
Fig 1. Evolutionary relationships of taxa.
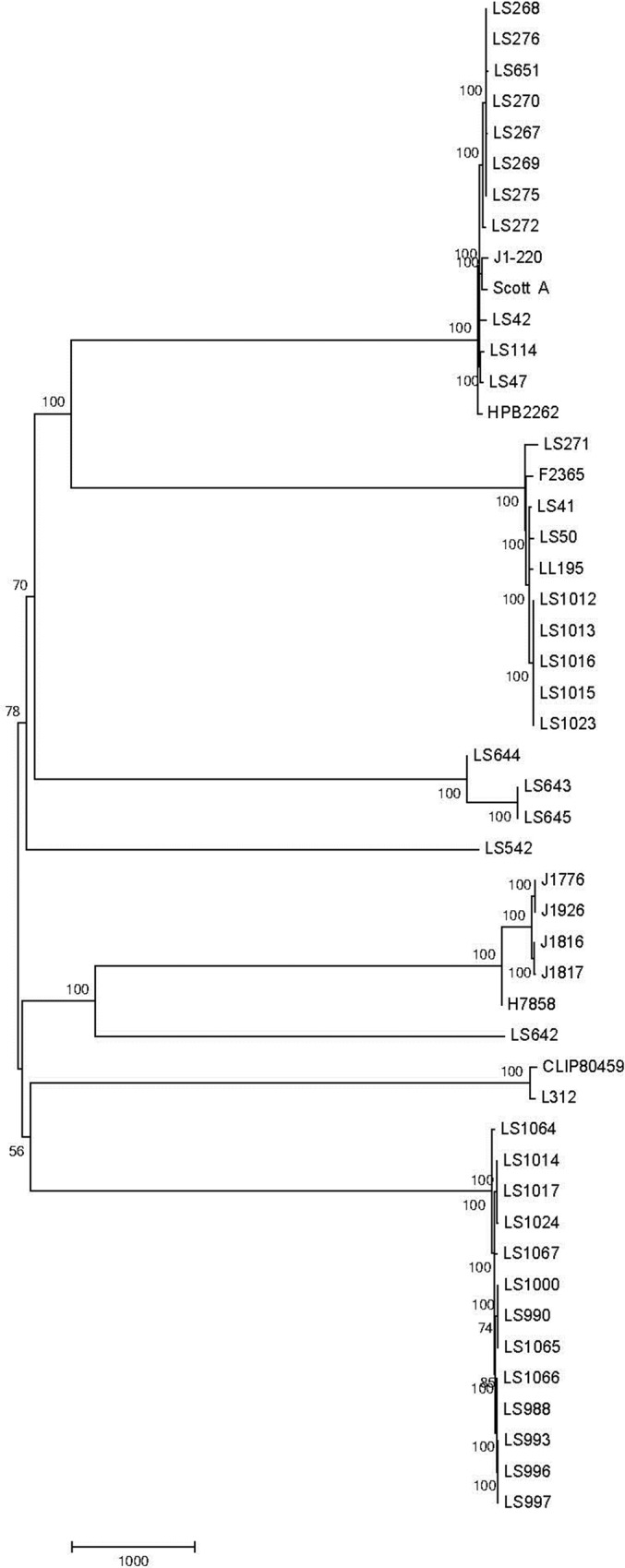
The evolutionary history was inferred using the Neighbor-Joining method based on the data from the BLAST-SNP analysis [22]. The optimal tree with the sum of branch length = 30986.27311707is shown. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) is shown next to the branches [32]. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the number of differences method [33] and are in the units of the number of base differences per site. The analysis involved 49 nucleotide sequences of 4b and 4bV strains indicated in Table 1. Codon positions included were 1st+2nd+3rd+Noncoding. All ambiguous positions were removed for each sequence pair. There were a total of 23545 positions in the final dataset. Evolutionary analyses were conducted in MEGA6 [27]. The strains highlighted in Table 1 are similarly noted here with cluster 1 in purple, cluster 2 in green, and cluster 3 in blue.