
Figure 1.
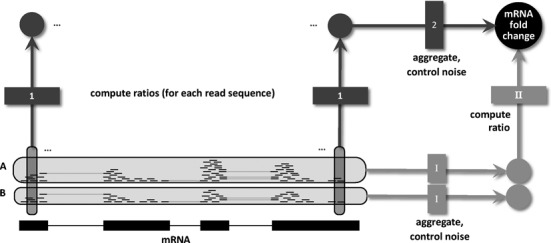
Workflows for differential NGS analysis. Differential analysis of NGS data starts with the aligned reads of two conditions, here exemplified as RNA-seq reads from samples A and B aligned to an mRNA. Existing models take one specific route through the necessary steps defined in the main text: (I) For each sample, reads are aggregated and an appropriate probabilistic model is used to control noise and estimate the sample specific mRNA abundance. (II) These abundance estimates are then divided to give an estimate of the mRNA fold change. Our approach takes a different route by first computing local ratios for all read sequences and then aggregating them using an appropriate noise model for count ratios to estimate the total mRNA fold change. Using a basic noise model for the second step makes both routes equivalent. However, using extensions to it leads to more accurate fold change estimates by exploiting the fact that bias cancels out when taking the ratio of counts of individual sequences. Note that two important aspects of NGS (replicate experiments and normalization) are left out in this figure and are analyzed and discussed below.