

Abstract
Background
Mouse models offer an essential tool to unravel the impact of genetic mutations on autism-related phenotypes. The behavioral impact of some important candidate gene models for autism spectrum disorder (ASD) has not yet been studied, and existing characterizations mostly describe behavioral phenotypes at adult ages, disregarding the developmental nature of the disorder. In this context, the behavioral influence of CNTN4, one of the strongest suggested ASD candidate genes, is unknown. Here, we used our recently established developmental test battery to characterize the consequences of disruption of contactin 4 (Cntn4) on neurological, sensory, cognitive, and behavioral phenotypes across different developmental stages.
Methods
C57BL/6J mice with heterozygous and homozygous disruption of Cntn4 were studied through an extensive, partially longitudinal, test battery at various developmental stages, including various paradigms testing social and restricted repetitive behaviors.
Results
Developmental neurological and cognitive screenings revealed no significant differences between genotypes, and ASD-related behavioral domains were also unchanged in Cntn4-deficient versus wild-type mice. The impact of Cntn4-deficiency was found to be limited to increased startle responsiveness following auditory stimuli of different high amplitudes in heterozygous and homozygous Cntn4-deficient mice and enhanced acquisition in a spatial learning task in homozygous mice.
Conclusions
Disruption of Cntn4 in the C57BL/6J background does not affect specific autism-related phenotypes in developing or adult mice but causes subtle non-disorder specific changes in sensory behavioral responses and cognitive performance.
Electronic supplementary material
The online version of this article (doi:10.1186/s11689-016-9140-2) contains supplementary material, which is available to authorized users.
Keywords: CNTN4, Autism spectrum disorder, 3p deletion syndrome, Developmental trajectories, Mouse model, Behavior, Reversal learning, Negative findings, Hyperreactivity, Schizophrenia
Background
Autism spectrum disorder (ASD) is a behaviorally defined developmental disorder with a strong genetic component [1, 2]. The identification of genetic risk factors such as common genetic variants, rare inherited and de novo mutations have lead to the implication of hundreds of different genes [3, 4]. These findings illustrate the complexity and heterogeneity of the genetic architecture of ASD.
Subsequently, mouse models are being used to unravel the functional impact of implicated genes on ASD phenotypes in a controlled genetic and environmental background. However, knowledge of the impact on cognitive and behavioral development of the majority of these genes is missing or incomplete [5], and most behavioral characterizations of animal models are limited to adult phenotypes disregarding the developmental nature of ASD [6].
Here, we characterize the impact of contactin 4 (Cntn4) null mutation on behavioral development, using our recently developed longitudinal test battery for mice that tests a wide array of neurological, cognitive, and behavioral parameters across development starting from 3 weeks of age [6].
CNTN4 is an axonal glycoprotein belonging to the contactin family, a six-member subgroup of the immunoglobulin superfamily of cell adhesion molecules [7]. CNTN4 is known to act as an axon guidance molecule in the establishment of olfactory neural circuitry during neural development [8] and promotes target-specific axon arborization of a subset of retinal ganglion cells onto the nucleus of the optic tract [9]. Knowledge of the neurobiological functions of CNTN4 in normal and abnormal development of brain systems are far from complete.
The CNTN4 gene has been implicated in ASD due to its presence in the genetic locus of the 3p-deletion syndrome, a mental retardation syndrome [10]. Subsequently, evidence for a role of CNTN4 has been accumulated [11–14] but has also been questioned [15, 16]. Three cases carrying a copy number variant (CNV) in the CNTN4 gene were reported by the Autism Genome Project Consortium (AGP) [12]. Deletions and duplications in the CNTN4 gene or its promoter region were found in 10 families of the Autism Genetic Resource Exchange (AGRE) collection [13]. In the Autism Case-Control cohort (ACC), deletion in the promoter region of CNTN4 was found in three cases but not in controls [13]. Association of CNTN4 with developmental disorders such as ASD further seems supported by the protein’s neurobiological functions [2, 7].
Follwoing these observations, we performed a careful longitudinal functional characterization of homozygous and heterozygous disruptions of Cntn4 to resolve the impact of this gene on behavioral and cognitive development.
Methods
Generation and breeding of Cntn4 mice
Cntn4-deficient mice were kindly provided by Dr. Yoshihiro Yoshihara (RIKEN, Japan) [8]. These mice were generated using a standard gene-targeting method as previously described. A targeting vector was designated to mutate the translation start codon (ATG) in the exon 2 of the Cntn4 gene into a stop codon (TAG) and introduce a pgk-neo selection marker. Consequently, these mice were backcrossed with C57BL/6 mice more than nine times. Upon arrival in the University Medical Center Utrecht, the mice were re-derived, followed by heterozygous breeding for the use in our experiments.
All animals were born and weaned at the University Medical Center Utrecht. Average nest size of Cntn4 litters was 7.2, and the litters larger than 10 animals per litter were culled back to average 7. The minimum litter size used for behavioral experiments was four animals per litter. Detailed information on the genotyping of Cntn4 mice is provided in Additional file 1.
Given the extensive number of tests, the mice were spread over four different batches. Table 1 provides the order of behavioral testing and the number of animals per genotype per batch. Phenotypic assessments in batches 1, 2, and 3 were performed at the University Medical Center Utrecht. Batch 4 was transported and tested at Sylics (Synaptologics BV, Amsterdam, The Netherlands).
Table 1.
Overview of the behavioral tests per batch
| Age | Task | Batch | Subjects (n) |
|---|---|---|---|
| 3 weeks | Juvenile social interaction | Batch 1 + 2 | 6–7 genotype-matched pairs per genotype |
| 4 weeks | Extended SHIRPA screen | Batch 1 + 2 | 19–26 per genotype |
| 6 weeks | Extended SHIRPA screen | Batch 1 + 2 | 19–26 per genotype |
| 8 weeks | Extended SHIRPA screen | Batch 1 + 2 | 19–26 per genotype |
| Adult | Extended SHIRPA screen | Batch 1 | 10–15 per genotype |
| Open field | Batch 1 | 10–15 per genotype | |
| Elevated plus maze | Batch 1 | 10–15 per genotype | |
| Social discrimination | Batch 1 | 10–15 per genotype | |
| Buried food test | Batch 2 | 9–11 per genotype | |
| Set shifting-reversal task | Batch 2 | 9–11 per genotype | |
| Social approach in 3-chamber | Batch 3 | 12 per genotype | |
| Novel object exploration task | Batch 3 | 12 per genotype | |
| Barnes maze-reversal task | Batch 4 | 16 per genotype | |
| Pre-pulse inhibition | Batch 4 | 16 per genotype |
Animals in batch 1 + 2 were weaned at post-natal day 21. Batch 3 + 4 animals were weaned at post-natal day 28
All experiments were approved by the ethical committee for animal experimentation of the University Medical Center Utrecht and Free University Amsterdam and performed according to the institutional guidelines that are in full compliance with the European Council Directive (86/609/EEC).
Developmental neurological and behavioral screening
Cntn4−/−, Cntn4+/−, and wild-type male littermates were subjected to our previously described longitudinal screening strategy (extended SHIRPA battery) testing an array of neurological, behavioral, and cognitive parameters at 4, 6, 8, and 11 weeks of age [6, 17]. The longitudinal test battery includes the assessment of autism-related traits such as motor stereotypies (e.g., self-grooming) and sensorimotor coordination (e.g., latency to fall from the rotarod). Detailed information on behavioral testing is provided in Additional file 1.
Screening of social behaviors and restricted repetitive behaviors
Abnormalities in social interaction behaviors were assessed in the juvenile social interaction test (3 weeks of age) [18], followed by a three-chamber social approach [18], and a 2-day social discrimination paradigm in adult age animals [19]. Stereotypic movements, restricted interests, and repetitive patterns of behavior were analyzed in the novel object investigation task during exposure to four novel toys [20]. Cognitive flexibility was assessed by multi-trial associative learning in an extensive set-shifting paradigm [6], as well as a Barnes maze spatial learning task including reversal [21]. Acoustic startle response and sensorimotor gating were assessed in the pre-pulse inhibition (PPI) test [19]. Anxiety-related behaviors were tested in the elevated plus maze and open field [6]. Statistical analyses are described in the Additional file 1.
Results
eSHIRPA assays
The eSHIRPA (extended SmithKline Beecham, Harwell, Imperial College and Royal London Hospital phenotype assessment) screen did not show differences between the Cntn4+/− mice, Cntn4−/− mice, and wild-type controls at 4, 6, 8, or 10 weeks of age in general health, body weight, and neurological reflexes nor in the development of various locomotor parameters including total distance moved, movement velocity, and movement duration (Figs. 1a–d, Table 2). Moreover, we found no developmental differences in the amount of self-grooming or sensorimotor coordination on the rotarod (Figs. 1e–f).
Fig. 1.
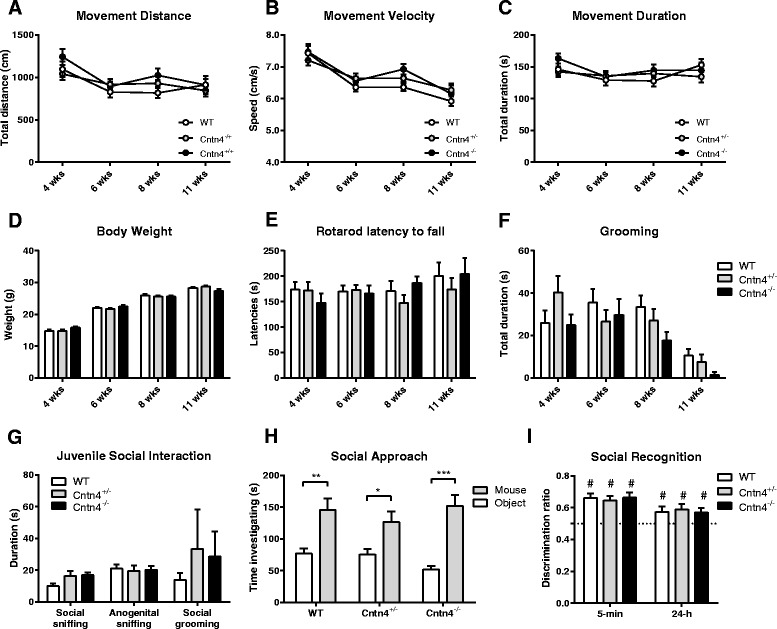
Developmental neurological and behavioral screen and analysis of social behaviors in Cntn4-deficient mice. a Distance moved (rmANOVA genotype, F (2,63) = 0.760, p = .472), b movement velocity (rmANOVA genotype, F (2,63) = 1.256, p = .292), c movement duration (rmANOVA genotype, F (2,63) = 0.342, p = .479), d body weight (rmANOVA genotype, F (2,63) = 0.588, p = .558), e latency to fall of the accelerating rotarod (rmANOVA genotype, F (2,64) = 0.110, p = .896), and f time spent self-grooming (rmANOVA genotype, F (2,64) = 1.038, p = .360) at pre-adolescence (4 weeks), adolescence (6 weeks), early adulthood (8 weeks), and adulthood (10 weeks) (n = 19–26 per genotype) during the eSHIRPA test. g Social sniffing (owANOVA, F (2,16) = 2.926, p = .083), anogenital sniffing (owANOVA, F (2,16) = 0.055, p = .946), and social grooming (owANOVA, F (2,16) = 0.334, p = .721) during the juvenile social interaction test in genotype-matched mice at post-natal day 21 (n = 6–7 pairs of genotype-matched interacting animals per genotype). h Social exploration (owANOVA between genotypes, F (2,32) = 0.599, p = .556) as a function of exploration of the cage with the novel mouse versus the empty cage during the three-chamber task (n = 12 per genotype). i Social exploration during the social discrimination test following a 5-min inter-trial interval (owANOVA between genotypes F (2,36) = 0.138, p = .872) and a 24-h inter-trial interval (owANOVA between genotypes F (2,36) = 0.096, p = .909), with exploration of the novel mouse as fraction of the total duration of social exploration (n = 10–15 per genotype). Data are presented as means ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; #p < 0.05
Table 2.
Physical and neurological features during the different phases of the extended SHIRPA primary screen.
| Age (weeks) | 4 | 6 | 8 | Adult | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Test | WT | +/− | −/− | Sig. | WT | +/− | −/− | Sig. | WT | +/− | −/− | Sig. | WT | +/− | −/− | Sig. |
| Subjects (n) | 19 | 26 | 22 | 19 | 26 | 22 | 19 | 26 | 22 | 13 | 15 | 10 | ||||
| Body position (active) | 95 | 96 | 100 | 0.22 | 100 | 100 | 100 | 1.00 | 100 | 96 | 100 | 0.46 | 100 | 100 | 100 | 1.00 |
| Body position (inactive) | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | ||||
| Body position (excessive activity) | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||||
| Tremor (absent) | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 |
| Palpebral closure (eyes open) | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 |
| Coat appearance tidy and groomed | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 |
| Whiskers (present) | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 |
| Lacrimation (absent) | 100 | 96 | 100 | 0.45 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 |
| Defecation (quantity) | 1.9 | 1.6 | 2.0 | 0.47 | 3.1 | 2.5 | 1.9 | 0.06 | 2.8 | 2.3 | 2.4 | 0.61 | 3.8 | 3.7 | 3.2 | 0.70 |
| ± SEM | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.3 | 0.4 | 0.3 | 0.4 | 0.7 | 0.7 | 0.9 | ||||
| Transfer arousal (brief freeze) | 11 | 15 | 14 | 0.70 | 11 | 19 | 9 | 0.71 | 11 | 8 | 0 | 0.16 | 8 | 0 | 10 | 0.50 |
| Transfer arousal (immediate movement) | 89 | 85 | 86 | 89 | 81 | 91 | 89 | 92 | 100 | 92 | 100 | 90 | ||||
| Gait (fluid) | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 |
| Tail elevation (horizontal extension) | 100 | 100 | 100 | 1.00 | 95 | 100 | 100 | 0.28 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 |
| Tail elevation (straub tail) | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||||
| Startle response (preyer reflex) | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 |
| Touch escape (flight prior to touch) | 79 | 73 | 77 | 0.88 | 89 | 85 | 73 | 0.35 | 63 | 88 | 68 | 0.05 | 85 | 87 | 100 | 0.45 |
| Touch escape (response to touch) | 21 | 27 | 23 | 11 | 15 | 27 | 37 | 12 | 32 | 15 | 13 | 0 | ||||
| Positional passivity (struggles) | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 |
| Skin color (pink) | 95 | 100 | 100 | 0.28 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 |
| Skin color (blanched) | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||||
| Trunk curl (absent) | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 |
| Limb grasping (absent) | 100 | 92 | 100 | 0.20 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 |
| Pinna reflex (present) | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 |
| Corneal reflex (present) | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 |
| Contact righting reflex (present) | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 |
| Evidence of biting (none) | 79 | 77 | 77 | 0.99 | 84 | 88 | 100 | 0.18 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 |
| Grip (OK) | 100 | 96 | 100 | 0.36 | 95 | 96 | 100 | 0.59 | 100 | 100 | 100 | 1.00 | 100 | 100 | 90 | 0.24 |
| Full puberty | 89 | 81 | 100 | 0.30 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 | 100 | 100 | 100 | 1.00 |
Cntn4 +/−, Cntn4 −/−, and wild-type (WT) control mice were screened in the Perspex jar for body position (active, inactive, or excessively active), tremor (present or not), palpebral closure (eyes open or not), coat appearance (well groomed or irregularities like piloerection), whiskers (intact or trimmed), and lacrimation (present or not). In the arena, the mice were screened for transfer arousal (freezing or immediate movement), gait (fluid or not), tail elevation (dragging, horizontal, or straub tail), startle response (preyer reflex, no response, or additional response), and touch escape (response to touch or flight prior to touch while finger approaches). Mice were transferred out of the arena to observe positional passivity (struggling by different types of handling), skin color (color of plantar surface of forelimbs), trunk curl (forward curling with head to abdomen), limb grasping (clasping of rear limbs), pinna reflex (presence of ear retraction), corneal reflex (presence of eyeblink), contact righting reflex, evidence of biting, grip (grasping of grid), vocalization, and puberty (presence of sex organs). Sig. represents statistical significance of between-genotype differences. Data are presented as a percentage of the total number of animals per genotypes, except for defecation (count of the fecal boli)
Social interaction behavior
The juvenile social interaction test revealed no differences in the amount of social sniffing, anogenital sniffing, or social grooming (Fig. 1g) between the Cntn4+/−, Cntn4−/−, and wild-type control mice. In the adult three-chamber test, all genotypes showed a clear preference for exploration of a mouse over an object, and no genotype differences in the amount of social exploration were observed (Fig. 1h). Furthermore, genotypes were equally capable to distinguish between a familiar mouse and a novel mouse in a social recognition paradigm, both at 5 min and 24 h after initial exposure (Fig. 1i).
Restricted and repetitive behaviors
There were no differences in the grooming behavior between the Cntn4+/−, Cntn4−/−, and wild-type control mice at adult age, in line with the amount of grooming observed in the longitudinal eSHIRPA screening (Fig. 2a). We also found no genotype differences in restricted interest or in repetitive patterns of behavior in the novel object investigation task during the exploration of the four novel toys (Fig. 2b, c).
Fig. 2.
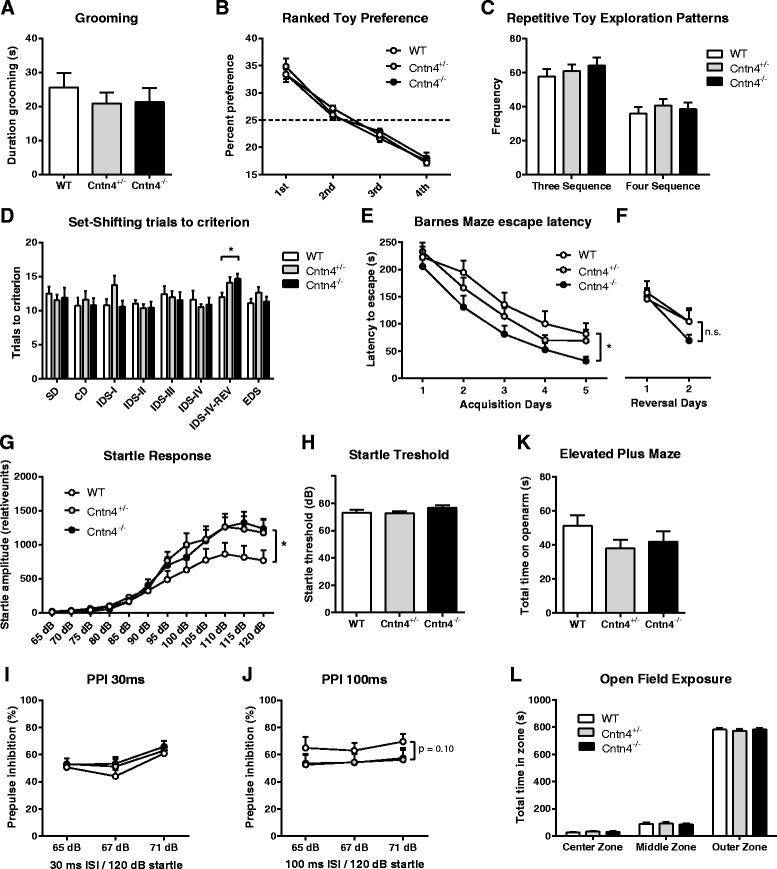
Restricted repetitive behaviors and sensory-sensitivity screening of adult Cntn4 mice. Restricted and repetitive behavior in the novel object investigation task. a Stereotypic movements as total time spent grooming (owANOVA, F (2,33) = 0.431, p = .653). b Restricted interest as frequency-based percentage preference of exploration of each of the four novel toys (1st preference owANOVA, F (2,33) = 0.446, p = .644; 2nd preference owANOVA, F (2,33) = 1.569, p = .223; 3rd preference owANOVA, F (2,33) = 1.208, p = .312; 4th preference owANOVA, F (2,33) = 0.236, p = .791). c Repetitive toy exploration patterns based on repetitive sequences of three elements (owANOVA, F (2,33) = 0.760, p = .476) and four elements (owANOVA, F (2,33) = 0.227, p = .798) (n = 12 per genotype). d Reversal learning during the set-shifting reversal-learning task. X-axis represents the different sub-tasks. Y-axis represents the total number of trials that were needed to reach the criterion of 8 correct digs in 10 consecutive trials (n = 9–11 per genotype). e–f Spatial learning and reversal learning during the Barnes maze paradigm. Y-axis represents the daily mean of latency to find the escape hole during e the acquisition phase (rmANOVA genotype, F (2,44) = 4.151, p = .022) and f the reversal-learning phase (rmANOVA genotype, F (2,43) = 0.830, p = .830) after replacing the escape to the other side of the maze (n = 16 per genotype). Startle and PPI results in Cntn4 mice, with g startle magnitude as function of startle stimulus in all genotypes (MANOVA, F (24,70) = 1.984, p = 0.014), h startle threshold (owANOVA, F (2,45) = 1.542, p = 0.225), i–j pre-pulse inhibition tested with different pre-pulse intensities with inter-stimulus interval (ISI) at 30 ms (two-way ANOVA, F (2,135) = 2.376, p = 0.096) and at 100 ms (two-way ANOVA, F (2,135) = 1.1927, p = 0.306; n = 16 per genotype). Anxiety behavior during the elevated plus maze test and open-field test measured as k elevated plus maze anxiety and as total time spent on the open arms (owANOVA, F (2,36) = 1.450, p = 0.248), l total time spent in the center (owANOVA, F (2,36) = 0.165, p = 0.848), middle (owANOVA, F (2,36) = 0.413, p = 0.665), and outer zones (owANOVA, F (2,36) = 0.125, p = 0.883) of the open field (n = 10–15 per genotype). Data are presented as means ± SEM; *p < 0.05. SD simple discrimination, CD compound discrimination, IDS I–IV intra-dimensional shift I–IV, IDS-reversal reversal of intra-dimensional shift IV (owANOVA, F (2,27) = 3.487, p = .045; Dunnett’s t, WT vs HET p = 0.037, WT vs KO p = 0.092), EDS extra-dimensional shift (owANOVA, F (2,27) = 1.416, p = .260)
We additionally analyzed cognitive flexibility through the assessment of reversal learning in a set-shifting task. Prior to this task, we ascertained intact olfactory capacities, as we found no genotype differences in the latency to find a buried piece of food. In the set-shifting task, all genotypes were equally able to associate a food reward with a specific digging material or odor, as was evident through the performance of simple, compound discrimination and intra- and extra-dimensional shifts (Fig. 2d).
The reversal-learning phase of this test yielded an inconclusive result, as the genotype effect that we observed on reversal learning was only significant (ANOVA p = 0.04) in one of the two outcome measures (i.e., the number of trials to reach the criterion but not in errors to reach criterion). Moreover, a shift cost in wild-type mice was observed in errors to reach criterion, although not in the number of trials to reach criterion. Given this inconclusive result, we tested reversal-learning performance in a different paradigm. In this Barnes maze reversal-learning paradigm, we confirmed that Cntn4 does not affect reversal learning, as all genotypes needed equal amount of time as well as distance before reaching the re-located escape hole (Fig. 2f). In contrast to the set-shifting test, we observed a shift cost that was observed in all genotypes in the reversal phase of the Barnes maze test (Fig. 2e, f).
Responses to sensory stimuli and anxiety-related behaviors
The startle response was consistently increased in the Cntn4+/− and Cntn4−/− mice at different high amplitudes, although the startle threshold was not significantly different between genotypes (Figs. 2g, h). No significant effects were found on pre-pulse inhibition at both inter-stimulus intervals of 30 and 100 ms (Figs. 2i, j).
The increased startle response to auditory stimuli of the different high amplitudes seemed not to result from increased anxiety levels, as Cntn4+/−, Cntn4−/− mice and wild-type controls did not differ in their elevated plus maze (Fig. 2k) and open-field exploratory behaviors (Figs. 2l).
Discussion
We present a comprehensive assessment of the impact of the ASD candidate gene CNTN4 on a variety of neurological, behavioral, and cognitive aspects of development. We found no effect of Cntn4 deficiency on ASD-related behavioral mouse paradigms such as the juvenile social interaction test, the three-chamber test, grooming behavior, and sensorimotor coordination. We also did not observe developmental neurological, behavioral, or cognitive abnormalities in the extended SHIRPA screen.
The reversal-learning phase of the set-shifting task yielded an inconclusive result, as no shift cost was observed in wild-type animals for the number of trials to reach criterion. Indeed, the genotype difference that was observed in the number of trials to reach the criterion in the reversal-learning phase was rather caused by lack of shift cost in wild-type animals than a reversal-learning deficit in the Cntn4−/− mice. In line with this reasoning, when we compared the outcome of Cntn4−/− mice in the reversal-learning phase with the results obtained for C57BL/6J mice obtained in a previous study, we find no genotype difference in this phase of the set-shifting task [6]. In the Barnes maze reversal-learning paradigm, we also did not find a reversal-learning deficit. Based on the summary of these data, we conclude that Cntn4 disruption has no effect on cognitive flexibility.
Non-ASD-specific effects at adult ages were indicated by increased startle response to auditory stimuli of different high amplitudes and by faster escape hole finding during subsequent days of acquisition in the Barnes maze.
Hyperresponsivity to acoustic stimuli is related to many neurodevelopmental disorders and has also been reported in ASD [22]. In addition, acoustic hyperresponsivity in patients with fragile X syndrome is known to be consistent with animal model data [23]. Similar to our findings, an increased startle response to acoustic stimuli was recently described in children with ASD [24], although these were in response to weak stimuli in contrast to the high amplitudes we found. The observed hyperresponsivity in Cntn4-deficient mice was unrelated to anxiety levels, as mice showed similar exploratory behaviors in classical anxiety behavioral tests, such as the elevated plus maze and open field. Although speculative, the increased startle response together with the enhanced acquisition in the Barnes maze could indicate that Cntn4 deficiency in the C57BL/6J background leads to a state of increased behavioral responsiveness without overt anxiety or avoidance behavior in mice [25].
Together, the findings show that in the C57BL/6J background, disruption of Cntn4 does not lead to substantial behavioral defects related to autistic development. A limited behavioral penetrance of CNTN4 mutations on autistic development may be consistent with a recent study that revisited the association of contactins, including CNTN4, with ASD [16]. The behavioral phenotype of Cntn4 mice contrasts other previously studied genetic ASD models [18]. For instance, Shank3 or Pten mice show extensive impairments in social interaction and sensorimotor phenotypes and are therefore regarded as translational models for ASD [26–28].
Although ASD-related phenotypes in Cntn4-deficient mice were observed, our findings do have relevance for neurodevelopmental disorder research. The specific phenotypes observed in Cntn4-deficient mice may be used to study the mechanisms underlying increased responsiveness or vigilance, a trait observed across many different human disorders such as attention-deficit hyperactivity disorder, post-traumatic stress disorder, and schizophrenia [29–31]. Cntn4-deficient mice may serve as a model to study the mechanistic underpinnings of behavioral states in which vigilance is altered. Indeed, common single-nucleotide variants in the CNTN4 locus have recently been associated with other neuropsychiatric disorders, such as schizophrenia [32], perhaps pointing to a non-disorder specific contribution of this cell adhesion gene in neuropsychiatric pathogenesis. In addition, a role for Cntn4, was recently shown in target-specific arborization during development of the accessory optic system [9]. Our study shows the importance of detailed developmental neurological, behavioral, and cognitive characterization of genetic animal models to complement human genetic studies in ASD and related disorders.
Conclusions
In our test battery, disruption of Cntn4, a prominent ASD candidate gene, had no effect on cognitive and behavioral development or ASD-specific phenotypes. At adult age, we could detect an effect of Cntn4 disruption on an adult sensory behavioral and a spatial cognitive feature.
Acknowledgements
We thank Y. Yoshihara for the generous gift of Cntn4-deficient mice, M. Brandt for the help with the three-chamber sociability test, H. Oppelaar and M. Broekhoven for the technical assistance, and A. Oguro-Ando for the discussion. This study was supported by a funding by the European Autism Interventions—A Multicentre Study for Developing New Medications (EU-AIMS) to Dr. Martien J. Kas. The research of EU-AIMS receives support from the Innovative Medicines Initiative Joint Undertaking under Grant agreement number 115300, resources of which are composed of financial contributions from the European Union’s Seventh Framework Programme (FP7/2007–2013), from the European Federation of Pharmaceutical Industries and Associations companies in-kind contributions, and from Autism Speaks.
Abbreviations
- ASD
autism spectrum disorder
- eSHIRPA
extended SmithKline Beecham, Harwell, Imperial College and Royal London Hospital phenotype assessment
- PPI
pre-pulse inhibition
Additional file
Detailed information on genotyping, behavioral testing and statistical analyses.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RTM participated in the design of the study, carried out the experiments and genotyping, analyzed the data, interpreted the results, and wrote the manuscript. HB provided input into the study design, interpreted the results, and wrote the manuscript. ER and LV participated in the design of the study and coordination of the experiments. ML participated in the design of the study, data analysis, and interpretation of the results. JPHB aid in the genotyping and participated in the interpretation of the results. MJHK contributed to the design of the study, development of the methods, coordination of the statistical analyses, and interpretation of the data. All authors contributed to the reviewing of the manuscript. All authors read and approved the final manuscript.
Contributor Information
Remco T. Molenhuis, Email: r.t.molenhuis-2@umcutrecht.nl
Hilgo Bruining, Email: h.bruining@umcutrecht.nl.
Esther Remmelink, Email: esther.remmelink@sylics.com.
Leonie de Visser, Email: leoniedv@gmail.com.
Maarten Loos, Email: maarten.loos@sylics.com.
J. Peter H. Burbach, Email: j.p.h.burbach@umcutrecht.nl
Martien J. H. Kas, Email: m.j.h.kas@umcutrecht.nl
References
- 1.Am. Psychiatr. Assoc.: Diagnostic and statistical manual of mental disorders. 5th ed. Washington, DC; 2013.
- 2.Chen JA, Peñagarikano O, Belgard TG, Swarup V, Geschwind DH. The emerging picture of autism spectrum disorder: genetics and pathology. Annu Rev Pathol: Mech Dis. 2015;10:111–144. doi: 10.1146/annurev-pathol-012414-040405. [DOI] [PubMed] [Google Scholar]
- 3.Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet. 2014;94:677–694. doi: 10.1016/j.ajhg.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Ercument Cicek A, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abrahams BS, Arking DE, Campbell DB, Mefford HC, Morrow EM, Weiss LA, et al. SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs) Mol Autism. 2013;4:36. doi: 10.1186/2040-2392-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Molenhuis RT, de Visser L, Bruining H, Kas MJ. Enhancing the value of psychiatric mouse models; differential expression of developmental behavioral and cognitive profiles in four inbred strains of mice. Eur Neuropsychopharmacol. 2014;24:945–954. doi: 10.1016/j.euroneuro.2014.01.013. [DOI] [PubMed] [Google Scholar]
- 7.Zuko A, Kleijer KTE, Oguro-Ando A, Kas MJH, Van Daalen E, Van Der Zwaag B, et al. Contactins in the neurobiology of autism. Eur J Pharmacol. 2013;719:63–74. doi: 10.1016/j.ejphar.2013.07.016. [DOI] [PubMed] [Google Scholar]
- 8.Kaneko-Goto T, Yoshihara S-I, Miyazaki H, Yoshihara Y. BIG-2 mediates olfactory axon convergence to target glomeruli. Neuron. 2008;57:834–846. doi: 10.1016/j.neuron.2008.01.023. [DOI] [PubMed] [Google Scholar]
- 9.Osterhout JA, Stafford BK, Nguyen PL, Yoshihara Y, Huberman AD. Contactin-4 mediates axon-target specificity and functional development of the accessory optic system. Neuron. 2015;86:985–999. doi: 10.1016/j.neuron.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fernandez T, Morgan T, Davis N, Klin A, Morris A, Farhi A, et al. Disruption of contactin 4 (CNTN4) results in developmental delay and other features of 3p deletion syndrome. Am J Hum Genet. 2004;74:1286–1293. doi: 10.1086/421474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roohi J, Montagna C, Tegay DH, Palmer LE, DeVincent C, Pomeroy JC, et al. Disruption of contactin 4 in three subjects with autism spectrum disorder. J Med Genet. 2009;46:176–182. doi: 10.1136/jmg.2008.057505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo H, Xun G, Peng Y, Xiang X, Xiong Z, Zhang L, et al. Disruption of contactin 4 in two subjects with autism in Chinese population. Gene. 2012;505:201–205. doi: 10.1016/j.gene.2012.06.051. [DOI] [PubMed] [Google Scholar]
- 15.Cottrell CE, Bir N, Varga E, Alvarez CE, Bouyain S, Zernzach R, et al. Contactin 4 as an autism susceptibility locus. Autism Res. 2011;4:189–199. doi: 10.1002/aur.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murdoch JD, Gupta AR, Sanders SJ, Walker MF, Keaney J, Fernandez TV, et al. No evidence for association of autism with rare heterozygous point mutations in contactin-associated protein-like 2 (CNTNAP2), or in other contactin-associated proteins or contactins. PLoS Genet. 2015;11:e1004852. doi: 10.1371/journal.pgen.1004852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rogers DC, Jones DNC, Nelson PR, Jones CM, Quilter CA, Robinson TL, et al. Use of SHIRPA and discriminant analysis to characterise marked differences in the behavioural phenotype of six inbred mouse strains. Behav Brain Res. 1999;105:207–217. doi: 10.1016/S0166-4328(99)00072-8. [DOI] [PubMed] [Google Scholar]
- 18.Silverman JL, Yang M, Lord C, Crawley JN. Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci. 2010;11:490–502. doi: 10.1038/nrn2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bruining H, Matsui A, Oguro-Ando A, Kahn RS, Spijker HM V’t, Akkermans G, et al. Genetic mapping in mice reveals the involvement of Pcdh9 in long-term social and object recognition, and sensorimotor development. Biol Psychiatry. 2015;78:485–495. doi: 10.1016/j.biopsych.2015.01.017. [DOI] [PubMed] [Google Scholar]
- 20.Pearson BL, Pobbe RLH, Defensor EB, Oasay L, Bolivar VJ, Blanchard DC, et al. Motor and cognitive stereotypies in the BTBR T+tf/J mouse model of autism. Genes Brain Behav. 2011;10:228–235. doi: 10.1111/j.1601-183X.2010.00659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seigers R, Loos M, Van Tellingen O, Boogerd W, Smit AB, Schagen SB. Cognitive impact of cytotoxic agents in mice. Psychopharmacology (Berl). 2015;232(1):17–37. [DOI] [PubMed]
- 22.Green S, Hernandez L, Tottenham N, Krasileva K, Bookheimer S, Dapretto M: Neurobiology of sensory overresponsivity in youth with autism spectrum disorders. JAMA Psychiatry. 2015;72(8):778-786. [DOI] [PMC free article] [PubMed]
- 23.Chen L, Toth M. Fragile X mice develop sensory hyperreactivity to auditory stimuli. Neuroscience. 2001;103:1043–1050. doi: 10.1016/S0306-4522(01)00036-7. [DOI] [PubMed] [Google Scholar]
- 24.Takahashi H, Nakahachi T, Komatsu S, Ogino K, Iida Y, Kamio Y. Hyperreactivity to weak acoustic stimuli and prolonged acoustic startle latency in children with autism spectrum disorders. Mol Autism. 2014;5:23. doi: 10.1186/2040-2392-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blanchard DC, Griebel G, Blanchard RJ. Mouse defensive behaviors: pharmacological and behavioral assays for anxiety and panic. Eur J Pharmacol. 2003;463:97–116. doi: 10.1016/S0014-2999(03)01276-7. [DOI] [PubMed] [Google Scholar]
- 26.Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, et al. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature. 2011;472:437–442. doi: 10.1038/nature09965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang YH, Ehlers MD. Modeling autism by SHANK gene mutations in mice. Neuron. 2013;78:8–27. doi: 10.1016/j.neuron.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ey E, Leblond CS, Bourgeron T. Behavioral profiles of mouse models for autism spectrum disorders. Autism Res. 2011;4:5–16. doi: 10.1002/aur.175. [DOI] [PubMed] [Google Scholar]
- 29.Parker JDA, Majeski SA, Collin VT. ADHD symptoms and personality: relationships with the five-factor model. Pers Individ Dif. 2004;36:977–987. doi: 10.1016/S0191-8869(03)00166-1. [DOI] [Google Scholar]
- 30.Dalgleish T, Moradi AR, Taghavi MR, Neshat-Doost HT, Yule W. An experimental investigation of hypervigilance for threat in children and adolescents with post-traumatic stress disorder. Psychol Med. 2001;31:541–547. doi: 10.1017/S0033291701003567. [DOI] [PubMed] [Google Scholar]
- 31.Freedman R, Waldo M, Bickford-Wimer P, Nagamoto H. Elementary neuronal dysfunctions in schizophrenia. Schizophr Res. 1991;4:233–243. doi: 10.1016/0920-9964(91)90035-P. [DOI] [PubMed] [Google Scholar]
- 32.Schizophrenia Working Group of the Psychiatric Genomics Consortium Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]