

Abstract
Noncoding regions of the genome play an important role in tumorigenesis of cancer. Using expression cloning, we have identified a cytotoxic T lymphocyte (CTL)–defined antigen that recognizes a protein sequence derived from an open reading frame transcribed from the reverse strand in the 3′ untranslated region of tRNA isopentenyltransferase 1 (TRIT1). A peptide derived from this open reading frame (ORF) sequence and predicted to bind to HLA-B57, sensitized HLA-B57+ tumor cells to lysis by CTL793. The peptide also induced a CTL response in peripheral blood mononuclear cells (PBMC) of patient 793 and in two other melanoma patients. The CTL lysed peptide-pulsed HLA-B57+ target cells and melanoma cells with endogenous antigen expression. The recognition of this antigen is not limited to HLA-B57-restricted CTLs. An HLA-A2 peptide derived from the ORF was able to induce CTLs in PBMC of 2 HLA-A2+ patients. This study describes for the first time a CTL-defined melanoma antigen that is derived from an ORF on the reverse strand of the putative tumor suppressor gene TRIT1. This antigen has potential use as a vaccine or its ability to induce CTLs in vitro could be used as a predictive biomarker.
Introduction
Metastatic melanoma patients have very limited treatment options as they invariably develop resistance to most available chemo- and targeted-therapy drugs including small molecule inhibitors such as vemurafenib and dabrafenib that are directed against BRAFV600E.1,2 Recent promising results with immune checkpoint reagents such as anti-CTLA4 antibody (Ipilimumab) and anti-PD1 antibody (Nivolumab and Lambrolizumab) in melanoma patients2 have renewed interest in using immune-based therapies of cancer patients. Therapy response to immune checkpoint blockade, unlike targeted-therapy with drugs, is more modest but long-lasting (reviewed in ref. 3). However, only a subset of melanoma patients responds to these reagents.3 Experimental therapies using combination of immune checkpoint inhibitors and targeted therapy are underway to increase the number of therapy responders and to delay the progression of the disease. Preliminary observations suggest that these approaches, though promising, can be accompanied by toxicities.4,5 Some of the toxicity issues are attributed to nonspecific activation of immune cells in the presence of targeted-therapy and immune check point inhibitors (see review6). In adoptive cell therapies, T-cell reactivity to melanoma-associated antigens and cross-reactive epitopes on normal tissues antigens are of a major concern. Studies from Rosenberg’s laboratory have shown that adoptive cell therapy using TILs reactive to mutated epitopes have durable tumor regression rates when compared to TILs reactive to nonmutated epitopes.7,8 Additionally, T cells from melanoma patients undergoing therapy with Ipilimumab and showing tumor regression are shown to be reactive to mutated and cryptic epitopes.9 Genome sequencing analyses have revealed that some of these epitopes are derived from the so called noncoding region of the DNA.8,10,11 Thus, the identification of unique mutated or tumor specific antigens recognized by T cells of melanoma patients is of significance in a clinical setting. Precisely, specific induction of T cells to mutated tumor antigens will minimize toxicity issues that are frequently observed with the use of immune checkpoint reagents or targeted therapy drugs due to nonspecific activation of immune cells. Also, the frequency of tumor-directed specific T-cell responses will be a useful tool in predicting better therapy responses.
In the present study, we have used a previously described antimelanoma reactive cytotoxic T lymphocyte (CTL) CD4+ clone 793 derived from peripheral blood mononuclear cells (PBMC) of a primary melanoma patient.12,13 The CTL clone recognized exclusively HLA-B57+ melanoma cell lines derived from different patients. Using expression cloning, we have identified a novel antigen recognized by the CTL. The antigen (referred as AS-TRIT (anti-sense TRIT1)) is encoded by an open reading frame (ORF) transcribed from the reverse strand in the 3′ untranslated region of the tRNA isopentenyltransferase 1 (TRIT1) gene. This was confirmed by the reactivity of CTL to HLA-B57 binding peptide derived from the ORF.
Several mechanisms (e.g., overlapping reading frames, coding intron sequences) that generate nontraditional T-cell epitopes have been described in the literature (reviewed in ref. 14). A renal cell carcinoma-related T-cell epitope was derived from a transcript of the reverse strand of a known ORF.15 Considering the vast majority of RNA (~90%) in eukaryotic cells consists of so called noncoding RNAs16,17 and some of them may be translated,17–19 a potential new source of antigens exits. Based on the completed sequencing of mammalian genomes and transcriptome data analyses, an increased number of potential transcriptional start sites and additional RNAs have been discovered in the last few years20–23 including overlapping bidirectional transcription in mammalian cells.24 Recent studies suggests that noncoding RNAs play a major role in tumorigenesis (see review17 ).Thus, understanding the biology of noncoding RNA and identification of tumor-reactive T-cell epitopes in these regions will be of potential benefits in cancer patients.
Results
Identification of the AS-TRIT cDNA clone
A standard expression cloning method was used to screen a cDNA expression library of WM793 cells for the antigen recognized by CTL793. A previously isolated HLA-B57 cDNA13 and a cDNA expression library from WM793 cells were cotransfected into COS7 cells and 100 cDNA clones per pool were screened for reactivity with CTL793. Positive pools were subdivided into smaller pools until a single clone (793A11) was isolated. This cDNA (793 cDNA, henceforth referred as “AS-TRIT”) clone had an insert size of 442 bp and the sequence was 100% identical to the published TRIT1 sequence (Genbank NM_017646.4). However, the cDNA sequence was located in the 3′ untranslated region of TRIT1 and therefore could not code for any portion of the TRIT1 protein. Additionally, the cDNA was in a reverse orientation to the TRIT1 reading frame.
Full-length AS-TRIT RNA
The AS-TRIT cDNA clone was isolated from an oligo-dT primed cDNA library expected to be located at the 3′ end of the RNA. The unknown 5′ end of AS-TRIT RNA was determined using the RACE approach. Only the decapped probe gave a 5′ RACE probe, which was not amplified using untreated RNA during RACE. This suggested again with an independent method that an antisense RNA to TRIT1 exists. The length of the AS-TRIT RNA is at least 890 nucleotides (Figure 1c). The 3′ end of the RNA is uncertain. The genomic DNA has a string of A’s, where the oligo-dT primer is located, and therefore, it is not certain if the polyA tail is the real 3′ end or if the oligo-dT had primed internally of the AS-TRIT RNA.
Figure 1.

Location and sequence of AS-TRIT. (a) Location of AS-TRIT cDNA in relation to TRIT1. (b) Nucleotide sequence of the AS-TRIT cDNA clone with the predicted amino acid sequence and the cytotoxic T lymphocyte–recognized epitope underlined. The three antisense oligonucleotides and the location of peptide 2 are shown. (c) Predicted full length sequence of 793 RNA and putative ORFs (either underlined or in bold) using ATG as start codon with a minimum length of 30 aa.
Identification of the T-cell antigen recognized by CTL793
To determine the peptide antigen recognized by CTL793, we analyzed the cloned cDNA sequence in both directions for the presence of ORFs with a minimum length of 20 amino acids and ATG as the start codon using ExPasy (http://web.expasy.org/translate/). A total of five putative ORFs on both strands matched our search criteria. Each deduced protein sequence was scanned for potentially HLA-B57 binding epitopes using the epitope prediction (x-(ATS)-x-x-x-x-x-x-(FWY)). Five potential HLA-B57 binding epitopes were determined (see Materials and Methods). The peptides (25 µmol/l) were loaded onto Ag−, HLA-B57+ 1205LU melanoma cells13 in presence of β2-microglobulin, incubated for 8 hours, excess peptide removed, and peptide-pulsed cells were tested for lysis by CTL793. 1205LU melanoma cells previously were not recognized by CTL793 and hence designated as Ag−.13 As a negative control, an A1 peptide potentially binding HLA-B57 was used. Peptide 2 (KSIWKLDNF, Figure 1b) induced the highest reactivity in this assay with a maximum specific cytotoxicity of 30% (Figure 2a). Other peptides, namely peptides 3 and 4 induced very low (~13%) target cell lysis, and lysis induced by peptides 1 and 5 was below 10% (data not shown). CTL793 did not significantly lyse 1205LU cells pulsed with the control peptide. Peptide 2 was derived from the longest ORF in the cDNA, which is in the reverse orientation of TRIT1 (Figure 1b). In an independent experiment, reactivity of the CTL to peptide 2 was validated by pulsing the HLA-B57+ 1205LU melanoma target with concentrations ranging from 0.7 to 50 µmol/l of the peptide (Figure 2b). Target cells pulsed with peptide concentrations as low as 3.12 µmol/l were lysed by the CTL (Figure 2b). To further confirm that peptide 2 is the tumor epitope recognized on WM793 cells, a cold target inhibition was performed. Increasing numbers of peptide 2-loaded 1205LU cells inhibited the lysis of 51Cr-labeled WM793 cells (Figure 2c), confirming that peptide 2 defines the epitope recognized by CTL793. These data support the finding that a peptide derived from an ORF located on the reverse strand of the 3′ untranslated region of TRIT1 is the antigen recognized by CTL793 (Figure 2a).
Figure 2.

Recognition of peptide-pulsed 1205LU and WM793 cells by cytotoxic T lymphocyte (CTL)793. (a) 1205LU cells were pulsed with the peptide 2 (∆) or control peptide (■) at 25 µmol/l concentration in presence of β2-microglobulin (1 µg/ml) for 8 hours, excess peptide removed and then 1205LU target cells were incubated with CTL793 at various effector to target (E:T) cell ratios. (b) As above 1205LU cells were pulsed with peptide 2 (∆) or control peptide (■) at peptide concentrations ranging from 0.7 to 50 µmol/l concentrations and then incubated with CTL 793 to determine the lysis at E:T ratio of 25:1. (c) CTL793 were incubated with 51Cr-labeled WM793 and increasing numbers of peptide 2-pulsed (∆) or control peptide-pulsed (■) 1205LU cold target cells. CTL activity of CTL793 was measured in a standard 51Cr-release assay at an E: T ratio of 2:1. Results are expressed as mean % cytoxicity ± SE (a–c) and CTL lysis of 1205LU cells pulsed with peptide 2 was significantly different (P < 0.01) from target cells pulsed with control peptide (a,b). In (c), CTL lysis of WM793 target cells was significantly (P < 0.01) inhibited when increasing number of peptide 2 pulsed cold 1205LU target cells were used. All experiments were repeated at least 2× for confirmation.
Confirmation of antigen recognition by CTL793
To confirm in a different assay that we had cloned the epitope recognized by CTL793 and that the antigen was derived from the reverse strand of TRIT1, we subcloned a shortened version of the cDNA (consisting mostly of the ORF only) into the pIRESbleo vector (Clontech, Palo Alto, CA) in both directions and transfected COS7 cells with B57 cDNA and either the sense or antisense cDNA. Only COS7 cells transfected with the cDNA in the antisense orientation in regards to TRIT1 mRNA were able to stimulate cocultured CTL793 cells to release interferon (IFN)-γ (689–1,250 pg/ml)). Finally, antisense oligonucleotides (see Materials and Methods and Figure 1b) were used to transiently downmodulate the expression of the AS-TRIT RNA in WM793 cells. WM793 cells treated with oligonucleotide 793chimeric-1 were no longer lysed by CTL793 (Figure 3). Since CTL lysis was lost after treatment of the cells with an oligonucleotide targeting the specific RNA, this further confirmed that the antigen recognized by CTL793 is derived from this hitherto undescribed ORF on the reverse strand of the tumor suppressor TRIT1.
Figure 3.
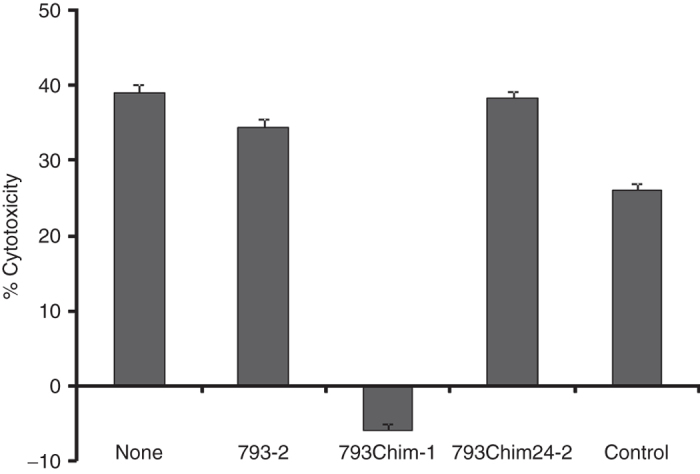
Recognition of WM793 cells transfected with antisense oligonucleotides by cytotoxic T lymphocyte (CTL)793. WM793 cells were transiently transfected with the three different antisense oligonucleotides or were mock-transfected and used as target cells in a cytotoxicity (Live/dead viability assay kit) assay with CTL793 (E:T ratio of 25:1) 2 days later. Untreated WM793 cells were included as a positive control. Results are expressed as mean % cytotoxicity ± SE from two independent experiments.
AS-TRIT RNA expression
We were unable to detect AS-TRIT RNA in RNA samples of different melanoma cell lines by conventional Northern blot analysis. Therefore, we performed a two-step reverse transcription polymerase chain reaction (see Materials and Methods) with several samples. In all four RNA samples derived from different cell lines (melanoma, colon carcinoma (CRC), and one Epstein Bar Virus (EBV)–transformed B cells), a strong TRIT1 signal of the expected size of 367 bp and varying levels of the 288 bp large AS-TRIT-specific signal were detected (Figure 4a). In general, AS-TRIT-specific signals were low when compared to TRIT1 signal (Figure 4b). We have further confirmed by sequencing that the 288 bp fragment encoded the correct sequence. The smaller band in the WM793 sample was an unspecific by-product (labeled + in Figure 4a). The highest expression level of AS-TRIT RNA was detected in the WM793 sample, the cell line which was efficiently lysed by CTL793 (Figure 4b).25 In these melanoma cells, high expression of the AS-TRIT RNA may downmodulate the expression of TRIT1 RNA (Figure 4a,b). Expression of AS-TRIT RNA was low in ME9874 and this correlates with low lysis of ME9874 by CTL793 that was previously reported in our study.13 HT29 (CRC) and 793EBV-B were not recognized by CTL793.13 In a recent study, expression of antisense RNA of TRIT1 was found in many cell lines derived from various cancer types including acute leukemia, basal cell, cervical, merkel cell and ovarian carcinomas, and spindle cell sarcoma with a high expression in some tumors.26
Figure 4.
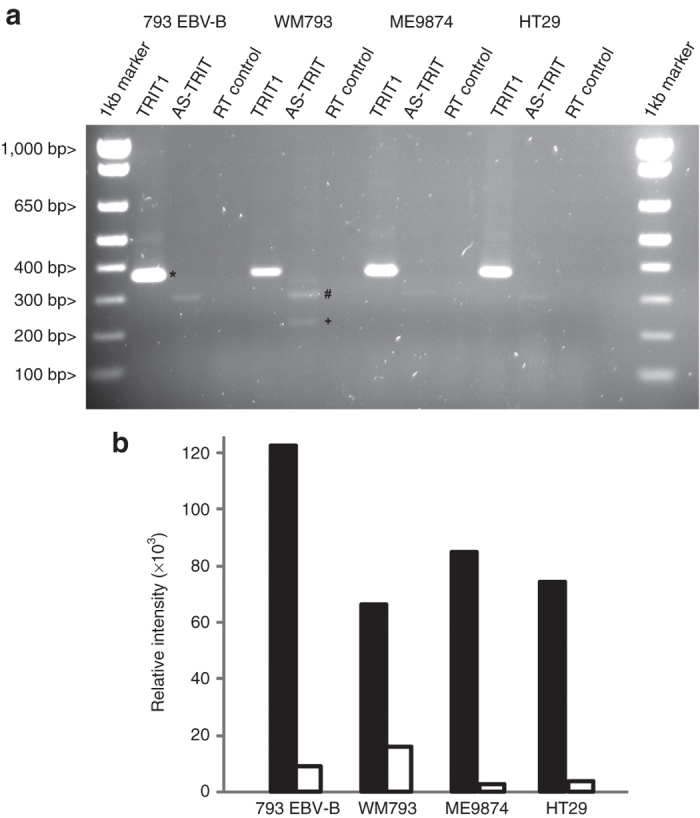
Expression analysis of AS-TRIT RNA (a) RT-PCR analyses of AS-TRIT RNA in cells which are recognized (WM793, ME9874) or not (793EBV-B, HT29) by cytotoxic T lymphocyte (CTL)793. The presence of TRIT1 or AS-TRIT RNA was analyzed by RT-PCR and a reverse transcriptase negative control was included to exclude residual genomic DNA contamination. Besides the 367 bp TRIT1 (*) and the 288 bp AS-TRIT (#) fragments an unspecific fragment (+) was seen in some samples. (b) Relative intensities of TRIT1 (■) or AS-TRIT (□) DNA bands were quantified using KODAK 1D image analysis software and shown as bar graph.
Recognition of AS-TRIT peptides by PBMCs of melanoma patients
To determine whether peptide 2 can induce CTL in the lymphocytes of melanoma patients, PBMC of patient 793 and 3 additional HLA-B57+ melanoma patients and 1 HLA-B57+ healthy donor were stimulated repeatedly with peptide 2 or with control peptide-pulsed autologous monocytes in vitro. Cytotoxic activities of stimulated cells were determined in 51Cr-release assays using unpulsed WM793 or Ag− HLA-B57+ 1205LU melanoma cells pulsed with peptide 2, control peptide or left unpulsed as target cells. Peptide 2-stimulated 793 PBMC lysed WM793 cells and peptide 2-pulsed 1205LU melanoma cells, but did not react with control peptide pulsed or unpulsed 1205LU cells (Figure 5a). Control peptide-stimulated 793 PBMC did not recognize any of the four target cells described above. Lymphocytes from two other HLA-B57+ melanoma patients showed moderate lysis of peptide-pulsed 1205 cells or WM793 cells (10–35% lysis at E: T ratio of 50:1; Figure 5b,c) and no lysis of unpulsed or control peptide-pulsed 1205 cells. Lysis of WM793 cells and peptide-2 pulsed 1205LU cells after in vitro stimulation with peptide 2 was reproducible (in three independent experiments). PBMC of a healthy HLA-B57-positive donor did not react with peptide 2-pulsed 1205LU cells or WM793 target cells after repeated in vitro stimulation with peptide 2 (Figure 5d).
Figure 5.
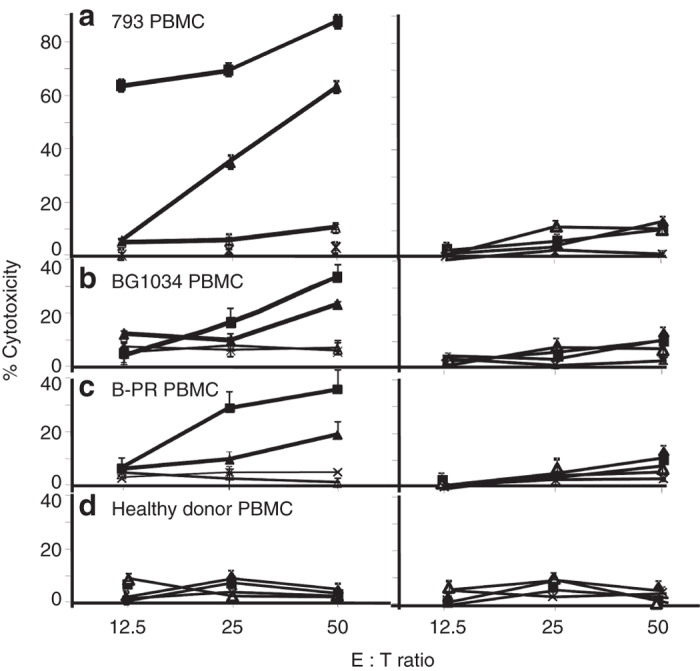
Cytotoxic activity of peptide 2 or control peptide-stimulated PBMC of HLA-B57+ melanoma patients and a healthy control donor against WM793 or peptide-pulsed 1205LU target cells. Cytolytic activity of short-term T-cell lines derived from peptide 2 (left panel) or control peptide (right panel) stimulation of PBMC were determined using WM793 (HLA-B57+, ■) or peptide 2-pulsed 1205LU target cells (▲) in a standard 51Cr release assay as in Figure 2. Control-peptide pulsed (∆) or unpulsed (+) 1205 LU cells were used as controls. Results are expressed as mean % cytoxicity ± SE (a–d) and cytotoxic T lymphocyte lysis of WM793 or 1205LU cells pulsed with peptide 2 was significantly different (P < 0.01) from 1205LU cells or control-peptide pulsed target cells. All experiments were repeated at least three times for confirmation.
The ORF of AS-TRIT contains several epitopes predicted to bind with other HLA alleles. To test whether the AS-TRIT protein is immunogenic for patients with an HLA type other than HLA-B57, we tested two candidate peptides predicted to bind to HLA-A2 (see Materials and Methods) for their capacity to stimulate PBMCs of HLA-A2 melanoma patients. In two of five HLA-A2+ PBMC samples tested, sufficient lymphocyte growth was obtained to perform CTL assays after repeated in vitro stimulation. In both cases, AS-TRIT-specific peptide A2_2 induced short-term CTLs, which lysed antigen-positive HLA-A2+ tumor cells (Figure 6a,c). The lysis of HLA-A2+ tumor target cells was confirmed in two other independent experiments. The inhibition of target cell lysis by anti-MHC class I antibody and anti-HLA-A2 antibodies suggested that lysis is HLA-A2 restricted (Figure 6b,d). None of the PBMC samples incubated with the HLA-A2 control peptide proliferated in vitro.
Figure 6.
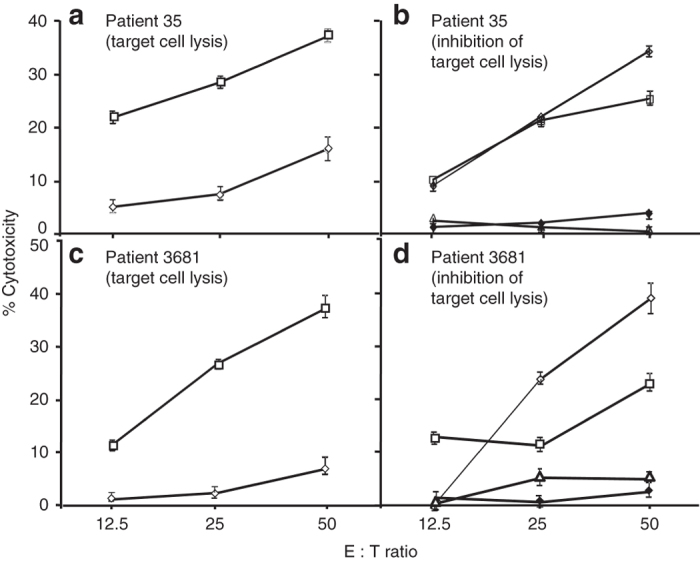
Cytotoxic activity of HLA-A2 peptide-stimulated PBMC of HLA-A2+ melanoma patients against target cells (a–d). Short-term T-cell lines derived from PBMC of HLA-A2+ melanoma patients after peptide-pulsed monocytes and IL-2 stimulation in vitro as in Figure 5 were tested for cytolytic activity against HLA-A2+ target (WM35) cells in a 51Cr release assay. PBMC from patient 35 (a) and patient 3681 (b) were stimulated with either peptide A2_1 (◊) or A2_2 (□) and were tested for cytotoxic activity against 51Cr-labeled WM35 tumor cells as targets. To determine MHC restriction of peptide A2_2 stimulated PBMC (35 PBMC (b); 3681 PBMC (d)), 51Cr release assay was performed in the presence of medium (◊), normal mouse IgG (□), anti-HLA class I antibody (∆) or anti-HLA-A2 antibody (♦). Results are expressed as mean % cytoxicity ± SE (a–d) and cytotoxic T lymphocyte lysis of WM35 (HLA-A2+) target cells was significantly inhibited (P < 0.001) in presence of anti-HLA class I or anti-HLA-A2 antibodies. All experiments were repeated at least twice for confirmation.
Homologues of AS-TRIT ORF
Using the determined protein sequence of AS-TRIT, no homologous protein sequence was found in published data bases (GenBank) with a blastp search. However, more careful analysis of homologous DNA sequences revealed homologous ORFs in the genome of Pan troglodytes, Macaca mulatta, and Sus scrofa (Table 1). In all three genomes, the ORFs are located in the 3′ region of TRIT1. No homologous ORFs were found in the genome of Rattus norvegicus or Mus musculus.
Table 1. Presence of ORFs homologous to AS-TRIT in the genome of various mammals and protein sequence identity to the human AS-TRIT protein.
| Organism | ORF (# of aa) | Homology with the human ORF (%) (size of homologous region, # of aa) |
|---|---|---|
| Pan troglodytes (chimpanzee) | 109 | 98 (109) |
| Macaca mulatta (rhesus monkey) | 138 | 90 (97) |
| Sus scrofa (pig) | 71 | 64 (59) |
| Rattus norvegicus (rat) | — | — |
| Mus musculus (mouse) | — | — |
ORF, open reading frame.
A function of the derived protein could not be predicted because there was no homology to any known protein or functional protein domain. Using computer algorithms (PSIPRED protein structure prediction, http://bioinf.cs.ucl.ac.uk/psipred/), the structure of the protein was determined as a helix-coil-helix-coil-helix structure.
Discussion
Melanoma patients’ responses to chemo- or targeted-therapy drugs are short-lived and one of the major reasons for therapy resistance could be due to the highly heterogeneity of tumor cells.27 Genetic analysis has revealed high frequency of mutations in melanoma tumors.27 Also, recent analysis of TILs from melanoma patients who undergo adoptive T cell therapy showed high persistence of T cells reactive to mutated antigens in peripheral circulation indicating T-cell sensitization to these antigens.8 Additionally, individuals with higher frequency of TILs reactive to mutations have shown better prognosis and thus, responses to mutated antigens could be used as a predictive biomarkers. In the present study, using expression cloning, we have identified a new ORF (AS-TRIT) on the reverse strand of tRNA isopentenyltransferase (TRIT1), which is recognized by antimelanoma reactive CTL793. The location of the CTL-reactive peptide/antigen sequence within the ORF (Figure 2) and the loss of tumor cell lysis after treatment of the cells with an antisense oligonucleotide (Figure 3) confirms that the peptide antigen recognized by the CTL is derived from the ORF transcribed from the reverse strand in the 3′ untranslated region of TRIT1. Further, our earlier study confirms that the peptide antigen is from an endogenous source as we have reported loss of tumor cell lysis by CTL793 when the tumor cells were subjected to low pH peptide stripping.13 In addition, in our unpublished observation, we have shown that HPLC-fractions of tumor cell surface membrane can stimulate CTL793 proliferation further confirming the endogenous transcription of TRIT1. In renal cell carcinoma, a similar CTL antigen derived from the reverse strand of a known gene has been described.15
The location of AS-TRTIT antigen is interesting for biological reasons. Human TRIT1, cloned by Golovko et al.28, is responsible for the addition of N6-isopentenyladenosine on residue 37 of certain tRNA molecules. Post-transcriptional modifications of tRNAs are essential for maintaining the correct reading frame during translation,29 which guarantees the fidelity of protein synthesis. Variations in tRNA modifications have been associated with malignancy.30 The chromosomal region of TRIT1 is amplified in some small-cell lung carcinoma cells resulting in an increase of TRIT1 mRNA expression.31 However in another study,32 TRIT1 mRNA levels were reduced in lung tumors compared to normal lung tissue. Transfection of lung carcinoma cells with TRIT1 resulted in smaller colonies in vitro. Furthermore, TRIT1 transfected tumor cells had a reduced tumorigenicity in nude mice. Based on these data, TRIT1 was proposed as a candidate tumor suppressor in lung tumors.32 Therefore, downregulation of TRIT1 may be enhancing tumor formation. Antisense transcription has been implied in the control of sense messenger RNA expression33 and could be one mechanism for downmodulation of RNA levels. Using overexpression or knock-down of AS-TRIT in melanoma cells or melanocytes by either transducing them with lentivirus expressing AS-TRIT or by blocking with oligonucleotide 793chim-1, we did not detect significant biological or phenotypic changes (data not shown). Transfecting cells with the oligonucleotide had a slight growth inhibitory effect. Although statistically significant, it was only minimal. However, it needs to be emphasized that the lack of any significant biological effect may be due to the fact that we tried to influence a cis-effect with a trans-acting approach.
Although the biological role of AS-TRIT transcription remains unclear, our results indicate that CTL induction in peripheral blood of melanoma patient is due to aberrant expression of AS-TRIT. Recognition of the AS-TRIT antigen by autologous CTLs was not restricted to patient 793. PBMC from two out of three additional HLA-B57 melanoma patients showed low but consistent lysis of partially matched HLA-B57+ target cells after in vitro stimulation with peptide 2, suggesting the sensitization of T cells to AS-TRIT antigen in vivo. As tumor cells were not available from the three patients, we were unable to confirm the expression of AS-TRIT RNA in these cells. Low frequency of sensitized T cells to AS-TRIT or decreased AS-TRIT RNA expression in tumor cells could be some of the reasons for diminished response in these patients. There was no induction of anti-AS TRIT T-cell responses observed in the PBMC of a HLA-B57+ healthy donor. Additionally, the AS-TRIT antigen is also recognized by the PBMC of two out of five HLA-A2+ patients after repeated in vitro stimulation with an A2_2 peptide with predicted binding epitopes for HLA-A2. Short-term CTLs stimulated with this A2_2 peptide in vitro lyse tumor cells in an HLA-A2-dependent manner. Again, tumor cells were not available from these patients to measure AS-TRIT RNA expression. Two of three melanoma patients (793 and 35) whose PBMC showed strong response to TRIT1-peptides binding to either HLA-A2 or HLA-B57, lived disease-free for more than 20 years after the surgical resection of their primary tumors. Also, both patients showed persistence of T cells reactive to AS-TRIT in peripheral blood 14–20 years after the removal of primary tumor lesions. Based on these results, the AS-TRIT antigen may provide a candidate vaccine in adjuvant therapy for HLA-B57+ and HLA-A2+ melanoma patients and boosts patients’ pre-existing immune response to the antigen. Recent encouraging results from preclinical studies and clinical trials in ovarian and pancreatic carcinoma suggest that the tumor antigen could be combined with strong adjuvants such as HSP70, GM-CSF, and/or mesothelin to attract innate immune cells and dendritic cells to augment antitumor T-cell vaccine responses.34,35 AS-TRIT antigen could be used in a similar manner to potentiate vaccine effect in melanoma patients.
Protein sequence comparison of the AS-TRIT ORF with published protein sequences using blastp did not reveal a homology to any known protein. However, detailed analysis revealed the presence of highly homologous ORFs in chimpanzee, rhesus monkey and pig genomes, also located in the 3′ untranslated region of TRIT1. No homologous ORF was found in rat and mouse (<50% homology), suggesting that the AS-TRIT ORF may be a late addition in evolution. The deduced protein sequence does not code for a known conserved domain and computer aided protein function predictions were negative (using Prosite (http://prosite.expasy.org/ and PFP http://kiharalab.org/web/pfp.php)). Therefore, it is unknown if this protein has any functions. There is a possibility that the protein has no function at all, and its expression in tumor cells is due to aberrant RNA translation, which has been described for tumors.10,36 Additionally, results by Ingolia et al. and others17,19 suggest that translation of “non-coding RNAs” may be much more prevalent than previously thought. Although newer results suggest ribosomal loading does not necessarily reflect translation of a functional protein, short translation products could arise from so-called noncoding RNAs,18 which are probably quickly degraded.
Ongoing combination therapies combining immune checkpoint inhibitors with targeted therapy are often accompanied by toxicities re-emphasizing the need for targeting unique tumor-specific antigens. In this context, the identification of our newly discovered ORF from the reverse strand of TRIT1 is a promising target for immunotherapeutic intervention in cancer patients. Thus, multidisciplinary approaches are needed to understand the role of noncoding RNA in tumorigenesis of melanoma, the potential use of derived protein epitopes as immunological targets and the measure of immune response against these proteins as a predictive biomarker.
Materials and Methods
Cell lines
WM793, 1205LU, WM35, and ME9874 melanoma cell lines were grown in appropriate melanoma or in RPMI 1640 media containing 2 or 10% fetal bovine serum.25 CRC line (HT29) was grown in CRC medium.37 EBV-B cell lines from patients were established as described.25 COS7 cells (Life Technologies, Carlsbad, CA) were maintained in Dulbecco's modified Eagle's medium, 10% fetal bovine serum. The CTL793 T-cell clone was established from a melanoma patient obtained 14 years after excision of the primary vertical growth phase lesion.13 CTL793 was grown in rIL-2-containing T-cell medium.13 The T-cell receptor of this clone expresses the Vα2 and Vβ8 chains. Melanocyte cell lines FOM158 and FOM173 were established from newborn foreskins and were grown in Medium 254CF with supplements (Life Technologies).
PBMC from cancer patients and from healthy donors
PBMC were obtained from the patients’ peripheral blood with informed consent and under a protocol approved by the Institutional Review Boards of both The Wistar Institute and the Hospital of University of Pennsylvania.
Identification of the recognized cDNA clone
The expression cDNA cloning method38 was used to identify the gene coding for the CTL793 melanoma antigen. In brief, polyA+ RNA (5 µg) from WM793 cells were reverse transcribed using the cDNA synthesis superscript plasmid system (Life Technologies). The cDNA library of 2.62 × 105 independent clones was divided into pools of 100 clones per well. For screening, 3 × 104 COS7 cells per well were cotransfected with 100 ng HLA-B57 cDNA (cloned from WM793 melanoma cell line and sequences verified)13 and 200 ng cDNA library pool DNA using Lipofectamine 2000 (Life Technologies). After transfection (12–16 hours), the medium was discarded and 1 × 104 T cells were added per well. After an additional 24–30 hours, IFN-γ levels in the supernatants were determined using an ELISA kit (Endogen-Pierce, Rockford, IL).
Confirmation that the antisense strand encodes the protein
To confirm that the antisense strand encodes the detected antigen COS7 cells were transfected with HLA-B57 DNA plus 793 cDNA (AS-TRIT) or a fragment of TRIT1. As a negative control, COS7 cells were transfected with HLA-B57 DNA alone. One day after transfection, CTL793 were added and supernatants were harvested after additional 24 hours incubation for determining IFN-γ concentrations using an ELISA kit.
Determination of the full length 793 RNA by RACE
The GeneRacer kit (Life Technologies) and an oligonucleotide based on the cDNA sequence of the positive clone 793A11 were used to determine the 5′ end of AS-TRIT RNA in WM793 cells. The sequence of the AS-TRIT-specific oligonucleotide was: 5′-AAAAGCAGCACAGATT CCACATTTTTATACATGAGGATCTTCTTTGTG-3′.
DNA sequencing and sequence analysis
DNA sequencing was performed by The Wistar DNA sequencing facility. The amino acid sequence was deduced from the DNA sequence using ExPASy (http://web.expasy.org/translate/). DNA and deduced amino acid sequence comparisons were performed with the BLAST program (http://blast.ncbi.nlm.nih.gov/Blast.cgi). For further predictions, additional programs of Genbank (http://www.ncbi.nlm.nih.gov/genbank/), ExPASy (http://ca.expasy.org/tools/) and PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/) were used.
Peptide design
The cloned cDNA including linkers had five putative ORFs (considering both reading directions) with a minimum length of 20 amino acids. Potential HLA-B57-binding epitopes were determined from the deduced amino acid sequences of the isolated cDNA clone using the HLA-B57 epitope prediction (x-(ATS)-x-x-x-x-x-x-(FWY)) of the HIV HLA anchor residue motifs search (http://www.hiv.lanl.gov/content/immunology/motif_scan/motif_scan). The following custom synthesized peptides (Life Technologies) predicted to bind to HLA-B57 were used: TSVIYKSIW (peptide 1), KSIWKLDNF (peptide 2), IASGRPFFF (peptide 3), STFLYMRIF (peptide 4), SSTDSTFLY (peptide 5), and IVEAEAMNY (unrelated HLA-A1 control peptide). Additional peptides of the AS-TRIT ORF predicted to bind to HLA-A2 were determined using the Rammensee epitope prediction model.39 The peptides were ILRTSLVHL (A2_1 peptide) and LRTSLVHLI (A2_2 peptide). As control, a HLA-A2 restricted peptide of epidermal growth factor receptor was used (KALEEKKGNY).
Stimulation of PBMC with peptides
Adherent monocytes (5 × 104 per well) were pulsed for 8 hours with peptides in PLG microspheres (1 µg/ml), excess peptides were removed, and then were cocultured with PBMC (105 cells/well) in T-cell medium supplemented with rIL-2 for 5–6 days.40 The peptide stimulation was repeated twice and cytotoxic activities of short-term T-cell lines (effectors) were measured in a 51Cr-release assay using peptide-pulsed or unpulsed melanoma target cells.
Cytotoxicity assay
CTL activity was tested in standard 4–18 hours 51Cr-release assays. In brief, 51Cr-labeled tumor targets12,41 were pulsed with various peptide concentrations (0.7–50 µmol/l) in the presence of β2-microglobulin (1 µg/ml) for 8 hours or left unpulsed and then incubated with T cells.40 At the end of the incubation, supernatants containing 51Cr were harvested and specific lysis of targets was determined.12,41 Counts per minute data were used for statistical analyses. Cytotoxicity assays were also done in the presence of HLA class I-specific mAb W6/32 (gift of B Perussia, Thomas Jefferson University) or anti-HLA-A2 mAb KS1 (obtained from S Ferrone, Massachusetts General Hospital, Boston, MA)42 to confirm the HLA dependency of lysis. All experiments were repeated more than once for independent confirmation.
Cold target inhibition
1205 LU cells (5 × 105 cells/0.5 ml) were pulsed with peptide 2 (25 µmol/l) in the presence of β2-microglobulin overnight at 37 °C in 5% CO2. Excess peptides were removed and graded numbers of cold targets were preincubated with CTL793 for 1 hour at 37 °C and 5% CO2, mixed with 51Cr prelabeled WM793 cells (1 × 103 cells plus 2.5 µCi 51Cr per well, overnight at 37 °C and 5% CO2) at an E: T ratio of 2:1 and were then further incubated for 6 hours. At the end of the incubation, supernatants were harvested and 51Cr-release was measured.
Reverse transcription PCR analysis
DNase-treated total RNA (10 µg) were reverse transcribed to obtain cDNA with specific primers (3′ AS-TRIT: 5′-AAAAGCAGCACAGATTCCAC-3′; 3′ TRIT-1: 5′-ACTCAATAGAAAGACAGCAGTG-3′) using thermoscript (Life Technologies). PCR reactions (35 cycles) were performed with diluted cDNA (1:10) with the following specific primers: For TRIT1 mRNA, the 3′ TRIT1 and 3′ AS-TRIT (=5′ TRIT1) primers were used (see above). For the AS-TRIT RNA PCR reaction, the 3′ AS-TRIT primer was combined with the 5′ AS-TRIT primer: 5′-ATGTCACAGCTTCCTCAGTC-3′. PCR products were loaded on 1.5% agarose gels, DNA bands imaged after ethidium bromide staining using GelLogic 200 (Eastman Kodak, Rochester, NY), and the intensity of each TRIT1 and AS-TRIT signal was determined using image analysis software. PCR products were sequenced to confirm their identity to the AS-TRIT RNA.
Antisense oligonucleotide treatment
Modified antisense oligonucleotides were custom designed (Integrated DNA Technology, Coralville, IA) and were reverse complementary to the predicted AS-TRIT ORF flanking the recognized epitope without any overlap (see Figure 1b). The three oligonucleotides with a phosphor-rothioate backbone were 793-2 (5′ T*T*T*A*T*G*T*C*C*C*T*G*A*C*T*C*T*G*G*C*T-3′), 793chim-1 (5′-mG*mG*mG*mA*mG*mU*T*C*T*A*G*A*T*T*T*G* A*G*T*G* mA*mA*mU*mG*mG*mC-3′) and 793chim24-2 (5′-mC*mU*mA*mC*mA*mG* A*G*A*A* G*G*A*G*G*G*A*A*mU*mC*mA*mG*mA*mC-3′). WM793 cells were transfected with the different antisense oligonucleotides using Oligofectamine (Life Technologies). After 2 days, cells were used in a cytotoxicity assay with CTL793 (live/dead viability assay kit).
Transduction of melanocytes and melanoma cells
Melanocytes (from newborn foreskins) were grown in melanocyte medium (Medium 254CF with supplements, Life Technologies) and they were transduced with lentivirus expressing either the AS-TRIT ORF or control vector using a standard protocol. Blasticidin-resistant cells were analyzed for colony formation (soft agar assay) and growth in melanocyte medium. Growth rates of transduced melanocytes were determined using the MTS assay (Promega, Madison, WI).
Statistical analysis
Data were analyzed using Student’s t-test.
Acknowledgments
The research was funded in part by grants CA60975, CA88193, CA25874, and CA10815. We thank Ling Li for supplying melanocytes. The authors declare no conflict of interest.
References
- Chapman, PB, Hauschild, A, Robert, C, Haanen, JB, Ascierto, P, Larkin, J et al. BRIM-3 Study Group. (2011). Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364: 2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi, FS, O’Day, SJ, McDermott, DF, Weber, RW, Sosman, JA, Haanen, JB et al. (2010). Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363: 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski, TF, Schreiber, H and Fu, YX (2013). Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 14: 1014–1022.24048123 [Google Scholar]
- Kaufman, HL, Kirkwood, JM, Hodi, FS, Agarwala, S, Amatruda, T, Bines, SD et al. (2013). The Society for Immunotherapy of Cancer consensus statement on tumour immunotherapy for the treatment of cutaneous melanoma. Nat Rev Clin Oncol 10: 588–598. [DOI] [PubMed] [Google Scholar]
- Wolchok, JD, Kluger, H, Callahan, MK, Postow, MA, Rizvi, NA, Lesokhin, AM et al. (2013). Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 369: 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanneman, M and Dranoff, G (2012). Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer 12: 237–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley, ME, Wunderlich, JR, Robbins, PF, Yang, JC, Hwu, P, Schwartzentruber, DJ et al. (2002). Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 298: 850–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins, PF, Lu, YC, El-Gamil, M, Li, YF, Gross, C, Gartner, J et al. (2013). Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med 19: 747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij, N, van Buuren, MM, Philips, D, Velds, A, Toebes, M, Heemskerk, B et al. (2013). Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol 31: e439–e442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen, RS, Andersen, SR, Hjortsø, MD, Lyngaa, R, Idorn, M, Køllgård, TM et al. (2013). High frequency of T cells specific for cryptic epitopes in melanoma patients. Oncoimmunology 2: e25374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, SM (2014). Everything old is new again: (linc)RNAs make proteins! EMBO J 33: 937–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somasundaram, R, Jacob, L, Swoboda, R, Caputo, L, Song, H, Basak, S et al. (2002). Inhibition of cytolytic T lymphocyte proliferation by autologous CD4+/CD25+ regulatory T cells in a colorectal carcinoma patient is mediated by transforming growth factor-beta. Cancer Res 62: 5267–5272. [PubMed] [Google Scholar]
- Somasundaram, R, Robbins, P, Moonka, D, Loh, E, Marincola, F, Patel, A et al. (2000). CD4(+), HLA class I-restricted, cytolytic T-lymphocyte clone against primary malignant melanoma cells. Int J Cancer 85: 253–259. [DOI] [PubMed] [Google Scholar]
- Mayrand, SM and Green, WR (1998). Non-traditionally derived CTL epitopes: exceptions that prove the rules? Immunol Today 19: 551–556. [DOI] [PubMed] [Google Scholar]
- Van Den Eynde, BJ, Gaugler, B, Probst-Kepper, M, Michaux, L, Devuyst, O, Lorge, F et al. (1999). A new antigen recognized by cytolytic T lymphocytes on a human kidney tumor results from reverse strand transcription. J Exp Med 190: 1793–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beiter, T, Reich, E, Williams, RW and Simon, P (2009). Antisense transcription: a critical look in both directions. Cell Mol Life Sci 66: 94–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheetham, SW, Gruhl, F, Mattick, JS and Dinger, ME (2013). Long noncoding RNAs and the genetics of cancer. Br J Cancer 108: 2419–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman, M, Russell, P, Ingolia, NT, Weissman, JS and Lander, ES (2013). Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell 154: 240–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia, NT, Lareau, LF and Weissman, JS (2011). Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147: 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carninci, P, Sandelin, A, Lenhard, B, Katayama, S, Shimokawa, K, Ponjavic, J et al. (2006). Genome-wide analysis of mammalian promoter architecture and evolution. Nat Genet 38: 626–635. [DOI] [PubMed] [Google Scholar]
- Engström, PG, Suzuki, H, Ninomiya, N, Akalin, A, Sessa, L, Lavorgna, G et al. (2006). Complex Loci in human and mouse genomes. PLoS Genet 2: e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frith, MC, Bailey, TL, Kasukawa, T, Mignone, F, Kummerfeld, SK, Madera, M et al. (2006). Discrimination of non-protein-coding transcripts from protein-coding mRNA. RNA Biol 3: 40–48. [DOI] [PubMed] [Google Scholar]
- Gingeras, TR (2006). The multitasking genome. Nat Genet 38: 608–609. [DOI] [PubMed] [Google Scholar]
- Sureau, A, Soret, J, Guyon, C, Gaillard, C, Dumon, S, Keller, M et al. (1997). Characterization of multiple alternative RNAs resulting from antisense transcription of the PR264/SC35 splicing factor gene. Nucleic Acids Res 25: 4513–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somasundaram, R, Swoboda, R, Caputo, L, Lee, A, Jackson, N, Marincola, FM et al. (2003). A CD4+, HLA-DR7-restricted T-helper lymphocyte clone recognizes an antigen shared by human malignant melanoma and glioma. Int J Cancer 104: 362–368. [DOI] [PubMed] [Google Scholar]
- Severin, J, Lizio, M, Harshbarger, J, Kawaji, H, Daub, CO, Hayashizaki, Y et al. FANTOM Consortium. (2014). Interactive visualization and analysis of large-scale sequencing datasets using ZENBU. Nat Biotechnol 32: 217–219. [DOI] [PubMed] [Google Scholar]
- Lawrence, MS, Stojanov, P, Polak, P, Kryukov, GV, Cibulskis, K, Sivachenko, A et al. (2013). Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499: 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golovko, A, Hjälm, G, Sitbon, F and Nicander, B (2000). Cloning of a human tRNA isopentenyl transferase. Gene 258: 85–93. [DOI] [PubMed] [Google Scholar]
- Urbonavicius, J, Qian, Q, Durand, JM, Hagervall, TG and Björk, GR (2001). Improvement of reading frame maintenance is a common function for several tRNA modifications. EMBO J 20: 4863–4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirheimer, G, Baranowski, W and Keith, G (1995). Variations in tRNA modifications, particularly of their queuine content in higher eukaryotes. Its relation to malignancy grading. Biochimie 77: 99–103. [DOI] [PubMed] [Google Scholar]
- Henderson, LJ, Coe, BP, Lee, EH, Girard, L, Gazdar, AF, Minna, JD et al. (2005). Genomic and gene expression profiling of minute alterations of chromosome arm 1p in small-cell lung carcinoma cells. Br J Cancer 92: 1553–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinola, M, Galvan, A, Pignatiello, C, Conti, B, Pastorino, U, Nicander, B et al. (2005). Identification and functional characterization of the candidate tumor suppressor gene TRIT1 in human lung cancer. Oncogene 24: 5502–5509. [DOI] [PubMed] [Google Scholar]
- Katayama, S, Tomaru, Y, Kasukawa, T, Waki, K, Nakanishi, M, Nakamura, M et al. RIKEN Genome Exploration Research Group; Genome Science Group (Genome Network Project Core Group); FANTOM Consortium. (2005). Antisense transcription in the mammalian transcriptome. Science 309: 1564–1566. [DOI] [PubMed] [Google Scholar]
- Le, D, Wang-Gilam, A, Picozzi, V, Greten, TF, Crocenzi, TS, Springett, GM et al. (2014). A phase 2, randomized trial of GVAX pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: updated results. J Clin Oncol 32:177. [Google Scholar]
- Yuan, J, Kashiwagi, S, Reeves, P, Nezivar, J, Yang, Y, Arrifin, NH et al. (2014). A novel mycobacterial Hsp70-containing fusion protein targeting mesothelin augments antitumor immunity and prolongs survival in murine models of ovarian cancer and mesothelioma. J Hematol Oncol 7: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandolfi, PP (2004). Aberrant mRNA translation in cancer pathogenesis: an old concept revisited comes finally of age. Oncogene 23: 3134–3137. [DOI] [PubMed] [Google Scholar]
- Jacob, L, Somasundaram, R, Smith, W, Monos, D, Basak, S, Marincola, F et al. (1997). Cytotoxic T-cell clone against rectal carcinoma induced by stimulation of a patient’s peripheral blood mononuclear cells with autologous cultured tumor cells. Int J Cancer 71: 325–332. [DOI] [PubMed] [Google Scholar]
- Brichard, V, Van Pel, A, Wölfel, T, Wölfel, C, De Plaen, E, Lethé, B et al. (1993). The tyrosinase gene codes for an antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J Exp Med 178: 489–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rammensee, H, Bachmann, J, Emmerich, NP, Bachor, OA and Stevanovic, S (1999). SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 50: 213–219. [DOI] [PubMed] [Google Scholar]
- Van Nuffel, AMT, Benteyn, D, Wilgenhof, S, Pierret, L, Corthals, J, Heirman, C et al. (2012). Dendritic cells loaded with mRNA encoding full-length tumor antigens prime CD4+ and CD8+ T cells in melanoma patients. Mol Therapy 20: 1063–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somasundaram, R, Swoboda, R, Caputo, L, Otvos, L, Weber, B, Volpe, P et al. (2006). Human leukocyte antigen-A2-restricted CTL responses to mutated BRAF peptides in melanoma patients. Cancer Res 66: 3287–3293. [DOI] [PubMed] [Google Scholar]
- Tsujisaki, M, Sakaguchi, K, Igarashi, M, Richiardi, P, Perosa, F and Ferrone, S (1988). Fine specificity and idiotype diversity of the murine anti-HLA-A2, A28 monoclonal antibodies CR11-351 and KS1. Transplantation 45: 632–639. [DOI] [PubMed] [Google Scholar]