

Abstract
Introduction
Delayed posthypoxic leukoencephalopathy (DPHL) may result from a variety of hypoxic insults, including respiratory depression from an opiate overdose. The underlying pathophysiological mechanism of DPHL remains uncertain. We describe a patient with a typical case of DPHL who responded clinically to antioxidant treatment.
Methods
Clinical, serological, and radiographic investigations were undertaken in the evaluation of the patient.
Results
A 63-year-old man developed altered mental status 10 days following recovery from an opiate overdose and aspiration pneumonia that required intubation. The clinical course and brain imaging were consistent with DPHL. Initiation of antioxidant therapy with vitamin E, vitamin C, B-complex vitamins, and coenzyme Q10 coincided with the prompt reversal of clinical deterioration.
Conclusions
The potential therapeutic effect of antioxidants on DPHL needs to be explored in future cases. If this relationship indeed holds true, it would be consistent with the hypothesis that formation of reactive oxygen species during reperfusion plays a role in the pathophysiology of this disorder.
Key Words: Delayed posthypoxic leukoencephalopathy, Antioxidants, Leukoencephalopathy, Hypoxia, Anoxia, Overdose
Introduction
Delayed posthypoxic leukoencephalopathy (DPHL) is a rare clinical entity characterized by initial recovery from hypoxic brain injury, followed by neurological deterioration [1]. The lucid interval usually lasts 1-2 weeks, and may be as long as 40 days [2,3]. Although DPHL can be caused by any hypoxic insult to the brain, carbon monoxide poisoning and opioid overdose are the two most commonly reported inciting events; other reported etiologies include complications of general anesthesia, cardiac arrest, gastrointestinal bleeding, drowning, and strangulation [4]. Neuropsychiatric manifestations predominate, with disorientation and deficits in attention and memory, and in severe cases, catatonia and psychosis [5]. Hyperreflexia is frequently seen early in the course, progressing to parkinsonism and akinetic mutism [6]. The mechanism of the delayed deterioration is unknown, and successful treatment options have not yet been developed. Treatment is supportive, and recovery is variable both in terms of the length of time to recovery and the severity of long-term deficits [4].
Case Report
A 63-year-old right-handed man with type 2 diabetes, hepatitis C virus treated with interferon several years ago, and alcohol and opiate use disorders presented with altered mental status. Twenty-one days prior to arrival at our institution, the patient had been admitted to the intensive care unit at an outside hospital for treatment of a morphine overdose, having been found unconscious and hypopneic by his wife. He was treated for opiate overdose, acute kidney injury and aspiration pneumonia, and discharged home after 1 week with a normal mental status. Head CT during that admission was normal. He was readmitted 10 days later for evolving neurobehavioral symptoms including walking around nude in his apartment, trying to use a remote control as a telephone, and having difficulty dressing himself. Workup included normal CBC, CMP, ESR, TSH, B12 and folate; Lyme and HIV were negative; urine toxicological assay was positive only for THC. Brain MRI 17 days after the overdose revealed bilateral cerebral white matter FLAIR hyperintensities, sparing the cerebellum and brainstem (fig. 1, top row).
Fig. 1.
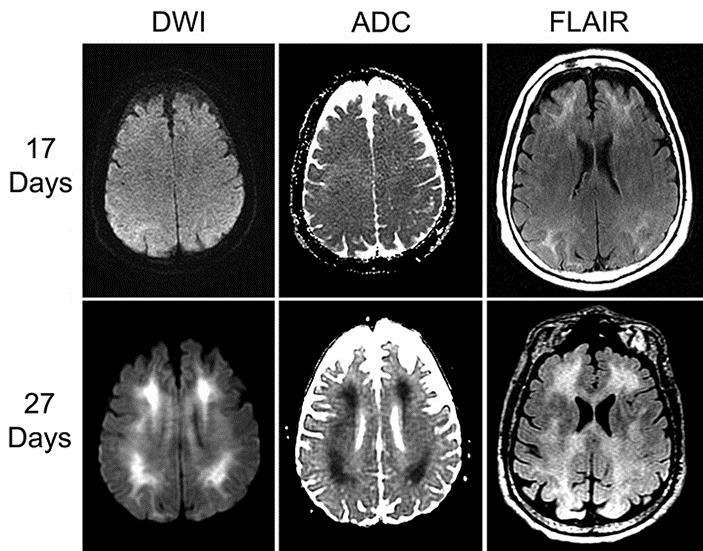
Brain MRI 17 days (top row) and 27 days after overdose (bottom row). From left to right: diffusion-weighted imaging (DWI), apparent diffusion coefficient (ADC), and fluid-attenuated inversion recovery sequences (FLAIR).
On transfer to our institution, the patient displayed signs of a florid disinhibition syndrome with facetiousness and inappropriate jocularity, as well as disorientation, confabulation, and perseveration. Attention and short-term memory were profoundly impaired. Cranial nerves and motor strength were normal. There was hyperreflexia, more notable in the lower extremities; gait was wide-based, with short steps. DPHL was suspected based on the radiographic findings and history of recent hypoxic injury.
Further evaluation at our institution included anti-thyroid peroxidase antibodies, RPR, and HIV RNA PCR, which were all negative; serum methylmalonic acid was normal. HCV PCR was undetectable, consistent with reported prior treatment. EEG showed diffuse theta and frontally predominant delta slowing. Lumbar puncture was bland, including bacterial cultures and HSV PCR. MRI with contrast, performed 6 days after admission to our hospital (27 days after overdose), showed worsening T2 hyperintensity in the cerebral white matter and corpus callosum, as well as new frontoparietal white matter diffusion restriction (fig. 1, bottom row); frontoparietal magnetic resonance spectroscopy displayed a mild decrease in the NAA to creatinine ratio, and a mild increase in choline to creatinine peaks, without a lactate peak.
His neuropsychiatric function continued to deteriorate, with loss of bowel and bladder control and emergence of parkinsonism with rigidity, cogwheeling, and bradyphrenia, progressing to marked apathy and mutism. This continuing decline prompted an initiation of antioxidant therapy for the reasons outlined below, comprising vitamin E 400 mg daily, vitamin C 1,000 mg daily, B-complex vitamins, and coenzyme Q10 400 mg three times a day. His neurocognitive status markedly improved after the initiation of antioxidant therapy, with increased spontaneous movement and speech, and gradual improvement in memory, orientation and motor function. He continued to make significant gains during inpatient and outpatient rehabilitation. Ten weeks after the initial date of admission, his gait and strength had returned to baseline, but he was left with residual emotional lability and impaired impulse control.
Discussion
The prognosis of DPHL is highly variable but is often characterized by a severe and prolonged clinical course, with a sustained deterioration phase following the relapse [7]. Full recovery has occasionally been described, but long-term or permanent disability is the usual outcome. Pathology includes selective and dramatic cerebral hemispheric demyelination, with preservation of other neural tissue and otherwise only occasional scattered perivascular lymphocytic cuffs [1]. The pathophysiology is thought to reflect demyelination following necrosis of vulnerable oligodendrocytes in arterial border zones from the hypoxic-ischemic insult, with resultant failure of continued synthesis of new myelin and slow rate of myelin turnover [1]. Relative deficiency of arylsulfatase A, important for myelin metabolism, has been implicated as a potential contributing factor in some reports [5,8].
Treatment is generally supportive, although pharmacologic agents including steroids, carbidopa/l-dopa, amantadine, and magnesium have been used with inconsistent results [5,9,10]. In one case report, antioxidants (coenzyme Q10, vitamin E, and vitamin B complex) were seemingly successful in reversing DPHL; however, conclusions were limited due to concomitant administration of methylprednisolone, l-dopa, and amantadine [5].
The rationale behind the use of antioxidant therapy in our case was the hypothesis that during ischemia and reperfusion, catecholamines are oxidized by molecular oxygen and monoamine oxidase resulting in the production of the superoxide anion and hydrogen peroxide, which lead to neuronal injury through the generation of oxygen free radicals [11]. We reasoned that if this hypothesis is correct, antioxidants may abort free radical-induced toxicity to oligodendroglia and neurons, and prevent ongoing injury. Support for this hypothesis comes from a double-blind randomized evaluation of hyperbaric oxygen therapy for carbon monoxide poisoning that showed a higher rate of neurological sequelae in the treatment group compared to controls, presumably through the generation of reactive oxygen species [12].
In a single case, it is impossible to conclude that the improvement was the direct result of the antioxidant regimen. The temporal relationship could have been coincidental, and we are unable to exclude the possibility that the initiation of antioxidant therapy simply coincided with the nadir of our patient's clinical course. Nevertheless, the immediate, profound and lasting improvement in this devastating and progressive condition suggested the possibility of therapeutic efficacy. This approach needs to be formally tested in future studies, but should be considered in patients with DPHL given the negligible risk of antioxidant therapy, and the potential for rescuing neurological function in these patients.
Opioid overdose is increasingly recognized as the initiating hypoxic-ischemic event in DPHL [5,6,7,8,13,14,15]. Given the current epidemic of opioid overdose in the United States, DPHL is an entity which clinicians should be aware of, particularly in the fields of emergency medicine, neurology, and psychiatry. This will prevent misdiagnosis and reduce the need for unnecessary and invasive diagnostic procedures.
Statement of Ethics
This research complies with the guidelines for human studies and animal welfare regulations. The subject gave his written, informed consent for the study to be published. Informed consent at the time of treatment was not required because treatment was provided as part of clinical care, did not constitute a research study, was not based on a study protocol, and did not require approval by our institution's committee on human research.
Disclosure Statement
All authors report that there are no financial relationships deemed relevant to the manuscript.
Acknowledgement
This study was supported in part by the Birmingham and MINDlink Foundations.
References
- 1.Plum F, Posner JB, Hain RF. Delayed neurological deterioration after anoxia. Arch Intern Med. 1962;110:18–25. doi: 10.1001/archinte.1962.03620190020003. [DOI] [PubMed] [Google Scholar]
- 2.Chen-Plotkin AS, Pau KT, Schmahmann JD. Delayed leukoencephalopathy after hypoxic-ischemic injury. Arch Neurol. 2008;65:144–145. doi: 10.1001/archneurol.2007.7. [DOI] [PubMed] [Google Scholar]
- 3.Weber W, Henkes H, Moller P, Bade K, Kuhne D. Toxic spongiform leucoencephalopathy after inhaling heroin vapour. Eur Radiol. 1998;8:749–755. doi: 10.1007/s003300050467. [DOI] [PubMed] [Google Scholar]
- 4.Shprecher D, Mehta L. The syndrome of delayed post-hypoxic leukoencephalopathy. NeuroRehabilitation. 2010;26:65–72. [PMC free article] [PubMed] [Google Scholar]
- 5.Carroll I, Heritier Barras AC, Dirren E, Burkhard PR, Horvath J. Delayed leukoencephalopathy after alprazolam and methadone overdose: a case report and review of the literature. Clin Neurol Neurosurg. 2012;114:816–819. doi: 10.1016/j.clineuro.2011.12.052. [DOI] [PubMed] [Google Scholar]
- 6.Shprecher DR, Flanigan KM, Smith AG, Smith SM, Schenkenberg T, Steffens J. Clinical and diagnostic features of delayed hypoxic leukoencephalopathy. J Neuropsychiatry Clin Neurosci. 2008;20:473–477. doi: 10.1176/jnp.2008.20.4.473. [DOI] [PubMed] [Google Scholar]
- 7.Bileviciute-Ljungar I, Haglund V, Carlsson J, von Heijne A. Clinical and radiological findings in methadone-induced delayed leukoencephalopathy. J Rehabil Med. 2014;46:828–830. doi: 10.2340/16501977-1820. [DOI] [PubMed] [Google Scholar]
- 8.Betts AM, Ritter JL, Kubal WS. Reversible delayed posthypoxic leukoencephalopathy after drug overdose: MRI findings in a collection of patients. Emerg Radiol. 2012;19:165–173. doi: 10.1007/s10140-011-1013-0. [DOI] [PubMed] [Google Scholar]
- 9.Lou M, Jing CH, Selim MH, et al. Delayed substantia nigra damage and leukoencephalopathy after hypoxic-ischemic injury. J Neurol Sci. 2009;277:147–149. doi: 10.1016/j.jns.2008.09.032. [DOI] [PubMed] [Google Scholar]
- 10.Rozen T. Rapid resolution of delayed post-hypoxic leukoencephalopathy with intravenous magnesium. Neurorehabilitation. 2012;30:329–332. doi: 10.3233/NRE-2012-0763. [DOI] [PubMed] [Google Scholar]
- 11.Phebus LA, Clemens JA. Effects of transient, global, cerebral ischemia on striatal extracellular dopamine, serotonin and their metabolites. Life Sci. 1989;44:1335–1342. doi: 10.1016/0024-3205(89)90390-1. [DOI] [PubMed] [Google Scholar]
- 12.Scheinkestel CD, Bailey M, Myles PS, et al. Hyperbaric or normobaric oxygen for acute carbon monoxide poisoning: a randomised controlled clinical trial. Med J Aust. 1999;170:203–210. doi: 10.5694/j.1326-5377.1999.tb140318.x. [DOI] [PubMed] [Google Scholar]
- 13.Moragas M, Gascón-Bayarri J, Cos M, Juncadella M, Reñé R, Rubio F. Delayed postanoxic encephalopathy secondary to acute cocaine and heroin intoxication. Eur J Radiol. 2009;71:e43–e45. [Google Scholar]
- 14.Molloy S, Soh C, Williams TL. Reversible delayed posthypoxic leukoencephalopathy. AJNR Am J Neuroradiol. 2006;27:1763–1765. [PMC free article] [PubMed] [Google Scholar]
- 15.Salazar R, Dubow J. Delayed posthypoxic leukoencephalopathy following a morphine overdose. J Clin Neurosci. 2012;19:1060–1062. doi: 10.1016/j.jocn.2012.01.001. [DOI] [PubMed] [Google Scholar]