

Abstract
High-capacity adenoviral vectors (HCAdV) devoid of all viral coding sequences represent one of the most advanced gene delivery vectors due to their high packaging capacity (up to 35 kb), low immunogenicity and low toxicity. However, for many laboratories the use of HCAdV is hampered by the complicated procedure for vector genome construction and virus production. Here, a detailed protocol for efficient cloning and production of HCAdV based on the plasmid pAdFTC containing the HCAdV genome is described. The construction of HCAdV genomes is based on a cloning vector system utilizing homing endonucleases (I-CeuI and PI-SceI). Any gene of interest of up to 14 kb can be subcloned into the shuttle vector pHM5, which contains a multiple cloning site flanked by I-CeuI and PI-SceI. After I-CeuI and PI-SceI-mediated release of the transgene from the shuttle vector the transgene can be inserted into the HCAdV cloning vector pAdFTC. Because of the large size of the pAdFTC plasmid and the long recognition sites of the used enzymes associated with strong DNA binding, careful handling of the cloning fragments is needed. For virus production, the HCAdV genome is released by NotI digest and transfected into a HEK293 based producer cell line stably expressing Cre recombinase. To provide all adenoviral genes for adenovirus amplification, co-infection with a helper virus containing a packing signal flanked by loxP sites is required. Pre-amplification of the vector is performed in producer cells grown on surfaces and large-scale amplification of the vector is conducted in spinner flasks with producer cells grown in suspension. For virus purification, two ultracentrifugation steps based on cesium chloride gradients are performed followed by dialysis. Here tips, tricks and shortcuts developed over the past years working with this HCAdV vector system are presented.
Keywords: Bioengineering, Issue 107, gene delivery, adenoviral vector, high-capacity, cloning, helper-dependent, spinner culture system, homing endonucleases, adenovirus, gene therapy
Introduction
For gene therapeutic applications it is of great importance to avoid cytotoxic and immunogenic side effects caused by expression of viral proteins, the transgene itself or by incoming viral proteins. Adenovirus vectors (AdV) are widely used to introduce foreign DNA into a wide variety of cells to investigate the impact of transgene expression 1,2. The most advanced version of AdV is represented by high-capacity adenovirus vectors (HCAdV) lacking all viral coding sequences 3,4 and thereby offering a packaging capacity up to 35 kb combined with low immunogenicity and low toxicity 5-8. Due to their high packaging capacity they allow delivery of large or multiple transgenes using a single vector dose. Therefore, they represent a valuable tool for the research community.
In contrast to first- or second-generation AdV lacking the early genes E1 and/or E3 that can be easily produced using commercial kits, vector genome construction and virus production of HCAdV is more complex. The system for the construction of HCAdV genomes is based on the plasmid pAdFTC carrying a HCAdV genome devoid of all viral coding sequences and the shuttle plasmid pHM5 9-12. Any gene of interest of up to 14 kilo bases (kb) can be cloned into the shuttle vector pHM5 in which the multiple cloning site is flanked by recognition/cleaving sites of the homing endonucleases PI-SceI and I-CeuI. Therefore, a cloned gene of interest can be released by consecutive PI-SceI and I-CeuI digests for subsequent directed insertion into the same restriction sites present in the HCAdV genome contained in the plasmid pAdFTC. In pAdFTC the transgene insertion site located between the PI-SceI and I-CeuI cleavage sites is flanked by stuffer DNA and the noncoding adenoviral sequences required for genome packaging such as the 5' and 3' inverted terminal repeats (ITRs) at both ends and the packaging signal downstream of the 5'ITR. The additional stuffer DNA provides optimal size of the final HCAdV genome ranging from 27 to 36 kb to ensure efficient packaging during virus production. Since pAdFTC is a large plasmid with up to 45 kb (dependent on the size of the inserted transgene) and the usage of homing endonucleases with comparably long DNA recognition sites exhibits strong DNA binding, several cleanup steps are necessary during transfer of the transgene from pHM5 to pAdFTC. Careful handling avoiding shearing forces is recommended.
The ITRs of the HCAdV genome are flanked by NotI restriction enzyme recognition sites located directly upstream of the 5'ITR and downstream of the 3'ITR 12. Therefore, HCAdV can be released by NotI digest for subsequent transfection of the viral genome into the HCAdV producer cell line. Note that the usage of the restriction enzyme NotI for release of the viral genome from the plasmid pAdFTC implies that the inserted transgene is devoid of NotI DNA recognition sites. The HEK293 cell based producer cells (116 cells) stably express Cre recombinase. For virus amplification 116 cells are co-infected with a helper virus (HV) providing all AdV genes needed for replication and packaging in trans 3,4. The HV is a first-generation AdV with a floxed packing signal which is removed during virus amplification by Cre recombinase expressed in 116 cells 4. This ensures that predominantly HCAdV genomes containing an intact packaging signal are encapsidated.
Pre-amplification of the HCAdV is performed by conducting serial passaging steps in 116 cells grown on surfaces in tissue culture dishes. After each passage viral particles are released from infected cells by conducting three consecutive freeze-thaw steps. With every passage increasing numbers of cells are infected with 1/3 of cell lysate from the preceding passage. Finally lysate from the last pre-amplification step is used to infect producer cells grown in suspension in a spinner flask for large scale amplification. Virions are purified from the suspension cells by performing ultracentrifugation in a cesium chloride density gradient 4,12. With this procedure empty particles and fully assembled particles are separated into two distinct bands. To further concentrate the HCAdV particles a second non-gradual ultracentrifugation step is performed. Subsequently the resulting band containing the HCAdV is collected and dialyzed against a physiological buffer. Final vector preparations are characterized with respect to numbers of absolute viral particles, infectious particles and HV contamination levels. Absolute viral particles can be determined by lysing viral particles and measuring the absorbance at 260 nm or by performing quantitative real-time PCR (qPCR) 12. Infectivity of the purified virus particles can be determined by qPCR measuring HCAdV genomes present within infected cells 3 hr post-infection.
Protocol
1. Construction of Recombinant HCAdV Genomes based on the Plasmid pAdFTC
Note: All plasmids have been described previously 11,12 and are available upon request. The cloning procedure is schematically shown in Figure 1.
Clone a gene of interest (GOI) including promoter and polyadenylation signal (pA) into the shuttle plasmid pHM5, using a cloning strategy of choice, to generate pHM5-GOI. NOTE: Since pAdFTC is a relatively large plasmid, classical plasmid preparation protocols are recommended to avoid sheering of the plasmid DNA by using commercial plasmid purification kits that are based on silica membranes. I-CeuI and PI-SceI strongly bind to the DNA and change the electrophoretic mobility of digested DNA. Therefore, a phenol-chloroform extraction and EtOH precipitation (step 1.3) is required before agarose gel electrophoresis.
Digest 20 µg of pHM5-GOI and 10 µg of pAdFTC by I-CeuI (10 U) for 3 hr at 37 °C in a total volume of 100 µl to linearize plasmids (~2.8 kb + GOI ORF sequence and ~31kb, respectively).
- Purify linearized plasmids using phenol-chloroform extraction followed by ethanol (EtOH) precipitation 13.
- Add 100 µl of phenol:chloroform:isoamyl alcohol and mix gently by inverting the tube several times. Centrifuge for 2 min at 15,000 x g.
- Transfer the supernatant to a new tube, add 20 µl of sodium acetate (pH 5, 3 M) and 400 µl of ice cold EtOH (99.8 %) and mix suspension vigorously. Centrifuge for 10 min at 15,000 x g.
- Remove the supernatant, add 300 µl of 70 % EtOH and centrifuge for 2 min at 15,000 x g. Then remove supernatant and air-dry DNA pellet. Dissolve the pellet in 10- 20 µl of dH2O. Do not dry DNA pellet too long, as it will be harder to get DNA in solution.
Digest I-CeuI -digested pHM5-GOI and pAdFTC plasmid from step 1.3) for at least 3 hr or O/N with the restriction enzyme PI-SceI (10 U) at 37 °C in a total volume of 50 µl and subsequently purify I-CeuI and PI-SceI digested plasmids using phenol-chloroform extraction followed by EtOH precipitation (see step 1.3). Dissolve DNA in 20 µl of nuclease free dH2O.
Separate pHM5 plasmid backbone (2.8 kb) and the GOI (from step 1.4) on a preparative agarose gel and gel-purify the GOI using a commercial gel- and PCR- clean-up kit. A representative agarose gel of the digest is shown in Figure 2A.
Dephosphorylate I-CeuIand PI-SceI-digested pAdFTC (from step 1.4) with Calf Intestinal Alkaline Phosphatase CIP (10 U) for 1 hr at 37 °C in a total volume of 25 µl and purify dephosphorylated plasmid pAdFTC by phenol-chloroform extraction and subsequent EtOH precipitation (step 1.3). Elute DNA in 20 µl of nuclease free dH2O.
Perform analytic gel electrophoresis from an aliquot of the dephosphorylated pAdFTC plasmid (from step 1.6) and the purified GOI-fragment (from step 1.5) to analyze whether digests are complete and the sizes of the expected fragments are correct (~31 kb for linearized pAdFTC). Note: The size of the GOI is variable depending on the size of the transgene expression cassette. Estimate the relative concentrations of the respective fragments for subsequent ligation. A representative agarose gel of purified fragments is shown in Figure 2B.
- Set up the ligation reaction in a total volume of 20 µl using 400 U of T4 DNA ligase and a molar ratio of vector to insert of 1:3. NOTE: In concordance with the agarose gel shown in Figure 2B we usually ligate in a total volume of 20 µl with 2- to 6-µl vector, 8- to 12 µl insert and 400 U of T4 DNA ligase. As the DNA concentration is usually low after several phenolization steps, use as much DNA as possible so that no additional H2O has to be added to reach the final volume of 20 µl. Ligate at 16 °C O/N and subsequently perform phenol-chloroform extraction followed by EtOH precipitation (step 1.3).
- Elute DNA in 15 µl of nuclease free dH2O and digest the purified ligation reaction with the restriction enzyme SwaI (10 U) for 2 hr at 25 °C in a total volume of 20 µl. Then concentrate and purify DNA, by performing phenol-chloroform extraction followed by EtOH precipitation (step 1.3). Elute DNA in 10 µl of nuclease free dH2O.
Transform 2 µl of the purified SwaI digested ligation product by electroporation into DH10B or DH5α electrocompetent E. coli and select clones for 16 to 24 hr at 37 °C on ampicillin-containing LB plates (50 mg/ml ampicillin). Select 5- 10 clones and prepare plasmid DNA mini-preparations using alkaline lysis followed by phenol-chloroform extraction and subsequent EtOH precipitation (step 1.3) 13.
Perform an analytical digest of plasmid mini-preparations followed by agarose gel electrophoresis to check the size of DNA fragments. Suggested restriction enzymes are for instance I-CeuI and PI-SceI releasing the insert, SpeI or HincII (Figure 2C).
Amplify a correct clone in ampicillin-containing LB medium (50 mg/ml ampicillin) and perform a midi- or maxi-plasmid preparation using any commercially available plasmid purification kit.
2. Release of HCAdV-genomes from pAdFTC Plasmid and Preamplification of HCAdV Vectors in the Producer Cell Line 116
Digest 20 µg of the pAdFTC-based adenoviral production plasmid containing the cloned GOI from step 1.11) in a total volume of 100 µl using NotI (20 U) at 37 °C for >2 hr and perform phenol-chloroform extraction twice followed by EtOH precipitation. Dissolve in 20- 30 µl sterile dH2O.
Check an aliquot of 1:10 diluted digested pAdFTC-GOI-DNA by gel electrophoresis. A 9 kb fragment for the plasmid backbone and, depending on the size of the GOI, a second DNA fragment for the HCAdV genome (size: 28- 36 kb) is detectable. NOTE: A representative example of the NotI digest is displayed in Figure 2D. Linearized DNA can be stored for several days at 4 °C or at -20 °C for several weeks.
Before proceeding with the protocol, make sure that the helper virus AdNG163R-2 has been amplified 4. Note: Cells transfected with pAdFTC derived HCAdV vectors are genetically modified organisms classified as Biosafety Level 2 (BSL-2), please use proper containment and waste handling measures, including personal protective equipment, and work under BSL-2 laminar flow hoods.
Culture 116 cells in MEM eagle medium supplemented with 10 % FBS and hygromycin B (100 µg/ml). Seed low passage (below passage 10) 116 cells (~0.4x 106 cells) in a 60 mm tissue culture dish one day before transfection so that they reach 50- 80 % confluency the next day.
- Transfect linearized HCAdV genome from step (2.1) into 116 cells by using methods, such as calcium phosphate transfection or other commercially available transfection reagents (Figure 3A). 16- 18 hr post-transfection, carefully remove the medium, add 3 ml fresh medium (MEM, 5 % FBS) and infect cells with the HV AdNG163R-2 4 applying 5 transducing units (TU) per cell (since a confluent 60 mm tissue culture dish contains ~3.2x 106 cells, add ~1.6x 107 TU of HV).
- Gently move the dish every 20 min during the first hour after infection to ensure equal HV distribution. Cultivate infected cells at 37 °C and 5 % CO2. In case of efficient virus amplification cytopathic effect (CPE) caused by virus replication (cells are rounded up and loosely attached or detached from the tissue culture dish) is observable 48 hr post-infection.If CPE is observed earlier the amount of HV has to be reduced. If CPE starts later, more helper virus has to be used.
- Harvest cells including supernatant 48 hr post-infection by flushing off the cells from the culture dish using the culture medium. Cells that are suspended in their culture medium are called passage 0 (P0). Split P0 into two fractions.
- From 0.5 ml of the detached cells spin down the cells for 3 min at 2,000 x g and remove medium from the cell pellet, that is later used for isolating genomic DNA (gDNA) for qPCR based analysis of the amplification process. Store cell pellets at -20 °C until further processing.
- From 2.5 ml of the detached cells release viral particles by freezing (at -80 °C or in liquid nitrogen) and thawing (at RT or in a 37 °C water bath) the cells that are resupended in their medium three to four times. This fraction is than called lysate of P0.
Ensure that tissue culture dishes with 116 cells that are grown to 90- 95 % confluency using MEM-medium supplemented with FBS (10 %) and hygromycin B (100 µg/ml) and HV are available for the following passaging steps.
Mix 1 ml of fresh media (MEM, 5 % FBS) with 2.5 ml of the lysate from the preceding passage (step 2.6.2) to a final volume of 3.5 ml, add HV applying 2 TU per cell (since a 60 mm tissue culture dish at a confluency of 90- 95 % contains ~3.2x 106 cells, add ~6.4x 106 TU of HV). (Figure 3B). Carefully remove the medium from a 60 mm dish of 116 cells and add the viral mixture to the cells. Repeat step 2.6)- 2.8) twice to obtain lysates of passages P1 and P2.
Mix 17.5 ml fresh media (MEM, 5 % FBS) with the 2.5 ml of the lysate from P2 and add HV applying 2 TU per cell (since a 150 mm tissue culture dish at a confluency of 80- 100 % contains ~2x 107 cells, add ~4x 107 TU of the HV). Remove the medium from a 150 mm dish of 116 cells and infect cells with the viral mixture. Cultivate infected cells at 37 °C and 5 % CO2.
- 48h post-infection harvest cells as described in step 2.6) to obtain passage P3 (Figure 3B).
- Repeat step 2.6)1.
- From the remaining 19.5 ml of P3 release viral particles from the resupended cells by freezing and thawing three to four times. This fraction is than called lysate of P3. Note: Some vectors eventually may require additional passages in 150 mm dishes until they are amplified sufficiently. Therefore effectivity of the pre-amplification process needs to be monitored during serial passaging. qPCR based analysis of the amplification process can be performed as described in section 3 (see also Figure 4A). Pre-amplification of HCAdV carrying an expression cassette for green fluorescent protein (GFP) can easily be evaluated by observing the GFP flourecscent signal using a fluorescent microscope (Figure 4B).
3. Monitoring of the Amplification Process using Quantitative-Real Time PCR (qPCR) (see also Figure 4A).
Isolate gDNA from cell pellets from each passage of the preamplifaction (steps 2.6)- 2.10) using any commercial DNA isolation kit for isolation of gDNA from cultured cells or another method of choice. For optimal PCR results use gDNA as fresh as possible. Otherwise gDNA can be stored at -20 °C until further use.
To determine the number of HCAdV genomes present in the cells of the respective passage by qPCR analysis generate a standard curve using 101- 109 copies of a plasmid carrying the GOI contained in the HCAdV genome.
Analyze the same amount of gDNA from P0- P3 (step 2.6- 2.10) applying qPCR using 400 nM of primers specific for the GOI contained in the HCAdV genome. For conducting qPCR follow manufacturer's instructions of respective qPCR chemicals. Set the qPCR program as follows: pre-incubation at 95 °C for 10 min, amplification in 40 cycles of 95 °C for 10 s, 55- 60 °C for 15 s and 72 °C for 20 s. On the basis of the standard curve the number of HCAdV genomes in the reaction can be interpolated.
4. Large Scale Amplification of HCAdV Vectors in 116 cells Growing in Suspension
Add 900 ml of prewarmed (37 °C) fresh MEM supplemented with 10 % FBS and hygromycin B (100 µg/ml) into a 3 l spinner culture flask (Figure 3B).
Remove medium from at least 10 individual 150 mm tissue culture dishes with 116 cells grown at a confluency of 90- 100 % and flush off cells with 10 ml of fresh pre-warmed (37 °C) MEM supplemented with FBS (10 %) and hygromycin B (100 µg/ml) using a serological pipette. Pipette up and down several times to get a homogeneous cell suspension. Transfer cells directly into the spinner flask already containing 900 ml from step 4.1). Note: Do not use Trypsin/EDTA as it may negatively affect growth of suspension cells. After removal of medium immediately transfer cells into the spinner flask. Do not handle more than two tissue culture dishes at a time. The longer the waiting time after adding fresh media the more difficult it will be to detached the cells from the tissue culture dish.
To ensure optimal cell growth incubate spinner flask on a magnetic stirrer in a tissue culture incubator for 24 hr at 37 °C and 5 % CO2. Adjust the magnetic stirrer to 70 rpm to avoid attachment of cells to the glass surfaces.
Monitor cell growth in spinner culture flask to examine whether cells grown in the spinner culture flask are viable and whether they grow to sufficient amounts. Each time prior to adding fresh medium transfer 2 ml from the bioreactor to a 60 mm tissue culture dish. Observe cell morphology under a microscope. Note: Cells forming clumps floating in the culture medium indicate optimal growth and viability (see also Figure 4C). After 24 hr they form colonies on the surface of the tissue culture dish. 30- 50 % confluence indicates optimal density.
24 hr after setting up the spinner culture flask add 500 ml of fresh media (MEM, 10 % FBS) supplemented with hygromycin B (100 µg/ml). 24 hr later repeat this step.
72 hr after setting up the spinner culture, add 1,000 ml of fresh MEM media (10 % FBS) with hygromycin B (100 µg/ml) resulting in a total volume of 3 L of cell suspension.
24 hr after reaching a volume of three litres, harvest 116 suspension cells by centrifugation for 10 min at 500 x g at RT. Discard the supernatant. Keep the emptied spinner culture flask under the tissue culture hood.
- Resuspend cell pellets in fresh MEM supplemented with 5 % FBS by pipetting up and down about 8- 10 times to get a homogeneous cell suspension. The volume of medium depends on whether lysate from Step 2.10)2. (19.5 ml, see step 4.8)1. option a) or a purified viral stock of HCAdV from step 5.9)2. (several µl depanding on viral titer, see step 4.8)2., option b) is used for infection of the cells. Use a total volume of 150 ml.
- Option a: For infection of 116 suspension cells with lysate from P3 (primary amplification) transfer cells from step 4.8) to a 250 ml storage bottle equipped with a sterile magnetic stir bar or a 250ml small scale spinner flask and co-infect cells with the virus within the lysate from P3 (step 2.10.2) and 2 TU of HV per cell. Assuming a density of 3x 105 cells per ml, the total cell number is 9x 108 cells. Thus add 1.8x 109 TU of the HV.
- Option b: For infection of 116 suspension cells with purified viral stock, transfer cells from step 4.8) to a 250 ml storage bottle equipped with a sterile magnetic stir bar or a 250ml small scale spinner flask and co-infect cells with the 100 viral particles per cell of formerly purified HCAdV (from step 5.9)2.). Thus add 9x 1010 VP according to physical titer (measured by absorbance at 260nm; step 6) of the purified HCAdV and 1.8x 109 TU of the HV.
Stir the cell-virus mixture from sep 4.8.1) or 4.8.2) in on a magnetic stirrer in the tissue culture incubator at 37 °C and 5 % CO2 for 2 hr at 60 rpm. Make sure that the storage bottle is not completely closed to allow air circulation.
2 hr post-infection transfer the total volume of the cell-virus mixture back into the 3 l spinner culture flask and add 1,850 ml of fresh pre-warmed (37 °C) MEM media supplemented with 5 % FBS to a total volume of 2 L and incubate in the tissue culture incubator at 37 °C and 5 % CO2 for 48 hr at 70 rpm.
Harvest cells by centrifugation for 10 min at 890x g at RT in 500 ml centrifuge tubes. Remove the medium and resuspend pelleted cells in a total volume of 28 ml of DPBS. Pipette up and down to obtain a homogeneous cell suspension. Freeze the cell-virus suspension in liquid nitrogen or at -80 °C making sure that the suspension is completely frozen. Store the cell-virus suspension at -80 °C until starting the virus purification.
5. Purification and Dialysis of HCAdV
To prepare viral lysate for cesium chloride (CsCl) gradients, freeze cell-virus suspension from step 4.11) in liquid nitrogen and thaw in a water bath at 37 °C four times.
Centrifuge the viral lysate at 500 x g for 8 min at RT and collect the supernatant containing the HCAdV.
- For Ultracentrifugation prepare CsCl solutions as follows.Weigh 37.5 g (for 1.5 g/cm3), 33.5 g (for 1.35 g/cm3), and 31,25 g (for 1.25 g/cm3) of CsCl powder respectively and fill up with dH2O to 25 ml.
- Stirr until solution becomes clear and sterile filter the solutions. Finally remove 1 ml of each solution and weigh it on a fine scale to check wether the density is correct. 1ml should weight 1.5 g, 1.35 and 1.25 g respectively.
- If the density is too high adjust it by stepwise addition of small volumes (µl) of sterile dH2O. After addition of dH2O measure the densitiy again. If necessary add more dH2O.
- Repeat the procedure until density is correct. If the density is too low adjust it by adding small amount of CsCl powder. Sterile filter it again and check the density. If the density is too high adjust it by adding dH2O as described before. If the density is still too low add mor CsCl, sterile filter it again and check the density. Repeat the procedure until the density is correct.
Prepare CsCl step gradients in six clear ultracentrifuge tubes. Carefully and slowly pipette CsCl solutions into the tubes using the following order: 0.5 ml of 1.5 g/cm3 CsCl solution, 3 ml of 1.35 g/cm3 CsCl solution and 3.5 ml of 1.25 g/cm3 CsCl solution (Figure 2C).
Overlay ~4.5 ml of cleared vector supernatant from step 5.2) on top of the 1.25 g/cm3 CsCl layer. Centrifuge the gradients in an ultracentrifuge using a swing out rotor (SW-41) at 12 °C for at least 2 hr at 226,000 x g (35,000 rpm) with slow acceleration and deceleration to separate HCAdV-genome containing viral particles from empty particles and cell debris. NOTE: Under optimal conditions a diffuse band of cell debris formes on top of the tubes. Below two white bands can be observed. The upper band contains empty particles whereas the lower band equals the HCAdV (Figures 2C and 5A).
Carefully remove the layers of cell debris and empty particles and collect 1 ml of the lower bands from each tube and transfer virus with a clean pipette tip into a sterile 50 ml tube. Add up to 24 ml of 1.35 g/cm3 CsCl solution to the collected virus particles and mix carefully.
Fill centrifuge tubes with 1.35 g/cm3 CsCl-virus solution to the top and centrifuge O/N (18- 20 h) at 226,000 x g (35,000 rpm) at 12 °C in an ultracentrifuge using a swing out rotor (SW-41) with slow acceleration and deceleration.
Collect the HCAdV present in the prominent lower band. As a potential upper band contains empty particles remove upper layers from the top using a pipette (see Figures 2C and 5B) Then take away the lower band using a pipette.
- Dialyze the collected virus particles for buffer exchange.
- Cut off a strip of dialysis tubing (MWCO: 50,000) of about 8 cm in length, wash it with sterile dH2O for three times. Then close one side of the dialysis tubing with a plastic clamp and transfer the collected virus from step 5.8) into tubing using a 1-ml pipette. Avoid air bubbles within the tubing and close it on the other side with a plastic clamp dialysis closures.
- Dialyze in 1 l of dialysis buffer (10 mM Tris-HCl (pH 7.5), 10 % glycerol and 1 mM MgCl2 in deionized H2O) for 2 hr at 4 °Cwith slow stirring. Exchange dialysis buffer with 2 l of dialysis buffer and dialyze O/N at 4 °C with slow stirring. Alternatively use a sucrosebuffer (140 mM NaCl, 5 mM Na2HPO4x2H2O, 1.5 mM KH2PO4 and 730 mM sucrose, pH 7.8).
- Collect dialyzed virus particles from step 5.9)2. using a 1 ml pipette. Prepare multiple aliquots of desired small volumes (25- 100 µl) and store purified virus at -80 °C.
6. Measuring the Physical Titer of Final HCAdV Preparations by Optical Density (OD)
Dilute 25 µl of the final vector preparation from step 5.9)2 with 475 µl of lysis buffer (10 mM Tris-HCl (pH 7.5), 10 mM EDTA (pH 8.0), 0.5 % SDS)), gently shake for 20 min at RT and finally centrifuge for 2 min at RT at 15,000 x g.
Since the absorbance values at 260 nm (A260) are usually low, measure absorbance four times using 100 µl of the supernatant and calculate mean value of the four measurements. Calculate the number of viral particles per ml (vp/ml-1) using the following formula: vp/ml-1 = (mean A260) x (20) x (1.1 x 1012) x (36 kb / HCAdV size in kb). NOTE: Results of a typical yield of absolute viral particles (OD titer) are shown in Figure 7A. Experience shows, that the OD titer overestimates the number of virus particles. Absolute viral particles measured by OD can be 20- 100 times higher than infectious viral particles measured by q-PCR. For more precise measurement of absolte and infectious HCAdV particles as well as HV contamination by q-PCR refer to section 7.
7. Measuring Total Particles, Infectious Units of the HCAdV and HV Contamination Levels in the Final Vector Preparation by qPCR. A Scheme of the Titration Procedure is Shown in Figure 6
Seed HEK293 cells in 6 or 12 well tissue culture plates so that cells reach 90 % confluency on the next day. To determine the number of infectious viral particles (infectious titer), infect cells of one well in a multi-well plate with 1 µl and a second well with 10 µl of purified virus from step 5.9)3.
Harvest cells 3 hr post infection. Remove medium and add trypsin to cover the whole well and incubate at 37 °C and 5 % CO2 for 5 min. Flush off the cells with the trypsin using a pipette and spin down the cells at 890 x g for 3 min at RT.
Resuspend the pelleted cells in 200 µl of DPBS and wash them thoroughly to remove free (non-infective) vector particles. Centrifuge for 3 min at 890 x g at RT. Discard the supernatant and resuspend cell pellets in 200 µl of fresh DPBS. Note: For accurate determination of the infectious titer, it is crucial to remove all non-infective viral particles from the cell surfaces by trypsin treatment and thorough washing.
To determine the total viral particle number (infective particles and non infective particles = physical titer), harvest non-infected HEK293 cells from two wells without trypsin by flushing off the cells with their culture medium using a pipette.
Spin down the cells for 3 min at 890 x g at RT. Discard the medium and resuspend pelleted cells in 200 µl of DPBS. Subsequently add 1 µl and 10 µl of purified HCAdV preparation directly to the cells, respectively. Use non infected cells as background, to ensure the same conditions for gDNA isolation and subsequent q-PCR.
- Isolate gDNA from HEK293 cells derived from steps 7.3) and 7.5)
- Vortex cells resuspended in 200 µl of DPBS (step 7.3 and 7.4) for 3 sec, add 200 µl of SDS solution. (10 mM Tris-HCl (pH 7.5), 10 mM EDTA (pH 8.0), 0.5 % SDS) and 20 µl Proteinase K (20 mg/ml) and vortex suspension for 3 sec. Incubate 12- 16 hr at 55 °C and shake slowly. Then add 2 µl of RNAse A (20 mg/ml) and incubate for 30 min at 37 °C.
- Add 350 µl of phenol:chloroform:isoamyl alcohol and centrifuge for 2 min at 15,000 x g. Transfer the supernatant to a new tube and repeat this step once
- Transfer the supernatant to a new tube, add 50 µl of sodium acetate (pH 5, 3 M) and 1 ml of ice cold EtOH (99.8 %) and mix suspension. Centrifuge samples for 10 min at 15,000 x g to pellet genomic DNA.
- Then remove the supernatant, add 500 µl of 70 % EtOH and centrifuge for 2 min at 15,000 x g. Remove the supernatant, add 500 µl of 70 % EtOH and shake for 30 min at RT. Then centrifuge for 2 min at 15,000 x g.
- Remove the supernatant and air-dry DNA pellet. Do not dry DNA pellets for a long time, as it will be harder to get gDNA in solution. Resuspend the DNA pellet in 120 µl of dH2O and incubate for ~1 hr at 37 °C while shaking. If DNA solution appears viscous, shake it for several hours at 37 °C, or incubate at 55 °C for ~1 hr.
To determine infectious HCAdV-particles and absolute HCAdV-particles, generate a standard curve of 101- 108 copies of a plasmid carrying the GOI contained in the HCAdV genome.
Determine total particles and infectious particles by analyzing the same amounts of gDNA of infected HEK293 cells from step 7.3) and 7.4) apply qPCR using 400 nM of primers specific for the GOI contained in the HCAdV genome.
Set the PCR program as follows: pre-incubation at 95 °C for 10 min, amplification in 40 cycles of 95 °C for 10 sec, 55 °C for 10 sec and 72 °C for 20 sec. On the basis of the standard curve (step 7.7), interpolate the total number of adenoviral genomes in the reaction.
Calculate the number of adenoviral genomes in the final vector preparation using the following formular: (Number of HCAdV / weight in ng of gDNA in the reaction) x (weight in ng of DNA of all cells in the infected dish/ volume virus in µl). NOTE: The typical range for absolute and infective viral particles yields are around 1x107-1x108 viral particles/ µl. Titers that are ten fold higher or lower than showed here can be considered as normal. Higher titers would be an improvement. With respect to the ratio of absolute HCAdV particles to infectious HCAdV particles, experience shows that approximately 5- 10 % of absolute HCAdV particles are infectious (Figure 7A).
To determine the contamination levels with HV, perform qPCR by amplifying a part of the adenoviral late gene 3 (L3) present in the HV genome. Use a plasmid carrying the adenoviral late gene 3 (L3) to generate a standard curve (step 7.7).
In this reaction apply 400 nM of the primers L3 forward 5'-AGA AGC TTA GCA TCC GTT ACT CGA GTT GG-3' and L3 reverse 5'-ATA AGC TTG CAT GTT GGT ATG CAG GAT GG-3' together with 300 nM of the L3-specific probe 5'-Fam-CCA CCC GTG TGT ACC TGG TGG ACA-Tamra-3'.
Set the PCR program as follows: pre-incubation/activation at 95 °C for 10 min, amplification during 40 cycles of 95 °C for 15 sec and 60 °C for 1 min. Use any universal probe PCR mastermix for the qPCR reaction. On the basis of the standard curve (step 7.7), calculate the number of infectious HV particles in the final vector preparation. See (Figure 7A) for typical yields for HV particles.
Finally calculate the ratio of absolute infectious particles of HCAdV to HV contamination levels to evaluate the quality of the vector preparation. NOTE: Until now a small portion of remaining HV cannot be excluded. Usually the percentage of HV contamination is ~5% of infectious particles or less, which is acceptable (Figure 7B). Of course as less HV as possible is desirable, especially if the vector preparation is going to be used for animal experiments.
Representative Results
Here representative examples for cloning, amplification and purification of HCAdV preparations are shown. An overview of the cloning strategy (Figure 1) and representative examples for cloning and release of the HCAdV genome by restriction enzyme digest are provided (Figure 2). A typical restriction pattern after release of the GOI-expression cassette from pHM5 by PI-SceI and I-CeuI digest and subsequent phenol-chloroform extraction and EtOH precipitation (see also steps 1.2- 1.5) is shown (Figure 2A). Typically a ~3 kb band corresponding to the pHM5 plasmid backbone and a second band corresponding to the expression cassette of the GOI can be observed. The band containing the GOI-expression cassette is then purified from the agarose gel. The purified GOI-expression cassette is analyzed on an agarose gel together with the purified, dephosphorylated pAdVFTC that was digested with PI-SceI and I-CeuI respectively (step 1.7) to verify the correct size and to estimate the relative amount of molecules for subsequent ligation (Figure 2B). After ligation (see step 1.8) followed by phenol-chloroform extraction and EtOH precipitation and SwaI digest to eliminate uncut pAdFTC and subsequent phenol-chloroform extraction and EtOH precipitation (step 1.9), ligation products are transformed into bacteria and clones are selected on ampicillin containing LB- agar plates. Usually dozens to hundreds of clones are obtained, from which 5- 10 clones are prepared for plasmid DNA mini-preparations. A restriction pattern of an analytical HincII digest of positive and negative clones together with an empty pAdFTC as negative control (see also step 1.10) is shown (Figure 2C). Here positive clones do not show a 5 kb band, that is present in empty pAdFTC and negative clones and a 1.7 kb fragment appears that is not present in the empty pAdFTC and negative clones. For this respective GOI this indicates tha cloning was successful. A positive clone that has been purified via midiprep was then digested with NotI to release the HCAdV-genome carrying the GOI from pAdVFTC-GOI (step 2.2). Finally NotI digested DNA ist purified twice using phenol-chloroform extraction and EtOH precipitation to ensure high purity of the DNA for subsequent transfection into 116 cells. Analysis of a small fraction of the NotI digested pAdVFTC-GOI on an agarose gel shows a ~9 kb band corresponding to the pAdFTC plasmid backbone and a large band of up to ~36 kb (depending on the size of the GOI expression cassette) corresponding to the HCAdV genome (Figure 2D). An overview of the amplification and purification pipeline is schematically shown (Figure 3). Representative examples of the monitoring of the vector pre-amplification process are provided (Figure 4). A typical amplification curve of viral genomes during the pre-amplification process by serial passaging of viral lysates, each time coinfecting with HV (section 2), that was monitored by qPCR (section 3) is shown (Figure 4A). Note that the number of vector genomes usually descreases during the first passages and strongly increases in later passages. A vector copy number of 106- 107 per 15000 cells is sufficient to infect 116 cells for large scale production in a 3 l spinner flask. If the GOI within the HCAdV vector genome contains a GFP expression cassette, the pre-amplification process (section 2) can also be monitored by fluorescent microscopy. Representative pictures of each passage are shown (Figure 4B). Note that the number of GFP positive cells usually decreases during first passages and strongly increases in late passages. If the number of GFP positive cells reaches ~100%, the vector was amplified sufficiently to infect 116 cells for large scale production in a 3 l spinner flask. The large scale amplification process of a HCAdV containing GFP expression cassette was monitored by microscopic analysis of culture medium and cells that was transfered from the spinner flask to a tissue culture dish. Microscopic pictures of cells producing HCAdV in suspension culture (see step 4.4 and 4.11) are provided (Figure 4C). Note that cells are growing in clumps indicating that the preceeding cell growth was sufficient and cells are healthy. When using fluorescent microscopy ~100 % of these cells were GFP positive, indicating efficient transduction of suspension cells in the bioreactor and efficient vector production. Vector particles were purified from the medium and cells of the 3 l suspension culture by CsCl ultracentrifugation. Representative pictures of the HCAdV vector purification using CsCl density gradient ultracentrifugation are shown (Figure 5). After an initial centrifugation using a CsCl step gradient usually two bands are observed. The upper band contains empty of partial capsids whereas the lower band is comprised of DNA containing vector particles (step 5.4- 5.5) (Figure 5A). DNA containing vector particles are subsequently harvested (step 5.6) and pooled for a second ultracentrifugation step to concentrate the vector particles. This step usually results in a single band of DNA containing vector particles that is finally harvested for subsequent dialysis (step 5.7- 5.8) (Figure 5B). Dialyzed vector preparations were characterized by measuring the number of absolute particles, infectious particles and HV contamination. A schematic outline of the titration procedure (section 6 and 7) is provided (Figure 6). Representative results for characterization of the final vector preparation are shown (Figure 7). The bar chart shows typical vector particle numbers that are obtained by the procedure that is described in this protocol. Absolute particles numbers of vector particles were determined by optical density (section 6), infectious vector particles and HV contamination were measured by qPCR (section 7) (Figure 7A). Experience shows, that the OD titer overestimates the number of virus particles. Absolute viral particles measured by OD can be 20- 500 times higher than infectious viral particles measured by q-PCR. Infectious vector particles usally range between 5- 10 % of absolute particles when both were measured by q-PCR (section 7). If the vector preparation has a very good quality, more than 20% of absolute particles are infectious. The typical ratio between HV and infectious HCAdV particles (step 7.14) usually ranges between 1-5 % of infectious particles (Figure 7B). This is acceptable as the HV is replication deficient. Nevertheless as less HV contamination as possible (< 1 % of infectious particles) would be preferred.
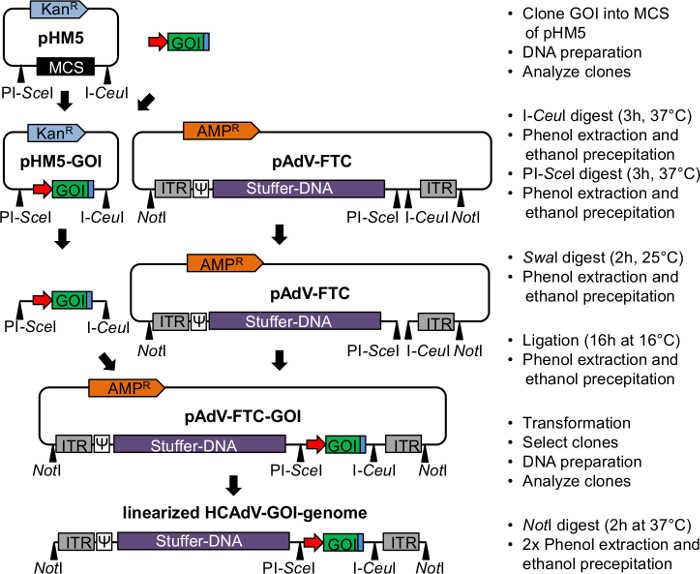 Figure 1. Flowchart of cloning the GOI into pAdFTC. The GOI is cloned into the MCS of the pHM5 shuttle plasmid. Plasmids pAdFTC and pHM5-GOI are digested with I-CeuI and PI-SceI and purified by phenol-chloroform extraction and EtOH precipitation. The digested pHM5 is loaded on a preparative agarose gel to purify the gene of interest (see also step 1.5), whereas the digested pAdFTC plasmid is dephosphorylated. Subsequently the ligation reaction with CIP-treated pAdFTC and the transgene expression cassette is set up (see step 1.8) and after completion purified by phenol-chloroform extraction and EtOH precipitation. The subsequent SwaI digest of the ligation product followed by phenol-chloroform extraction and EtOH precipitation prevents growth of clones carrying pAdFTC without insert. Black triangles indicate restriction enzyme recognition sites; grey boxes indicate AdV5 inverted terminal repeats (ITR), white boxes marked with Ψ indicate AdV5 packaging signal, red arrow indicates promoter, green boxes indicate transgene (gene of interest, GOI) to be expressed, blue boxes indicate polyadenylation signal, orange or blue arrows indicate antibiotic resistance genes of the plasmid backbone, respectively. Please click here to view a larger version of this figure.
Figure 1. Flowchart of cloning the GOI into pAdFTC. The GOI is cloned into the MCS of the pHM5 shuttle plasmid. Plasmids pAdFTC and pHM5-GOI are digested with I-CeuI and PI-SceI and purified by phenol-chloroform extraction and EtOH precipitation. The digested pHM5 is loaded on a preparative agarose gel to purify the gene of interest (see also step 1.5), whereas the digested pAdFTC plasmid is dephosphorylated. Subsequently the ligation reaction with CIP-treated pAdFTC and the transgene expression cassette is set up (see step 1.8) and after completion purified by phenol-chloroform extraction and EtOH precipitation. The subsequent SwaI digest of the ligation product followed by phenol-chloroform extraction and EtOH precipitation prevents growth of clones carrying pAdFTC without insert. Black triangles indicate restriction enzyme recognition sites; grey boxes indicate AdV5 inverted terminal repeats (ITR), white boxes marked with Ψ indicate AdV5 packaging signal, red arrow indicates promoter, green boxes indicate transgene (gene of interest, GOI) to be expressed, blue boxes indicate polyadenylation signal, orange or blue arrows indicate antibiotic resistance genes of the plasmid backbone, respectively. Please click here to view a larger version of this figure.
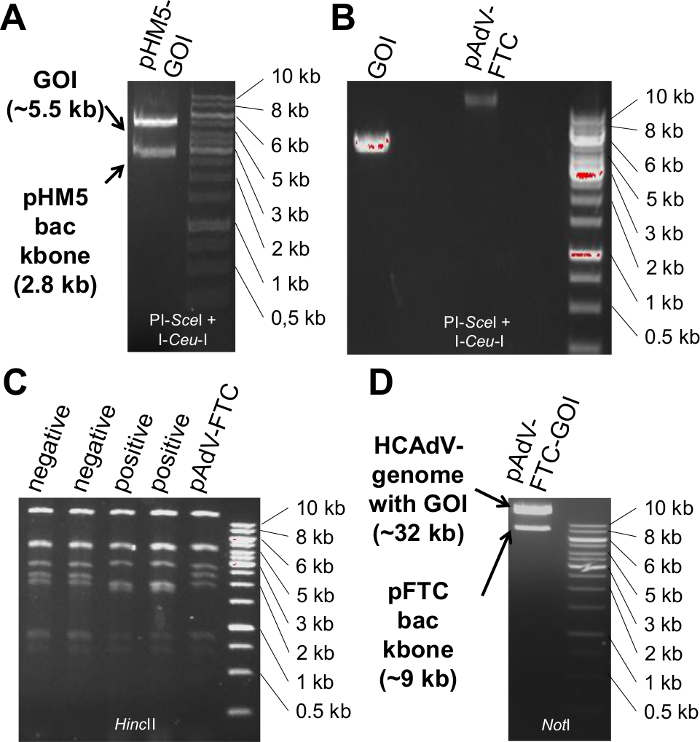 Figure 2. Representative results for the cloning procedure and release of the HCAdV genome from pAdVFTC. (A) Release of GOI-expression cassette from pHM5 by PI-SceI and I-CeuI digest (see also steps 1.2- 1.5). (B) purified GOI-expression cassette and purified, dephosphorylated pAdVFTC digested with PI-SceI and I-CeuI respectively (step 1.7). (C) Analytical HincII digest of pAdVFTC clones with and without GOI (see also step 1.10). (D) Release of the HCAdV-genome carrying the GOI from pAdVFTC-GOI by NotI digest (step 2.2) Please click here to view a larger version of this figure.
Figure 2. Representative results for the cloning procedure and release of the HCAdV genome from pAdVFTC. (A) Release of GOI-expression cassette from pHM5 by PI-SceI and I-CeuI digest (see also steps 1.2- 1.5). (B) purified GOI-expression cassette and purified, dephosphorylated pAdVFTC digested with PI-SceI and I-CeuI respectively (step 1.7). (C) Analytical HincII digest of pAdVFTC clones with and without GOI (see also step 1.10). (D) Release of the HCAdV-genome carrying the GOI from pAdVFTC-GOI by NotI digest (step 2.2) Please click here to view a larger version of this figure.
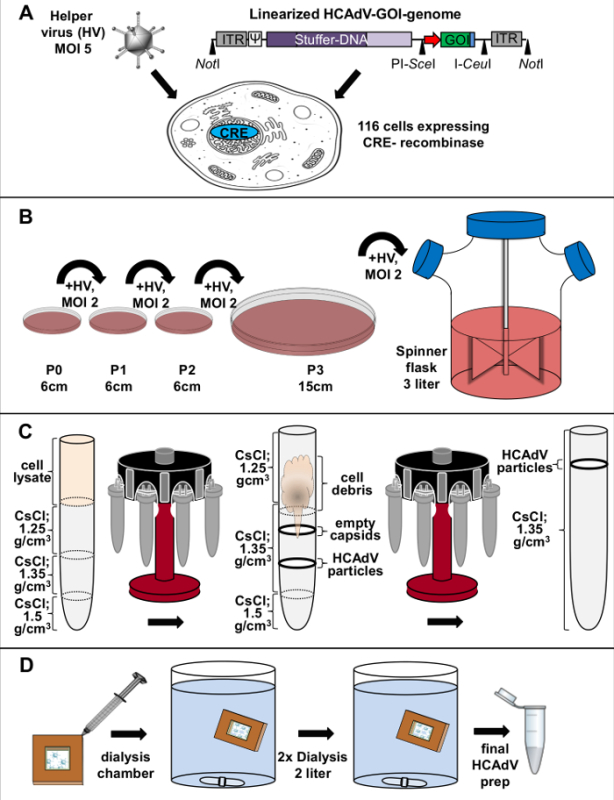 Figure 3. Schematic diagram for high-capacity adenoviral vector amplification and purification. (A) HCAdV DNA transfection and helper virus (HV) infection: Transfection of linearized HCAdV genome carrying the GOI into the HCAdV producer cell line (116 cells [4]) and subsequent infection with the HV AdNG163R-2 [4]. (B) Amplification of HCAdV: After pre-amplification steps by serially transferring cell lysate to a new tissue culture dish and co-infecting with HV, large-scale production is performed in a 3 L suspension culture. (C) HCAdV purification: For purification, virus is isolated by ultracentrifugation using cesium chloride gradients. (D) Dialysis: Purified HCAdV particles are dialyzed against 2 L storage buffer. HCAdV, high-capacity adenoviral vector; ITR, adenovirus serotype 5 inverted terminal repeat; Ψ, packaging signal; HV, helper virus; MOI, multiplicity of infection. Please click here to view a larger version of this figure.
Figure 3. Schematic diagram for high-capacity adenoviral vector amplification and purification. (A) HCAdV DNA transfection and helper virus (HV) infection: Transfection of linearized HCAdV genome carrying the GOI into the HCAdV producer cell line (116 cells [4]) and subsequent infection with the HV AdNG163R-2 [4]. (B) Amplification of HCAdV: After pre-amplification steps by serially transferring cell lysate to a new tissue culture dish and co-infecting with HV, large-scale production is performed in a 3 L suspension culture. (C) HCAdV purification: For purification, virus is isolated by ultracentrifugation using cesium chloride gradients. (D) Dialysis: Purified HCAdV particles are dialyzed against 2 L storage buffer. HCAdV, high-capacity adenoviral vector; ITR, adenovirus serotype 5 inverted terminal repeat; Ψ, packaging signal; HV, helper virus; MOI, multiplicity of infection. Please click here to view a larger version of this figure.
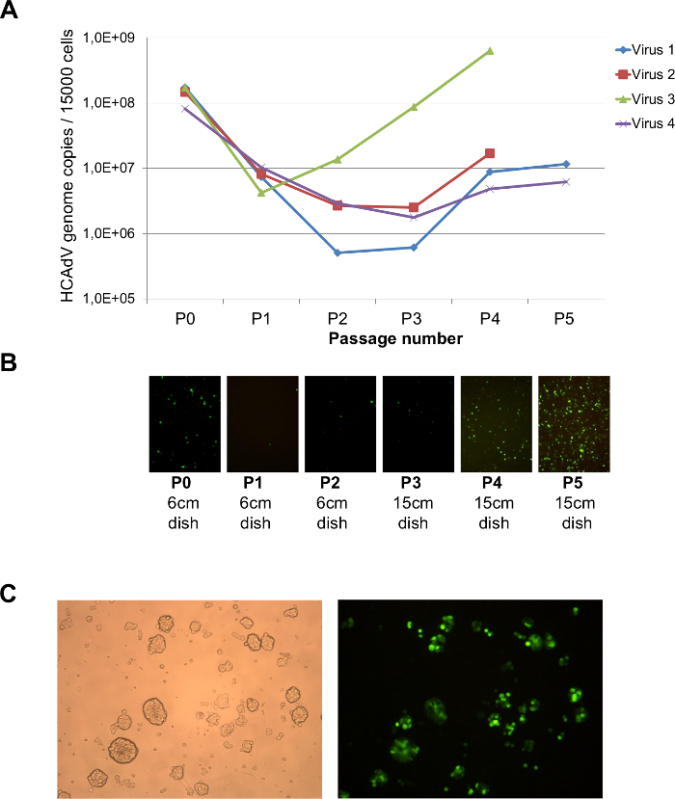 Figure 4. Representative results of the amplification process. (A) Monitoring of the pre-amplification process by amplification of viral genomes using qPCR (see also section 3.); (B) If the GOI contains GFP as a transgene, the pre-amplification process can be monitored by fluorescent microscopy. (C) Representative results of the large scale amplification process in a spinner flask examplified for HCAdV encoding GFP. The left panel shows a microscopic picture of infected 116 cells from the spinner flask. A sample of culture medium and cells were transferred to a 60 mm tissue culture dish. Note that clumps of amplified cells are visible. The right panel shows the same picture using fluorescent microscopy. Almost 100 % of cells are transduced with HCAdV carrying GFP. Please click here to view a larger version of this figure.
Figure 4. Representative results of the amplification process. (A) Monitoring of the pre-amplification process by amplification of viral genomes using qPCR (see also section 3.); (B) If the GOI contains GFP as a transgene, the pre-amplification process can be monitored by fluorescent microscopy. (C) Representative results of the large scale amplification process in a spinner flask examplified for HCAdV encoding GFP. The left panel shows a microscopic picture of infected 116 cells from the spinner flask. A sample of culture medium and cells were transferred to a 60 mm tissue culture dish. Note that clumps of amplified cells are visible. The right panel shows the same picture using fluorescent microscopy. Almost 100 % of cells are transduced with HCAdV carrying GFP. Please click here to view a larger version of this figure.
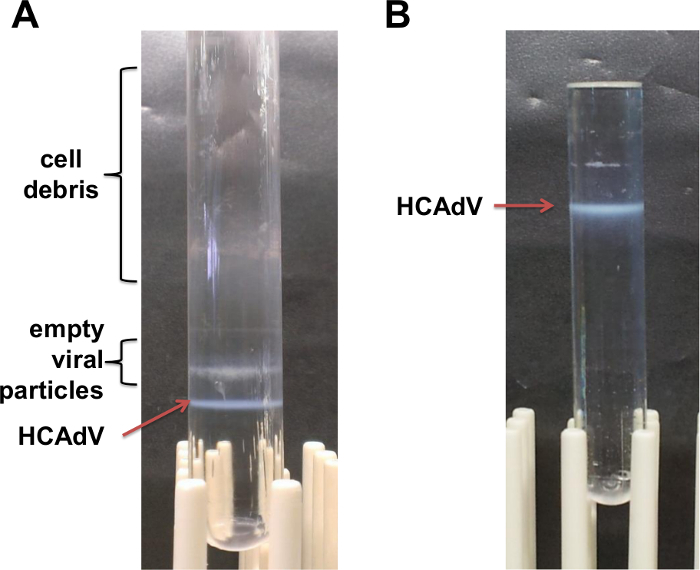 Figure 5. Representative results after CsCl gradient centrifugation. (A) After performing a step gradient (see also step 5.5); (B) After continuous gradient (see also step 5.7). Please click here to view a larger version of this figure.
Figure 5. Representative results after CsCl gradient centrifugation. (A) After performing a step gradient (see also step 5.5); (B) After continuous gradient (see also step 5.7). Please click here to view a larger version of this figure.
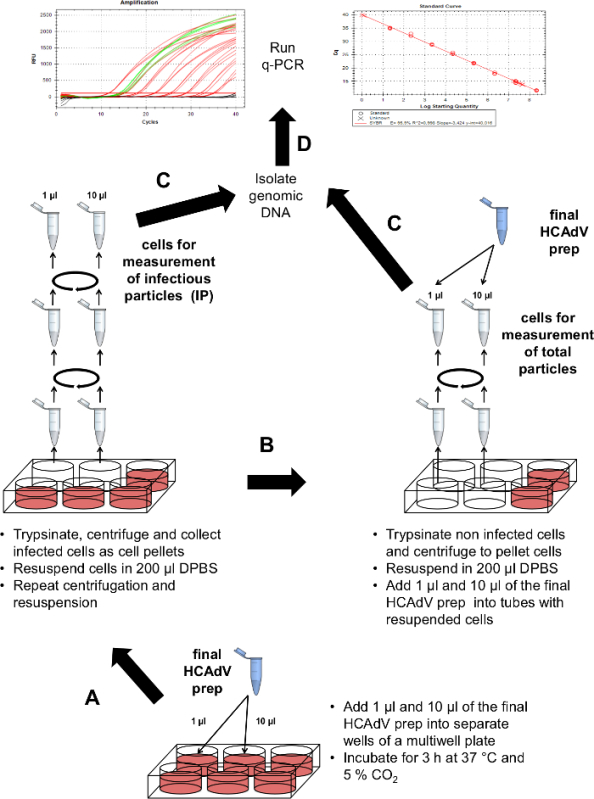 Figure 6. Schematic outline of the titration procedure. (A) Measurement of infectious particles: Cells in a multi-well plate are infected with different volumes of purified virus and collected as cell pellets. (B) Measurement of absolute particles: Unifected cells form a multi-well are collected by trypsinization and centrifugation. After resuspension purified virus is added and genomic DNA is isolated. (C) Isolation of genomic DNA. (D) To quantify viral genome copy numbers a quantitative PCR (q-PCR) is performed. Please click here to view a larger version of this figure.
Figure 6. Schematic outline of the titration procedure. (A) Measurement of infectious particles: Cells in a multi-well plate are infected with different volumes of purified virus and collected as cell pellets. (B) Measurement of absolute particles: Unifected cells form a multi-well are collected by trypsinization and centrifugation. After resuspension purified virus is added and genomic DNA is isolated. (C) Isolation of genomic DNA. (D) To quantify viral genome copy numbers a quantitative PCR (q-PCR) is performed. Please click here to view a larger version of this figure.
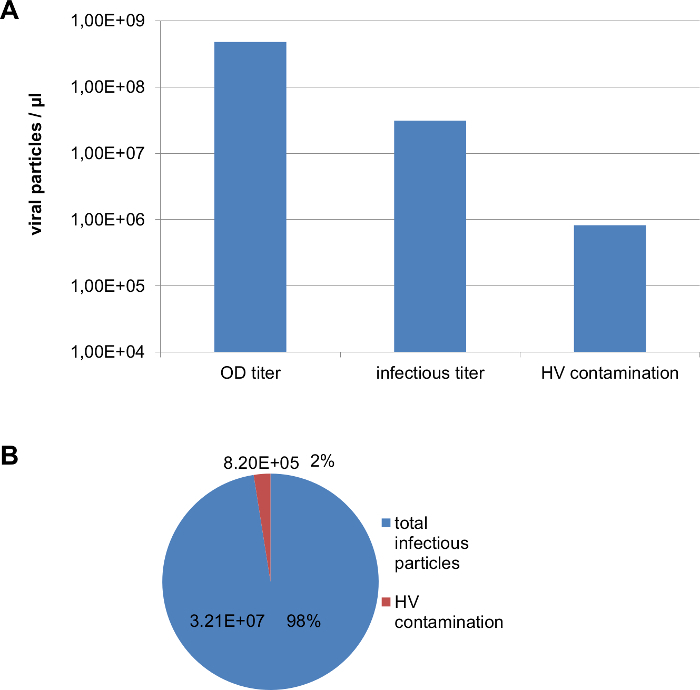 Figure 7. Representative results for characterization of the final vector preparation. (A) Absolute numbers of particles of a vector preparation according to the presented protocol determined with different methods: OD titer measured by optical density (section 6), infectious titer and HV contamination measured by qPCR (section 7). (B) Ratio between total infectious HCAdV particles and HV contamination levels measured by qPCRs (section 7). Please click here to view a larger version of this figure.
Figure 7. Representative results for characterization of the final vector preparation. (A) Absolute numbers of particles of a vector preparation according to the presented protocol determined with different methods: OD titer measured by optical density (section 6), infectious titer and HV contamination measured by qPCR (section 7). (B) Ratio between total infectious HCAdV particles and HV contamination levels measured by qPCRs (section 7). Please click here to view a larger version of this figure.
Discussion
The protocol presented here allows purification of HCAdV vectors based on human adenovirus type 5 based on previously described procedures 4,12. The HCAdV genome within the pAdFTC plasmid is devoid of all adenovirus genes and only carries the 5'- and 3'- ITRs and the packaging signal. In this strategy the HV AdNG163R-2 4 provides all necessary genes for efficient virus production in trans. This offers a packaging capacity of up to 35 kb, which clearly outcompetes first and second generation AdVs or the widely used lentivirus (LV) - or adeno associated virus (AAV) based vectors. A second advantage of HCAdV especially for in vivo applications is the fact that in contrast to first- or second generation AdVs they display reduced levels of cytotoxic and immunogenic side effects caused by the expression of viral proteins 5-8.
Nevertheless the protocol described here is more time and work intensive than for other viral vectors such as LV- or AAV-vectors as well as first- or second generation AdVs. A major obstacle is the cloning of large transgenes and the subsequent transfer to the virus production plasmid pAdV-FTC. The use of homing endonucleases PI-SceI and I-CeuI offer precise and directed insertion of the transgene from the shuttle plasmid pHM5, but have the disadvantage of strongly sticking to the cleaved DNA. For that reason many phenol-chloroform cleanup steps are necessary when preparing plasmid- and insert-DNA during the cloning procedure. Therefore, rigorously following the provided cloning protocol is strictly recommended. After cloning the HCAdV genome is released from the plasmid pAdFTC by NotI restriction enzyme digest and subsequently transfected into the producer cell line. Note that initial transfection efficiencies are crucial for achieving sufficient virus amplification. Importantly, the protocol offers several steps from which the procedure can be restarted in case subsequent steps fail. During pre-amplification one third of the lysates can be stored as backup to start the procedure from that point again or to shorten a repetition in case more virus needs to be produced.
HCAdV carrying large, complex or several transgenes or certain stuffer DNA may be amplifying slower than expected. Therefore, it is crucial to carefully follow the pre-amplification process before suspension culture is grown in a spinner flask. If pre-amplification is not successful, the procedure should be stopped and started from the beginning again. If pre-amplification is inefficient, additional passaging may be necessary until the amount of virus is high enough to infect the cells grown in a 3 L spinner flask. As an alternative to suspension culture in a spinner flask, 20 to 30 150 mm tissue culture dishes can be used for the final passage. However, this may be more time and work intensive.
During CsCl gradient centrifugation it is be beneficial to split the lysate from the spinner flask into two fractions and spin them separately as this will not dramatically reduce virus titers. As soon as infective HCAdV are successfully produced it is relatively easy to re-amplify the viral stock by co-infecting a suspension culture from a spinner flask with purified viral particles and HV.
For qPCR measurements gDNA can either be purified via the protocol mentioned here or any commercially available kit for the isolation of gDNA from cultured cells. By using commercially available kit the protocol can be shortened one day but a varying portion of the HCAdV genome copies that are present in infected cells may be lost during purification, depending on the kit used. Therefore HCAdV genomes will be slightly underrepresented in following q-PCR measurements when compared to the method presented in step 7.4).
Obtained total infectious titers in one final vector preparation derived from one spinner flask range from 1 x 1010 to 5 x 1011 infectious particles. This amount is sufficient to perform numerous in vitro experiments. Dependent on the target cells, also several in vivo experiments can be conducted. For instance, for achieving sufficient liver transduction after systemic administration 1 x 109 infectious particles are required.
In summary, the wide cell tropism and the possibility to produce capsid-modified adenovirus together with the improved safety profile of HCAdV vectors devoid of all viral genes render these HCAdV vector system a highly valuable tool for gene therapeutic applications.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
This work was supported by DFG grant EH 192/5-1 (A.E.), the EU (E-rare-2) project Transposmart (A.E.), the UWH Forschungsförderung (E.S. and W.Z), and the PhD programme of the University Witten/Herdecke (P.B.). J.L. was supported by a stipend of the Chinese Scholarship council and T.B. and M.G by the Else Kröner-Fresenius foundation (EKFS).
References
- Benihoud K, Yeh P, Perricaudet M. Adenovirus vectors for gene delivery. Curr Opin Biotechnol. 1999;10(5):440–447. doi: 10.1016/s0958-1669(99)00007-5. [DOI] [PubMed] [Google Scholar]
- Crystal RG. Adenovirus: the first effective in vivo gene delivery vector. Hum Gene Ther. 2014;25(1):3–11. doi: 10.1089/hum.2013.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks RJ, et al. A helper-dependent adenovirus vector system: removal of helper virus by Cre-mediated excision of the viral packaging signal. Proc Natl Acad Sci U S A. 1996;93(24):13565–13570. doi: 10.1073/pnas.93.24.13565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer D, Ng P. Improved system for helper-dependent adenoviral vector production. Mol Ther. 2003;8(5):846–852. doi: 10.1016/j.ymthe.2003.08.014. [DOI] [PubMed] [Google Scholar]
- Morral N, et al. High doses of a helper-dependent adenoviral vector yield supraphysiological levels of alpha1-antitrypsin with negligible toxicity. Hum Gene Ther. 1998;9(18):2709–2716. doi: 10.1089/hum.1998.9.18-2709. [DOI] [PubMed] [Google Scholar]
- Muruve DA, et al. Helper-dependent adenovirus vectors elicit intact innate but attenuated adaptive host immune responses in vivo. J Virol. 2004;78(11):5966–5972. doi: 10.1128/JVI.78.11.5966-5972.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrhardt A, et al. A gene-deleted adenoviral vector results in phenotypic correction of canine hemophilia B without liver toxicity or thrombocytopenia. Blood. 2003;102(7):2403–2411. doi: 10.1182/blood-2003-01-0314. [DOI] [PubMed] [Google Scholar]
- Cots D, Bosch A, Chillon M. Helper dependent adenovirus vectors: progress and future prospects. Curr Gene Ther. 2013;13(5):370–381. doi: 10.2174/156652321305131212125338. [DOI] [PubMed] [Google Scholar]
- Mizuguchi H, Kay MA. Efficient construction of a recombinant adenovirus vector by an improved in vitro ligation method. Hum Gene Ther. 1998;9(17):2577–2583. doi: 10.1089/hum.1998.9.17-2577. [DOI] [PubMed] [Google Scholar]
- Mizuguchi H, Kay MA. A simple method for constructing E1- and E1/E4-deleted recombinant adenoviral vectors. Hum Gene Ther. 1999;10(12):2013–2017. doi: 10.1089/10430349950017374. [DOI] [PubMed] [Google Scholar]
- Ehrhardt A, Kay MA. A new adenoviral helper-dependent vector results in long-term therapeutic levels of human coagulation factor IX at low doses in vivo. Blood. 2002;99(11):3923–3930. doi: 10.1182/blood.v99.11.3923. [DOI] [PubMed] [Google Scholar]
- Jager L, et al. A rapid protocol for construction and production of high-capacity adenoviral vectors. Nat Protoc. 2009;4(4):547–564. doi: 10.1038/nprot.2009.4. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 1-3. New York: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]