The striking structural features of cingulins are their repetitive structures and the high abundance of tryptophan and tyrosine. They have a high content of hydrophilic amino acid residues and are predicted to be intrinsically disordered proteins (29). Lysine- and serine-rich domains (i.e. silaffin-like regions) alternate with domains that are acidic and rich in tyrosine (Y-type cingulins) or both tryptophan and tyrosine (W-type cingulins) (29). We hypothesized that cingulins may have the following properties: (i) the capability to induce silica precipitation at mildly acidic pH (i.e. pH 5–6) due to the presence of silaffin-like domains and (ii) a high propensity to form supramolecular aggregates through ionic interactions between the polycationic silaffin-like domains and the polyanionic acidic domains and through interactions between the aromatic amino acid residues.
It has not yet been possible to investigate these properties with native cingulins because of the insolubility of the organic microrings, which prevents the extraction of intact proteins. To obtain first insight into the properties of cingulins, we have recombinantly expressed a W-type cingulin, rCinW2, and a Y-type cingulin, rCinY2, in E. coli (supplemental Fig. S2). Given the high similarity in amino acid composition and domain arrangement among all cingulins, we expect the properties of rCinY2 and rCinW2 to be representative of the properties of all Y-type and W-type cingulins, respectively.
During the purification of the recombinant cingulins, it became evident that both proteins required the presence of sufficient concentrations of NaCl to remain soluble in aqueous buffers. This solubility behavior is different from that of recombinant and native silaffins, which formed very stable aqueous solutions also in the absence of salt ions (24, 26, 35, 44). Dynamic light scattering revealed that both rCinY2 and rCinW2 had a tendency to self-aggregate, which was dependent on the pH and salt concentration of the solution. At pH 7.7 and low salt concentration (150 mm NaCl), the hydrodynamic radii of rCinW2 and rCinY2 were determined as 4.4 ± 1.0 and 3.8 ± 0.6 nm, respectively (Fig. 1A). These radii were about twice as large as the predicted hydrodynamic radii of 2.3 nm (CinW2) and 1.9 nm (CinY2) that were calculated according to Dill et al. (45), assuming spherical shapes of the proteins. This seemed to suggest that recombinant cingulins are present in solution as dimers. However, cingulins are predicted to be intrinsically disordered, non-spherical proteins (29), and thus the monomers of rCinY2 and rCinW2 should exhibit larger radii than predicted for spherical proteins of the same molecular mass. Therefore, we assume that rCinW2 and rCinY2 are monomers at pH 7.7. When the cingulin solutions were acidified to pH 5.5, aggregates with a radius of 171 ± 9 nm were formed in rCinW2 solutions, whereas rCinY2 remained monomeric (Fig. 1A). The aggregation behavior of rCinW2 and rCinY2 was reversed in solutions with high salt concentration (1,000 mm NaCl); at both pH conditions, rCinW2 was monomeric (radius, 4.4 ± 1.0 nm), whereas aggregates of 356 ± 116 nm were observed in the rCinY2 solution at pH 5.5 (Fig. 1B).
FIGURE 1.
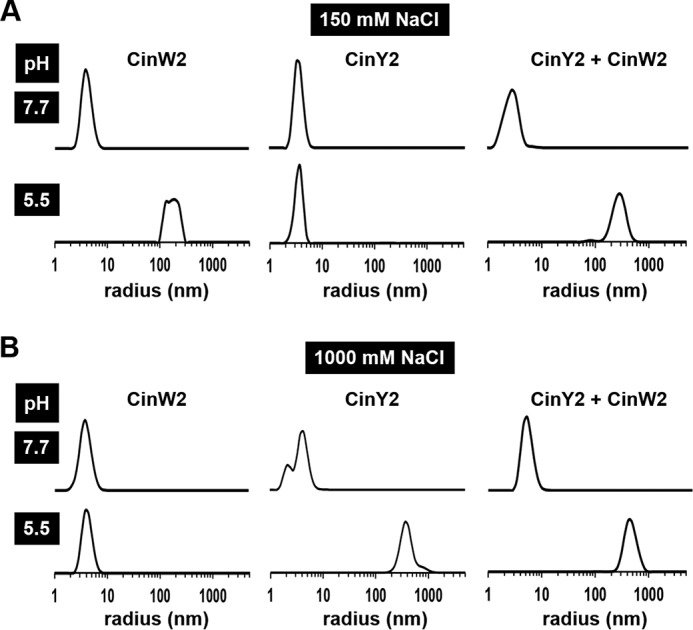
pH-induced aggregation of recombinant cingulins. Each cingulin was present at a concentration of 4 μm at pH 7.7 in the presence of 150 mm NaCl (A) or 1,000 mm NaCl (B). Solutions were titrated to pH 5.5 by the addition of acetic acid. The protein sizes in pure solutions of rCinY2 and rCinW2 and in a 1:1 mixture of the cingulins (rCinY2 + rCinW2) were determined by dynamic light scattering.
Dynamic light scattering was then further employed to investigate whether rCinY2 and rCinW2 interact with each other in solution. In an equimolar mixture of rCinW2 and rCinY2 at pH 7.7 (concentration of each protein was 4 μm), only one type of particle was detected, which exhibited a radius of 3.2 ± 0.7 nm at low salt concentration and 5.9 ± 1.4 nm at high salt concentration (Fig. 1, A and B). The particle sizes were within the error range essentially identical to those of the individual recombinant proteins in solution, suggesting that rCinW2 and rCinY2 remained monomeric at pH 7.7. When the cingulin mixtures were acidified to pH 5.5, 266 ± 67 nm (at 150 mm NaCl) and 412 ± 107 nm (at 1,000 mm NaCl) sized aggregates were observed, which was expected from the measurements with the pure proteins (Fig. 1, A and B). Surprisingly, monomeric proteins were entirely absent at pH 5.5 (Fig. 1, A and B), although pure rCinY2 and pure rCinW2 were incapable of aggregation at low salt concentration and high salt concentration, respectively (see above). These data suggest that rCinY2 and rCinW2 interact with each other at pH 5.5 and form large mixed aggregates. Aggregation of cingulins triggered by a pH shift down to pH 5.5 is of physiological relevance, because the silica deposition vesicles (SDVs) of diatoms, which are the intracellular site for biological silica formation, are believed to be acidic compartments (46, 47).
To investigate whether cingulins may be directly involved in silica biogenesis, rCinY2 and rCinW2 were tested for silica formation activity in vitro. Recombinant silaffin rSil3, which had previously been shown to possess silica formation activity (35), was used as a reference. Silica formation was studied at pH 5.5 in the presence of 150 mm NaCl. The low pH was meant to mimic the pH of the SDVs, and the presence of NaCl was required for solubility of the cingulins. Due to the low solubility of rCinW2, its silica formation activity could only be measured up to a protein concentration of 4 μm. Both rCinW2 and rCinY2 possessed silica formation activities, but they were substantially lower than the silica formation activity of rSil3 (Fig. 2A). The silica formation activity of rCinY2 and rCinW2 was ∼60 and ∼15%, respectively, of the rSil3 activity. To study the silica formation activity of a mixture of cingulins, the concentration of rCinY2 was varied between 1 and 20 μm in the presence of a constant concentration of CinW2 (3 μm). Interestingly, when the cingulin mixture was composed of equimolar amounts or rCinW2 and rCinY2, the silica formation activity was not the sum of the activities of the individual cingulins but instead about the same or even lower than the activity of rCinY2 on its own (Fig. 2A, inset). The result suggested that the rCinY2 and rCinW2 molecules interacted with each other and exerted a slight inhibitory effect on each other's silica formation activity. This interpretation is consistent with the dynamic light scattering data, which indicated the formation of mixed aggregates composed of both rCinY2 and rCinW2 molecules (see Fig. 1, A and B). Scanning electron microscopy analysis of the cingulin-induced silica particles revealed that the solutions of pure cingulins (each at 3 μm) produced spherical silica particles with average diameters of 674 ± 122 nm (n = 117) for rCinW2 (Fig. 2B) and 185 ± 34 nm (n = 171) for CinY2 (Fig. 2C). The silica particles produced by the mixture of cingulins (each cingulin at 3 μm) were also mainly spherical but exhibited a rather heterogeneous size distribution, and fused particles were much more abundant than in the material obtained from pure cingulins (Fig. 2D). Very similar silica structures were obtained when a 3.3:1 ratio of CinY2 (10 μm) and CinW2 (3 μm) was used. The silica particles produced by recombinant cingulins are in the same size range as those that were previously produced by native silaffins, silacidins, and long-chain polyamines (24, 25, 27, 44). Although these silica spheres are too large to be physiologically relevant, they are testimony to the recombinant cingulins' ability to promote rapid silica growth in vitro.
FIGURE 2.
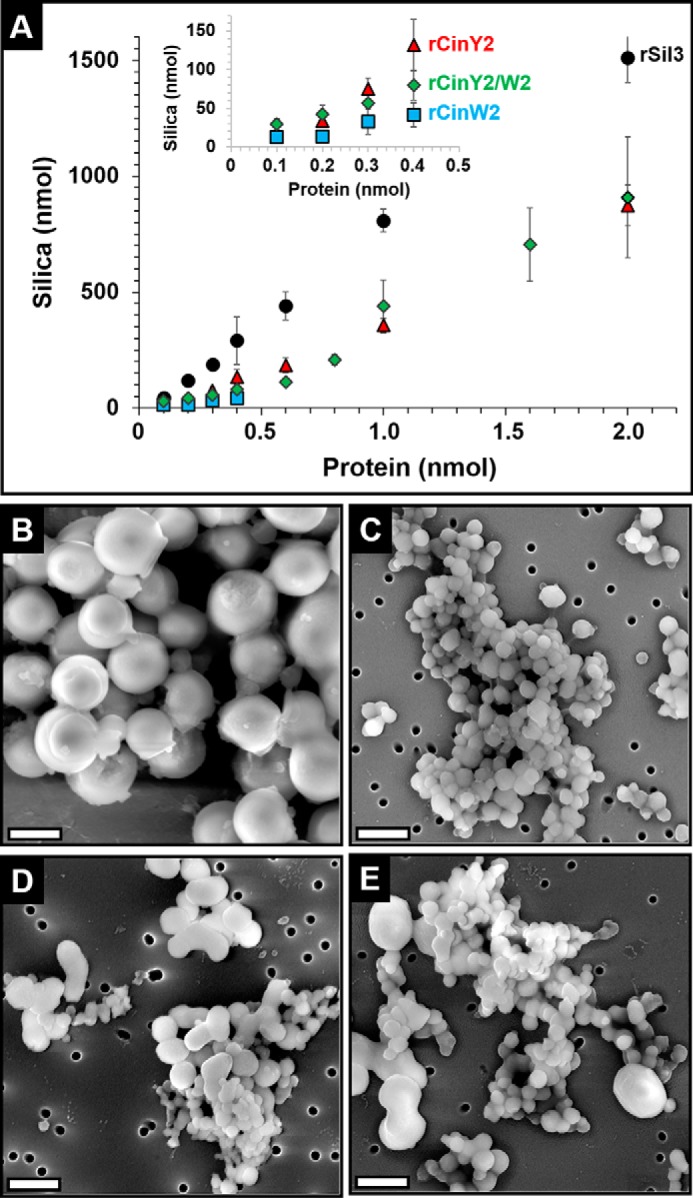
Silica formation by recombinant cingulins. Silica formation assays were performed in 50 mm sodium acetate/acetic acid, pH 5.5, in the presence of 150 mm NaCl. A, quantification of the silica formed in the presence of rSil3 (black circles), rCinY2 (red triangles), rCinW2 (blue squares), and a cingulin mixture (green diamonds) containing a constant concentration of rCinW2 (3 μm) and varying amounts of rCinY2. B–E, scanning electron microscopy images of precipitates formed by 3 μm rCinW2 (B), 3 μm rCinY2 (C), a cingulin mixture containing 3 μm rCinY2 + 3 μm rCinW2 (D), and 10 μm rCinY2 + 3 μm rCinW2 (E). Scale bars, 500 nm.
Accessibility of Organic Microrings in Biosilica
To gain insight into the role of the cingulin containing organic microrings in vivo, we have investigated their association with the silica of the girdle bands. It has been debated whether the organic microrings are located on the proximal surface of the girdle band silica (30) or embedded inside it (29). Therefore, we analyzed the accessibility of microrings in cingulin-GFP-expressing transformant strains using a polyclonal anti-GFP antibody as a probe. To maximize the possibility that the location of cingulin-GFP fusion proteins properly reflects the location of the endogenous cingulins and thus the organic microrings, CinY2-GFP and CinW2-GFP were expressed under the control of their native promoters Pciny2 and Pcinw2, respectively.
Pciny2-driven expression of CinY2-GFP resulted in a much more confined location of the fusion protein compared with the previously reported expression of CinY2-GFP under the control of the nitrate reductase promoter (Pnr2) (29) (Fig. 3A). Under the control of its native promoter, CinY2-GFP was located only at the last girdle band of a complete theca (the width of a single girdle band is ∼700 nm (48)). In contrast, expression of CinW2-GFP under control of its native promoter, Pcinw2, resulted in incorporation of the fusion protein into all parts of the girdle band region (Fig. 3A). This location was essentially identical to the previously reported location of CinW2-GFP expressed under the control of the nitrate reductase promoter, Pnr2 (29) (Fig. 3A).
FIGURE 3.

Localization of CinY2 and CinW2 in T. pseudonana biosilica. A, promoter-dependent location of cingulin-GFP fusion proteins expressed in T. pseudonana. Shown are confocal fluorescence microscopy images of live cells (girdle band view) at different stages of the cell cycle expressing CinY2 and CinW2 under control of the nitrate reductase promoter/terminator cassette (Pnr) or their native promoters Pciny2 and Pcinw2, respectively. Green, location of the cingulin-GFP fusion proteins; red, chloroplast autofluorescence. For orientation, the middle column shows schematic images of cross-sections of T. pseudonana cells at the respective cell cycle stages: silica (black symbols), plasma membranes (blue lines), and cytosol (gray). For clarity, intracellular organelles were omitted in the schematic drawings. B, accessibility of organic microrings in T. pseudonana biosilica. Biosilica and insoluble organic matrices were isolated from transformants expressing CinY2-GFP or CinW2-GFP under control of their native promoters. The biosilica and the organic matrices were subjected to immunolabeling using anti-GFP as primary antibody and an Alexa647-labeled secondary antibody. GFP and Alexa647 fluorescence was quantified in individual biosilica and organic microring particles. The right column shows the overlays of the images from the other three columns. Scale bars, 2 μm.
The accessibility of microrings in biosilica containing CinY2-GFP or CinW2-GFP was investigated by incubating the isolated biosilica with anti-GFP antibodies as the primary antibody, followed by an AlexaFluor647-labeled secondary antibody. The AlexaFluor647 fluorescence intensity in individual valves (Fig. 3B) was determined and served as a quantitative measure for accessibility of the microrings for the antibody. The same immunolocalization experiment was performed with isolated microrings, and the resulting Alexa647 fluorescence intensity (Fig. 3B) served as a reference value for maximum microring accessibility under the assay conditions. For each analyzed individual object (i.e. biosilica particle, microring), the ratio of the RFIs of AlexaFluor647 and GFP was calculated, thereby normalizing the immunolabeling intensity to the amount of antigen that was present in each object (Table 1). Therefore, the ratio of the RFI for biosilica (RFIBS) and the RFI for microrings (RFIMR) indicates which area fraction of a biosilica-associated microring is accessible to the antibody molecules. For CinY2-GFP and CinW2-GFP, the RFIBS/RFIMR ratios had similar values of 0.25 and 0.33, respectively (Table 1), which indicated that in biosilica, 66–75% of the organic microring area is shielded from the binding of antibody molecules.
TABLE 1.
Quantification of microring accessibility in T. pseudonana biosilica
The fluorescence intensities were determined from antibody binding experiments using epifluorescence microscopy. n = number of biosilica and microring particles analyzed. For RFI data, the S.E. is provided. BS, biosilica; MR, microring.
| Sample | RFI | RFIBS/RFIMR | |
|---|---|---|---|
| CinY2 | Biosilica | 0.40 ± 0.05 (n = 17) | 0.25 |
| Microrings | 1.63 ± 0.12 (n = 12) | 0.25 | |
| CinW2 | Biosilica | 0.78 ± 0.04 (n = 19) | 0.33 |
| Microrings | 2.33 ± 0.18 (n = 15) | 0.33 |
Identification of a Valve-derived Insoluble Organic Matrix
Insoluble organic matrices were previously shown to be associated with both types of diatom biosilica building blocks: girdle bands and valves (29–31). It has therefore been puzzling that T. pseudonana appeared to contain only a girdle band-associated insoluble organic matrix and no organic matrix that is specifically associated with the valve. Upon careful inspection of the insoluble organic matrix material with scanning electron microscopy, occasionally objects could be seen that were clearly structurally different from the organic microrings (compare Fig. 4 (A–C) with Fig. 7A) and the chitin meshwork (28). Each of these distinctive objects had a plate-like shape defined by a single ring-shaped filament and 9–15 regularly spaced dots close to the filament ring. One dot was positioned close to the circle's center, and variable amounts of irregularly shaped material inside the ring were often present (Fig. 4, A–C). The diameters of the filament rings (5.9 ± 0.5 nm, n = 34) and the positioning of the dots in the plate-like insoluble organic matrix matched the diameter (5.4 ± 0.7 nm, n = 24) and fultoportulae pattern of the of biosilica valves (Fig. 4D). Electron-dispersive x-ray analysis confirmed that the plate-like structures were free of silicon demonstrating demineralization of the organic material (Fig. 4, E and F). The central part of the plate-like organic matrix did not resemble the porous, ridged biosilica pattern of the T. pseudonana valve. Instead, a non-continuous, randomly structured organic material was present (Fig. 4, A–C). Due to its shape, size, and the characteristic fultoportulae-like dot pattern, we regard it as evidence that the plate-like insoluble organic matrix is associated with the valve biosilica in vivo.
FIGURE 4.

Electron microscopy and elemental analysis of biosilica and biosilica-derived organic material from T. pseudonana. A–C, scanning electron microscopy images of plate-like structures that were present in the organic material obtained after NH4F treatment of biosilica adsorbed onto platinum-coated filter membranes (note that the ∼100-nm sized holes that are scattered throughout each image are the pores in the membrane). The white arrowheads in A indicate regularly spaced dark spots that are arranged in a circular fashion close to the rim of the plate, and the white arrow points to the dark spot near the center of the plate. D, transmission electron microscopy image of a biosilica valve. The yellow arrowheads indicate the position of the fultoportulae (i.e. tubular structures near the edge of the valve), and the yellow arrows point to the position of two central fultoportulae (note that a T. pseudonana valve can contain 0, 1, or 2 central fultoportulae (48)). E and F, electron dispersive x-ray analysis of the material retained on the membrane surface after NH4F treatment of the biosilica (E) and of the biosilica adsorbed on the membrane before NH4F treatment (F). Scale bars, 1 μm.
FIGURE 7.

Proposed mechanism for biogenesis of the organic microrings and the girdle band biosilica. A, scanning electron microscopy image of an organic microring on a gold-coated filter membrane (note that the dark dots are the pores of the filter membrane). B, it is assumed that the organic microrings are constructed from organic nanorings. Each nanoring (top) is composed of a 2-nm-thick organic layer (orange) that is delineated by a 6-nm-thick filament (green) and would be responsible for templating the morphogenesis of a single girdle band (bottom). The filamentous rim would template the pore-free regions, whereas the organic layer would direct formation of silica with pore nanopatterns. C, detail of a cell in cross-section containing developing girdle band SDVs and mature girdle bands. (Other organelles are omitted for clarity.) The cytosol is depicted in gray. The color code of the organic nanoring inside the girdle band is the same as in B. The organic nanoring templates the morphogenesis of girdle band silica. After exocytosis, the newly synthesized girdle band becomes attached to the existing girdle band region of the hypotheca, presumably through covalent cross-linking between surface-exposed domains of the filaments.
Biochemical Composition
To gain insight into the biochemical composition of the insoluble organic microrings and microplates, we analyzed their amino acid and monosaccharide composition and performed proteomics analysis of the material. To avoid contamination of this material by the silica-associated chitin meshwork (28), exhaustive chitinase treatment of the insoluble organic material was performed prior to the biochemical analysis. The total amino acid composition was determined after acid hydrolysis of the chitinase-treated insoluble organic matrices (supplemental Table S1). Glycine and serine were by far the most dominant amino acids, which together constituted more than 50% of the amino acids in the insoluble organic matrix material. Previous ζ potential measurements (at pH 7.0) indicated a predominance of negatively charged groups in the organic matrices (29), which is consistent with the observations that (i) aspartate and glutamate were drastically more abundant than lysine, histidine, and arginine (supplemental Table S1), and (ii) O-phosphoserine and O-phosphothreonine were present (note that phosphorylated amino acids were detected after limited acid hydrolysis, as described under “Experimental Procedures”). However, the surplus of negative charges in the organic matrices was not as drastic as suggested by the composition of standard amino acids. First, mass spectrometric analysis indicated that post-translationally modified lysines that carry polyamine chains are highly abundant (Fig. 5, A and B). Second, a fraction of aspartate and glutamate may have been derived from glutamine and asparagine residues, which become completely deaminated during acid hydrolysis.
FIGURE 5.

Mass spectrometric analysis of post-translationally modified lysines in the insoluble organic matrices. A, electrospray ionization mass spectrum of a total acid hydrolysate of the insoluble organic matrices (positive ion mode). The italicized numbers indicate the m/z values of singly positively charged polyamine-modified lysines. The numbers highlighted with a gray background are m/z values for the singly positively charged amino acids lysine (147.11), ϵ-N-dimethyl-δ-lysine (175.15), arginine (175.15), ϵ-N-trimethyl-δ-lysine (189.16), and ϵ-N-trimethyl-δ-hydroxylysine (205.19). B, proposed chemical structures of the two polyamine-modified δ-hydroxylysine isomers with m/z = 333. The position of the methyl group that differs in isomers 1 and 2 is highlighted in gray. C, chemical relationships between the polyamine-modified lysine residues from the insoluble organic matrices.
The chemical structures of the polyamine-modified lysines (Fig. 5B) were determined by mass spectrometry (MS2 and MS3) using collision-induced fragmentation (supplemental Fig. S4). This revealed that the m/z = 333 ion is a mixture of two constitutional isomers that are derived from δ-hydroxylsine (Fig. 5B). Isomer 1 has previously been identified in native silaffin tpSil3 from T. pseudonana (49) and differs from isomer 2 only by the position of one methyl group (Fig. 5B). The three ions with m/z values of 315, 413, and 431 are closely related to isomers A and B and were probably generated in vitro during acid hydrolysis. Ion m/z = 315 can be explained by loss of the hydroxy group due to water elimination (−18 Da) from isomers 1 and 2 (Fig. 5C). Complexation of a single phosphoric acid molecule (+98 Da) by each of the dehydrated isomers generated ion m/z = 413 (Fig. 5C). Ion m/z = 431 is a mixture of isomers 1 and 2 each in a complex with one phosphoric acid molecule (Fig. 5C). Phosphate was abundantly present in the insoluble organic matrices amounting to 0.075 mg of phosphate/mg of matrix, which corresponds to ∼0.5 mol of phosphoric acid/mol of amino acid in the hydrolysate.
Monosaccharide analysis revealed a complex carbohydrate content of the insoluble organic matrices, including hexoses, pentoses, deoxy sugars, amino sugars, and uronic acids (supplemental Table S2). By comparing the amino acid and monosaccharide content, it could be estimated that in the insoluble organic matrices, the molar ratio of protein to carbohydrate was ∼3:1.
We hypothesized that the insolubility of the organic microrings and organic miocroplates is due to covalent cross-links between their protein components, which may include O-phosphoester bonds and O-glycosidic bonds. Such bonds can be efficiently cleaved by anhydrous HF without affecting the polypeptide backbone (50). Indeed, anhydrous HF treatment of the insoluble organic matrices extracted several components (supplemental Fig. S5). The HF extract was subjected to proteomics analysis using in-solution digests with site-specific endoproteases and nano-LC-MS/MS, which led to the identification of 19 proteins (supplemental Table S3). Six of these were regarded as obvious contaminants because they were predicted to be components of the plastidal photosynthesis machinery (supplemental Table S3). Another protein, TP-ID21233, was also regarded as a likely contaminant, because it lacked an N-terminal signal peptide for import into the secretory pathway (supplemental Table S3), which is a hallmark of all biosilica-associated proteins identified so far (21, 27, 29). Each of the remaining 12 proteins contained a predicted N-terminal signal peptide. Among these are five previously identified proteins: four cingulins (CinY1, CinY2, CinY3, and CinW3) and the silaffin tpSil1 (Table 2 and Supplemental Table S3). Obviously, the presence of cingulins was expected, but the presence of tpSil1 was somewhat surprising, because silaffins are known to be solubilized by ammonium fluoride treatment of the biosilica (21, 29). Silaffin tpSil1 has previously been shown to be specifically associated with the valve biosilica (51). Therefore, it is conceivable that a subset of tpSil1 may become covalently cross-linked to the valve-derived insoluble organic matrix during silica biogenesis in vivo. The remaining seven proteins in Table 2 have no predicted functions, share no significant sequence similarity with non-diatom proteins, and were named SiMat1–7 (for silica matrix).
TABLE 2.
Properties of predicted extracellular/cell surface proteins in the HF extract from the insoluble organic matrices (organic microrings and organic microplates)
Y-domain, tyrosine-rich, acidic domain; W-domain, tryptophan-rich, acidic domain; Y/W-domain, tyrosine- and tryptophan-rich, acidic domain. The protein identification number for the Uniprot database (UP-ID) and the T. pseudonana JGI database version 3.0 (TP-ID) are shown. For all proteins, the predicted molecular masses (kDa) and predicted isoelectric points (pI) after removal of the N-terminal signal peptide are provided. SLFP, silaffin-like protein.
| Name | UP-ID | TP-ID | Mass (kDa) | pI | Sequence features |
|---|---|---|---|---|---|
| SiMat1 (SFLP44) | B8BT90 | 21094 | 24.5 | 5.2 | 7 × KXXK, 3 × Y-domain |
| SiMat2 (SFLP84) | B8CGN0 | 25898 | 43.9 | 5.0 | 12 × KXXK, 2 × RXL |
| SiMat3 (SFLP52) | B8BZU6 | 22349 | 81.0 | 4.7 | 9 × KXXK, 3 × Y/W-domain, 1 × W-domain, 4 × RXL |
| SiMat4 | B8CGR0 | 25912 | 54.8 | 5.2 | 3 × KXXK, 1 × RXL, 11% P |
| SiMat5 | B8BXT7 | 21757 | 73.9 | 4.3 | 1 × RXL |
| SiMat6 | B8CBZ7 | 24761 | 19.0 | 4.7 | 1 × RXL, 7% Y |
| SiMat7 | B8CBQ8 | 24710 | 45.3 | 4.5 | 1 × RXL, 11% N |
| CinY1 | F2YBR7 | 12168 | 28.1 | 8.8 | 9 × KXXK, 1 × RXL, 3 × Y-domain |
| CinY2 | F2YBR8 | 25910 | 23.4 | 9.4 | 10 × KXXK, 1 × RXL, 3 × Y-domain |
| CinY3 | F2YBR9 | 25917 | 27.8 | 5.7 | 10 × KXXK, 1 × RXL, 7 × Y-domain |
| CinW3 | B8CEX1 | 25649 | 58.0 | 7.8 | 30 × KXXK, 1 × RXL 9 × Y/W-domain, 10 × W-domain |
| tpSil1 | Q5Y2C2 | 11360 | 48.9 | 5.6 | 2 × KXXK, 2 × RXL |
SiMat1–4 contain several KXXK tetrapeptide motifs (where X represents Ser, Gly, or Ala) (supplemental Fig. S6) that are also present in cingulins and silaffins (29). KXXK motifs have been shown (i) to exhibit silica formation activity whenthe lysine residues carried their natural post-translational modifications (52) and (ii) to be important for intracellular targeting to the silica (51). SiMat1, -2, and -3 have previously been identified in a bioinformatics screen for silaffin-like proteins due to the presence of domains with high serine and lysine content (29) (supplemental Table S2). SiMat5–7 are devoid of KXXK motifs, but each protein contains an RXL motif, which is a proteolytic cleavage site in many diatom biosilica-associated proteins, including silaffins and cingulins (29). All other SiMat proteins except for SiMat1 contain at least one RXL motif (Table 2). SiMat6 has a high content of tyrosine (7%), similar to SiMat1 (8%) and the Y-type cingulins, but otherwise is quite different from the cingulins regarding amino acid composition. SiMat7 is remarkably rich in asparagine (11%), including several clusters with three or more asparagine residues (supplemental Fig. S6). SiMat5 exhibits no striking features regarding amino acid composition or domain arrangement.
Localization of SiMat1
To investigate whether all SiMat proteins are bona fide components of the insoluble organic matrices rather than contaminants, we intend to study their locations in vivo through expression as GFP fusion proteins. Here we have initiated such analysis by expressing a SiMat1-GFP fusion protein (GFP fused to the C terminus) under control of the SiMat1 promoter. Confocal fluorescence microscopy imaging of live cells revealed that SiMat1-GFP was exclusively located in the girdle band region of the cell both during interphase and cell division. The location of the GFP fluorescence during different stages of the cell cycle indicates that SiMat1 is associated with 3–4 girdle bands that are close to the region where the epi- and hypotheca overlap (Fig. 6A). Strong GFP fluorescence is also observed in isolated biosilica and in the insoluble organic matrix material (Fig. 6B). Altogether, these data demonstrate that SiMat1 is a bona fide component of the biosilica-associated, insoluble organic microrings.
FIGURE 6.

Localization of SiMat1-GFP in T. pseudonana. A, confocal fluorescence microscopy images of individual live cells (girdle band view) at different stages of the cell cycle expressing SiMat1-GFP under control of its own promoter. The left column shows an overlay of the GFP fluorescence (green) with the bright field image of the cells. The right column shows the fluorescence image only including the red chloroplast autofluorescence. Scale bars, 2 μm. B, epifluorescence microscopy (top row) and bright field images (bottom row) of biosilica and insoluble organic matrices isolated from SiMat1-GFP-expressing transformant cells. Scale bars, 3 μm.
Discussion
In the present study, we have investigated the molecular composition, assembly, and function of the biosilica-associated nanopatterned organic matrices from T. pseudonana. We have demonstrated that T. pseudonana biosilica contains a second type of biosilica-associated insoluble organic matrix: nano-/micropatterned “organic microplates” that are specifically associated with valves. This observation is in agreement with the findings in other diatom species (29, 30, 31), and it seems likely that all diatoms may contain both valve-specific and girdle band-specific insoluble organic matrices.
The insoluble material of the organic microrings and microplates appears to contain covalent cross-links that are composed of HF-sensitive bonds (e.g. O-phosphoesters and O-glycosides), because protein constituents of the insoluble organic matrices became solubilized using anhydrous HF (see supplemental Fig. S5 and Table 2). In agreement with this, phosphoamino acids and a complex variety of carbohydrates are constituents of the insoluble organic matrices. HF treatment solubilized ∼60% (w/w) of the insoluble organic matrix material, suggesting that HF-insensitive covalent links (e.g. isopeptide bonds) also contribute to the insolubility of the organic microrings and microplates.
Proteomics analysis of the HF extract from the insoluble organic matrix material found four of the six previously identified cingulins, a previously identified silaffin, and seven proteins that have a predicted extracellular/cell surface location (SiMat1–7). SiMat1–4 share the characteristic KXXK (where X represents Ser or Gly) tetrapeptide motif with cingulins and silaffins (see Table 2), and therefore an involvement in silica biomineralization seems likely. This assumption has been further supported for SiMat1 by demonstrating its association with girdle bands and its incorporation into the silica forming organic microrings (see Fig. 6). SiMat1 does not share sequence similarity with cingulins, but it closely resembles the Y-type cingulins regarding the repetitive alternation of KXXK-bearing domains and tyrosine-rich acidic domains (see supplemental Fig. S6). Due to these similarities and because SiMat1 is present in the same subcellular compartment as cingulins (i.e. organic microrings), we regard this protein as the seventh member of the cingulin protein family, and it is therefore renamed CinY4.
The N-terminal 180 amino acids of SiMat3 share similarity with W-type cingulins, but the remaining part of the protein (625 amino acids) is clearly different from cingulins. SiMat2 and SiMat4–7 have no similarities regarding amino acid composition or domain arrangement to cingulins or any other known diatom biosilica-associated proteins from T. pseudonana or other diatom species (21, 27, 31, 53). Previous transcriptomics and proteomics screens that aimed at the identification of genes involved in diatom silica metabolism identified many dozens of candidates (54–56). Among these are the genes encoding SiMat2 and SiMat6, which show a 16-fold down-regulation and 36-fold up-regulation, respectively, upon silicic acid limitation (55). Further experimental data are required (e.g. through GFP tagging) to determine whether the SiMat2–6 proteins are bona fide components of the insoluble organic matrices or accidental contaminants.
Based on the results from the amino acid analyses (see supplemental Table S1), we hypothesize that cingulins are major components of the organic microrings and organic microplates of T. pseudonana. The content of the four most abundant amino acids (glycine, serine, aspartate, and tyrosine) matches quite well the amino acid composition of cingulins (25% Gly, 25% Ser, 8% Asp, and 5% Tyr) but not the amino acid compositions of the other SiMat proteins. A striking exception is the lysine content, which is drastically lower in the organic microrings and organic microplates than in the predicted sequences of cingulins (10% Lys). An explanation for this discrepancy is that the cingulins' lysine residues undergo post-translational modification in vivo, yielding the polyamine-modified lysine derivatives that were identified in the hydrolysate (see Fig. 5).
Regarding monosaccharide composition, the organic microrings and organic microplates of T. pseudonana differ significantly from the previously studied insoluble organic matrices from diatoms. Hexoses (especially mannose) constitute 74–89% of the insoluble organic matrix carbohydrates of the diatoms Coscinodiscus radiatus, Nitzschia curvilineata, Amphora salina, and Triceratium dubium) (30) but only 32% in T. pseudonana (see supplemental Table S2). The T. pseudonana insoluble organic matrices contain high amounts of uronic acids (27%) and pentoses (20%) (see supplemental Table S2), which are far less abundant in the other diatoms (1–12% uronic acids, 1–6% pentoses) (30). A common theme in the biosilica-associated insoluble organic matrices of all diatoms is the striking complexity in monosaccharide composition. This may reflect different biosynthetic origins of the carbohydrate moieties, which may become incorporated into the insoluble organic matrix as polysaccharides, glycoproteins, or glycolipids. Additionally, it is possible that the insoluble organic matrices from valves and girdle bands may differ substantially in carbohydrate composition.
The organic microrings were previously shown to have silica formation activity at pH 5.5 (29), a pH level that is intended to mimic the acidic pH conditions of the SDV lumen (46, 47). Here we demonstrate that the polypeptide backbones of cingulins CinY2 and CinW2 are capable of silica formation at pH 5.5, thus providing a molecular explanation for the silica formation activity of the organic microrings. Given their close structural similarity, we regard it as likely that the polypeptide backbones of all other cingulins, including the newly identified CinY4 (SiMat1), have silica formation activities. The reason for the cingulins' silica formation activities probably resides within the silaffin-like lysine-rich domains, which are always present in multiple copies (see Table 2 and supplemental Figs. S2 and S6). Proteins and synthetic polymers rich in amino groups generally have the capability to promote silica formation in vitro (58–61). Furthermore, it has been shown that the lysine residues of the silaffin-derived peptide R5 are essential for silica formation activity (62).
However, silica formation by recombinant polypeptides at pH 5.5 was somewhat unexpected, because silica formation by native silaffin natSil1A1 at pH 5.5 was absolutely dependent on the presence of post-translational modifications (polyamine-modified lysines and phosphorylated serines) (23, 24). This raises the question of why the recombinant cingulins, which entirely lack post-translational modifications, were able to form silica at pH 5.5. The silica-forming activity of natSil1A1, which contains only 15 amino acid residues, has been ascribed to the presence of a high number of positive charges and the ability to form large soluble aggregates (24). Aggregation of natSil1A1 was accomplished by intramolecular ionic interactions most likely between the polyamine-modified lysines and the phosphoryl groups (24). It is believed that within such aggregates, the silicic acid molecules become concentrated and their condensation becomes catalyzed through interaction with the polyamine moieties (57). At salt and pH conditions that were intended to mimic the interior of SDVs, only rCinW2 and not rCinY2 was able to form large aggregates (see Fig. 1A), yet rCinY2 had a significantly higher silica-forming activity than rCinW2 (see Fig. 2A). Therefore, we conclude that the number and density of lysine residues (23.5 mol %) within the KXXK-containing domains of an rCinY2 monomer (240 amino acids; see supplemental Fig. S2) are sufficient to accelerate silica formation. The lower silica-forming activity of rCinW2 compared with rCinY2 (see Fig. 2A) may be due to the higher density of negative charges in rCinW2 (note that the ratio of positive/negative charge is 2.2 in rCinY2 and 1.2 in rCinW2), which may partially screen the lysine residues from interacting with the silicic acid molecules. Intramolecular inhibition of silica formation activity by negatively charged groups has previously been observed for silaffin natSil2 (26).
For reasons explained above, we regard it as likely that most of the lysine residues in native cingulins carry additional positive charges in vivo due to post-translational modification with polyamine residues. This would be expected to enhance the silica formation activities of cingulin aggregates. On the other hand, native cingulins may contain many additional negative charges due to O-phosphorylation of serine and threonine residues and the glycosylation with uronic acids (see supplemental Table S2), which could diminish silica-forming activity due to screening of the positive charges. Assessing the silica formation activity of native cingulins has to await the development of a method for their isolation. Nevertheless, the information about the silica formation activities of recombinant cingulins that has been obtained in the present work provides the baseline for determining the influence of the post-translational modifications on the silica formation activities of native cingulins.
The experiments on the properties of recombinant cingulins, GFP tagging of cingulins in vivo, and cingulin accessibility in biosilica provide information regarding the mechanism of biogenesis of the organic microrings. The assembly of mixed cingulin aggregates (see Fig. 1) triggered by the acidic pH conditions inside the girdle band SDVs may be the first step in the formation of insoluble organic matrices. Aggregates may then become stabilized by covalent cross-linking, which likely involves (but is probably not restricted to) O-phosphoester and/or O-glycosidic bonds. It was previously hypothesized that the organic microrings are assembled in a stepwise fashion by extracellular fusion of organic nanorings (29) (Fig. 7). Each organic nanoring would be associated with the formation of a specific girdle band inside the SDV and remain attached to the girdle band after exocytosis. On the cell surface, the nanoring of the newly exocytosed girdle band would become chemically cross-linked (catalyzed by yet unknown enzymes) to the growing edge of the girdle band region (29) (Fig. 7, B and C). Alternatively, the microrings might be synthesized en bloc as a continuous organic matrix (i.e. not from nanoring subunits) and be added on the proximal surface of the biosilica after exocytosis of the last girdle band. The discovery that CinY2 is associated only with a specific subset of 1–2 girdle bands at all stages of the cell cycle (see Fig. 3A) is fully consistent with a stepwise assembly of the organic microrings from individual nanorings (Fig. 7).
Are nanorings present during silica formation inside the girdle band SDVs (and possibly actively involved in the mineralization process), or are they assembled on the silica surface after completion of this process? The strongly limited accessibility of biosilica-associated microrings for antibody molecules (see Fig. 3B and Table 1) and proteases (29) is consistent with silica covering the majority (at least two-thirds) of the surface of the organic microrings. This would imply that nanorings are present in SDVs during silica formation rather than being added later onto the silica surface. The silica-forming capability of recombinant cingulin aggregates (see Fig. 2) and of microrings (29) is in agreement with an active role of nanorings in silica deposition. However, currently, the possibility cannot be ruled out that layers of biosilica-associated biomolecules (silaffins, silacidins, long-chain polyamines, or chitin) rather than silica are responsible for screening of the organic microrings. Resolving this issue should be feasible through simultaneous high resolution localization of cingulins and the other biosilica-associated biomolecules using electron microscopy or superresolution fluorescence microscopy.
In the present study, we have attempted the structural and functional characterization of a chemically complex and completely insoluble organic matrix. Proteomics analysis of the HF extract identified only four of the six previously known cingulins, and ∼40% of the organic matrix material remained insoluble after HF treatment. Therefore, it is conceivable that the insoluble organic matrices contain additional SiMat proteins besides those that were discovered in the present study. To enable identification of the full set of SiMat proteins, further extraction procedures need to be developed. In addition to studying the locations and in vitro properties of SiMat proteins through the methods described here, it would be desirable to analyze their functions and the functions of cingulins in vivo using gene knockdown or knock-out techniques. We expect that diatom mutants with strongly decreased levels of a silica-forming SiMat protein/cingulin should exhibit reduced silica content and/or altered silica morphology. Combined information from in vitro and in vivo analyses is crucial for elucidating the role of the insoluble organic matrices in the morphogenesis of diatom silica. Such insight could then be utilized for generating genetically engineered diatom mutants with designed silica nanopatterns for a wide variety of applications, including catalysis, chemical sensing, and drug delivery (9, 10, 15, 21, 63).
Author Contributions
N. K. and N. P. conceived the study, and N. K. wrote the paper. A. K., D. P., A. M., and A. Scheffel conducted experiments and analyzed data. N. K., N. P., Anna Shevchenko, and Andrej Shevchenko analyzed data. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We are grateful to Dr. Martina Lachnit and Julia Palm (both from B CUBE, Technische Universität Dresden (TU Dresden)) for expert technical assistance and to the Light Microscopy Facility (BIOTEC/CRTD, TU Dresden) for help with confocal fluorescence microscopy. We are indebted to Thomas Kurth (Electron Microscopy Facility, BIOTEC/CRTD, TU Dresden) and Markus Günther (Biology, TU Dresden) for help with electron microscopy and to Philip Gröger (B CUBE, TU Dresden) for help with epifluorescence microscopy.
This work was supported by the Deutsche Forschungsgemeinschaft through Research Unit 2038 NANOMEE (Grants KR 1853/6-1 and SH 94/4-1). Work in the laboratory of Andrej Shevchenko was supported by the Max-Planck-Gesellschaft. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Tables S1–S3 and Figs. S1–S7.
- RFI
- relative fluorescence intensity
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- SDV
- silica deposition vesicle.
References
- 1. Raven J. A. (1983) The transport and function of silicon in plants. Biol. Rev. 58, 179–207 [Google Scholar]
- 2. Assmy P., Smetacek V., Montresor M., Klaas C., Henjes J., Strass V. H., Arrieta J. M., Bathmann U., Berg G. M., Breitbarth E., Cisewski B., Friedrichs L., Fuchs N., Herndl G. J., Jansen S., Krägefsky S., Latasa M., Peeken I., Röttgers R., Scharek R., Schüller S. E., Steigenberger S., Webb A., and Wolf-Gladrow D. (2013) Thick-shelled, grazer-protected diatoms decouple ocean carbon and silicon cycles in the iron-limited Antarctic Circumpolar Current. Proc. Natl. Acad. Sci. U.S.A. 110, 20633–20638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kröger N., and Sumper M. (1998) Diatom cell wall proteins and the cell biology of silica biomineralization. Protist 149, 213–219 [DOI] [PubMed] [Google Scholar]
- 4. Bäuerlein E. (2003) Biomineralization of unicellular organisms: an unusual membrane biochemistry for the production of inorganic nano- and microstructures. Angew. Chem. Int. Ed. Engl. 42, 614–641 [DOI] [PubMed] [Google Scholar]
- 5. Wilt F. H. (2005) Developmental biology meets materials science: morphogenesis of biomineralized structures. Dev. Biol. 280, 15–25 [DOI] [PubMed] [Google Scholar]
- 6. Sumper M., and Brunner E. (2006) Learning from diatoms: nature's tools for the production of nanostructured silica. Adv. Funct. Mater. 16, 17–26 [Google Scholar]
- 7. Hildebrand M. (2008) Diatoms, biomineralization processes, and genomics. Chem. Rev. 108, 4855–4874 [DOI] [PubMed] [Google Scholar]
- 8. Parkinson J., and Gordon R. (1999) Beyond micromachining: the potential of diatoms. Trends Biotechnol. 17, 190–196 [DOI] [PubMed] [Google Scholar]
- 9. Foo C. W., Huang J., and Kaplan D. L. (2004) Lessons from seashells: silica mineralization via protein templating. Trends Biotechnol. 22, 577–785 [DOI] [PubMed] [Google Scholar]
- 10. Gordon R., Losic D., Tiffany M. A., Nagy S. S., and Sterrenburg F. A. S. (2009) The glass menagerie: diatoms for novel applications in nanotechnology. Trends Biotechnol. 27, 116–127 [DOI] [PubMed] [Google Scholar]
- 11. Losic D., Mitchell J. G., and Voelcker N. H (2009) Diatomaceous lessons in nanotechnology and advanced materials. Adv. Mater. 21, 2947–2958 [Google Scholar]
- 12. Sandhage K. H. (2010) Materials “alchemy”: shape-preserving chemical transformation of micro-to-macroscopic 3-D structures. JOM 62, 32–43 [Google Scholar]
- 13. Nassif N., and Livage J. (2011) From diatoms to silica-based biohybrids. Chem. Soc. Rev. 40, 849–859 [DOI] [PubMed] [Google Scholar]
- 14. Clayton J., Campbell J., Li H., Jiao J., and Rorrer G. (2011) The potential of diatom nanobiotechnology for applications in solar cells, batteries, and electroluminescent devices. Energy Environ. Sci. 4, 3930–3941 [Google Scholar]
- 15. Yang W., Lopez P. J., and Rosengarten G. (2011) Diatoms: self assembled silica nanostructures, and templates for bio/chemical sensors and biomimetic membranes. Analyst 136, 42–53 [DOI] [PubMed] [Google Scholar]
- 16. Kröger N., and Brunner E. (2014) Complex-shaped microbial biominerals for nanotechnology. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 6, 615–627 [DOI] [PubMed] [Google Scholar]
- 17. Nakajima T., and Volcani B. E. (1969) ϵ-N-Trimethyl-l-δ-hydroxylysine phosphate and its nonphosphorylated compound in diatom cell walls. Science 164, 1400–14015783709 [Google Scholar]
- 18. Hecky R., Mopper K., Kilham P., and Degens T. (1973) The amino acid and sugar composition of diatom cell-walls. Mar. Biol. 19, 323–331 [Google Scholar]
- 19. Lowenstam H., and Weiner S. (1989) On biomineralization, Oxford University Press, Oxford [Google Scholar]
- 20. Kröger N., Bergsdorf C., and Sumper M. (1994) A new calcium-binding glycoprotein family constitutes a major diatom cell wall component. EMBO J. 13, 4676–4683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kröger N., and Poulsen N. (2008) Diatoms: from cell wall biogenesis to nanotechnology. Annu. Rev. Genet. 42, 83–107 [DOI] [PubMed] [Google Scholar]
- 22. Sumper M., and Brunner E. (2008) Silica biomineralisation in diatoms: the model organism Thalassiosira pseudonana. Chembiochem 9, 1187–1194 [DOI] [PubMed] [Google Scholar]
- 23. Kröger N., Deutzmann R., and Sumper M. (1999) Polycationic peptides from diatom biosilica that direct silica nanosphere formation. Science 286, 1129–1132 [DOI] [PubMed] [Google Scholar]
- 24. Kröger N., Lorenz S., Brunner E., and Sumper M. (2002) Self-assembly of highly phosphorylated silaffins and their function in biosilica morphogenesis. Science 298, 584–586 [DOI] [PubMed] [Google Scholar]
- 25. Kröger N., Deutzmann R., Bergsdorf C., Sumper M. (2000) Species-specific polyamines from diatoms control silica morphology. Proc. Natl. Acad. Sci. U.S.A. 97, 14133–14138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Poulsen N., Sumper M., and Kröger N. (2003) Biosilica formation in diatoms: characterization of native silaffin-2 and its role in silica morphogenesis. Proc. Natl. Acad. Sci. U.S.A. 100, 12075–12080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wenzl S., Hett R., Richthammer P., and Sumper M. (2008) Silacidins: highly acidic phosphopeptides from diatom shells assist in silica precipitation in vitro. Angew. Chem. Int. Ed. Engl. 47, 1729–1732 [DOI] [PubMed] [Google Scholar]
- 28. Brunner E., Richthammer P., Ehrlich H., Paasch S., Simon P., Ueberlein S., and van Pée K.-H. (2009) Chitin-based organic networks: an integral part of cell wall biosilica in the diatom Thalassiosira pseudonana. Angew. Chem. Int. Ed. Engl. 48, 9724–9727 [DOI] [PubMed] [Google Scholar]
- 29. Scheffel A., Poulsen N., Shian S., and Kröger N. (2011) Nanopatterned protein microrings from a diatom that direct silica morphogenesis. Proc. Natl. Acad. Sci. U.S.A. 108, 3175–3180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tesson B., and Hildebrand M. (2013) Characterization and localization of insoluble organic matrices associated with diatom cell walls: insight into their roles during cell wall formation. PLoS One 8, e61675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Buhmann M. T., Poulsen N., Klemm J., Kennedy M. R., Sherrill C. D., and Kröger N. (2014) A tyrosine-rich cell surface protein in the diatom Amphora coffeaeformis identified through transcriptome analysis and genetic transformation. PLoS One 9, e110369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bidle K. D., and Azam F. (1999) Accelerated dissolution of diatom silica by marine bacterial assemblages. Nature 397, 508–512 [Google Scholar]
- 33. Hamm C. E., Merkel R., Springer O., Jurkojc P., Maier C., Prechtel K., and Smetacek V. (2003) Architecture and material properties of diatom shells provide effective mechanical protection. Nature 421, 841–843 [DOI] [PubMed] [Google Scholar]
- 34. Davis A. K., Hildebrand M., and Palenik B. (2005) A stress-induced protein associated with the girdle band region of the diatom Thalassiosira pseudonana (Bacillariophyta). J. Phycol. 41, 577–589 [Google Scholar]
- 35. Kröger N., Dickerson M. B., Ahmad G., Cai Y., Haluska M. S., Sandhage K. H., Poulsen N., and Sheppard V. C. (2006) Bioenabled synthesis of rutile (TiO2) at ambient temperature and neutral pH. Angew. Chem. Int. Ed. Engl. 45, 7239–7243 [DOI] [PubMed] [Google Scholar]
- 36. Iler R. K. (1979) The Chemistry of Silica, p. 97, John Wiley & Sons, Inc., New York [Google Scholar]
- 37. Poulsen N., Chesley P. M., and Kröger N. (2006) Molecular genetic manipulation of the diatom Thalassiosira pseudonana (Bacillariophyceae). J. Phycol. 42, 1059–1065 [Google Scholar]
- 38. Poulsen N., Kröger N., Harrington M. J., Brunner E., Paasch S., and Buhmann M. T. (2014) Isolation and biochemical characterization of underwater adhesives from diatoms. Biofouling 30, 513–523 [DOI] [PubMed] [Google Scholar]
- 39. Buss J. E., and Stull J. T. (1983) Measurement of chemical phosphate in proteins. Methods Enzymol. 99, 7–14 [DOI] [PubMed] [Google Scholar]
- 40. Schägger H., and von Jagow G. (1987) Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166, 368–379 [DOI] [PubMed] [Google Scholar]
- 41. Wielsch N., Thomas H., Surendranath V., Waridel P., Frank A., Pevzner P., and Shevchenko A. (2006) Rapid validation of protein identifications with the borderline statistical confidence via de novo sequencing and MS BLAST searches. J. Proteome Res. 5, 2448–2456 [DOI] [PubMed] [Google Scholar]
- 42. Junqueira M., Spirin V., Balbuena T. S., Thomas H., Adzhubei I., Sunyaev S., and Shevchenko A. (2008) Protein identification pipeline for the homology-driven proteomics. J. Proteomics 71, 346–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Habermann B., Oegema J., Sunyaev S., and Shevchenko A. (2004) The power and the limitations of cross-species protein identification by mass spectrometry-driven sequence similarity searches. Mol. Cell Proteomics 3, 238–249 [DOI] [PubMed] [Google Scholar]
- 44. Poulsen N., and Kröger N. (2004) Silica morphogenesis by alternative processing of silaffins in the diatom Thalassiosira pseudonana. J. Biol. Chem. 279, 42993–42999 [DOI] [PubMed] [Google Scholar]
- 45. Dill K. A., Ghosh K., and Schmit J. D. (2011) Physical limits of cells and proteomes. Proc. Natl. Acad. Sci. U.S.A. 108, 17876–17882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vrieling E. G., Beelen T. P. M., van Santen R. A., and Gieskes W. W. C. (1999) Diatom silicon biomineralization as an inspirational source of new approaches to silica production. J. Biotechnol. 70, 39–51 [Google Scholar]
- 47. Shimizu K., Del Amo Y., Brzezinski M. A., Stucky G. D., and Morse D. E. (2001) A novel fluorescent silica tracer for biological silicification studies. Chem. Biol. 8, 1051–1060 [DOI] [PubMed] [Google Scholar]
- 48. Hildebrand M., York E., Kelz J. I., Davis A. K., and Frigeri L. G. (2006) Nanoscale control of silica morphology and three-dimensional structure during diatom cell wall formation. J. Mater. Res. 21, 2689–2698 [Google Scholar]
- 49. Sumper M., Hett R., Lehmann G., and Wenzl S. (2007) A code for lysine modifications of a silica biomineralizing silaffin protein. Angew. Chem. Int. Ed. Engl. 46, 8405–8408 [DOI] [PubMed] [Google Scholar]
- 50. Mort A. J., and Lamport D. T. A. (1977) Anhydrous hydrogen fluoride deglycosylates glycoproteins. Anal. Biochem. 82, 289–309 [DOI] [PubMed] [Google Scholar]
- 51. Poulsen N., Scheffel A., Sheppard V. C., Chesley P. M., and Kröger N. (2013) Pentalysine clusters mediate silica targeting of silaffins in Thalassiosira pseudonana. J. Biol. Chem. 288, 20100–20109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wieneke R., Bernecker A., Riedel R., Sumper M., Steinem C., and Geyer A. (2011) Silica precipitation with synthetic silaffin peptides. Org. Biomol. Chem. 9, 5482–5486 [DOI] [PubMed] [Google Scholar]
- 53. Nemoto M., Maeda Y., Muto M., Tanaka M., Yoshino T., Mayama S., and Tanaka T. (2014) Identification of a frustule-associated protein of the marine pennate diatom Fistulifera sp. strain JPCC DA0580. Mar. Genomics 16, 39–44 [DOI] [PubMed] [Google Scholar]
- 54. Frigeri L. G., Radabaugh T. R., Haynes P. A., and Hildebrand M. (2006) Identification of proteins from a cell wall fraction of the diatom Thalassiosira pseudonana: insights into silica structure formation. Mol. Cell Proteomics 5, 182–193 [DOI] [PubMed] [Google Scholar]
- 55. Mock T., Samanta M. P., Iverson V., Berthiaume C., Robison M., Holtermann K., Durkin C., Bondurant S. S., Richmond K., Rodesch M., Kallas T., Huttlin E. L., Cerrina F., Sussman M. R., Armbrust E. V. (2008) Whole-genome expression profiling of the marine diatom Thalassiosira pseudonana identifies genes involved in silicon bioprocesses. Proc. Natl. Acad. Sci. U.S.A. 105, 1579–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shrestha R. P., Tesson B., Norden-Krichmar T., Federowicz S., Hildebrand M., and Allen A. E. (2012) Whole transcriptome analysis of the silicon response of the diatom Thalassiosira pseudonana. BMC Genomics 13, 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kröger N., and Sandhage K. H. (2010) From diatom biomolecules to bioinspired syntheses of silica- and titania-based materials. MRS Bull. 35, 122–126 [Google Scholar]
- 58. Mizutani T., Nagase H., Fujiwara N., and Ogoshi H. (1998) Silicic acid polymerization catalyzed by amines and polyamines. Bull. Chem. Soc. Jpn. 71, 2017–2022 [Google Scholar]
- 59. Belton D., Paine G., Patwardhan S. V., and Perry C. C. (2004) Towards an understanding of (bio)silicification: the role of amino acids and lysine oligomers in silicification. J. Mater. Chem. 14, 2231–2241 [Google Scholar]
- 60. Lopez P. J., Gautier C., Livage J., and Coradin T. (2005) Mimicking biogenic silica nanostructures formation. Curr. Nanosci. 1, 73–83 [Google Scholar]
- 61. Lechner C. C., and Becker C. F. W. (2015) Silaffins in silica biomineralization and biomimetic silica precipitation. Mar. Drugs 13, 5297–5333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lechner C. C., and Becker C. F. W. (2014) A sequence-function analysis of the silica precipitating silaffin R5 peptide. J. Pept. Sci. 20, 152–158 [DOI] [PubMed] [Google Scholar]
- 63. Kröger N. (2007) Prescribing diatom morphology: toward genetic engineering of biological nanomaterials. Curr. Opin. Chem. Biol. 11, 662–669 [DOI] [PubMed] [Google Scholar]
- 64. Dionex (2009) Product Manual for AAA-Direct, Dionex Amino Acid Analyzer, p. 33, Document 031481-13, Dionex, Sunnyvale, CA [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.