
Figure 1.
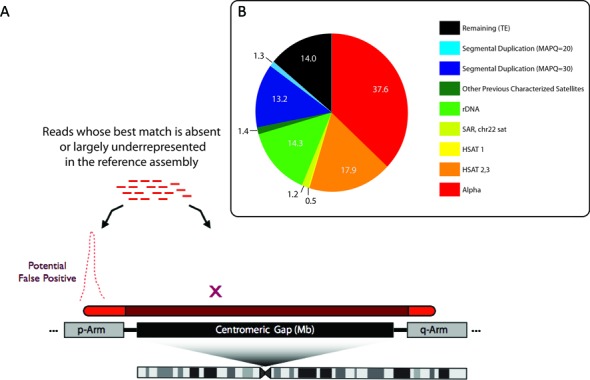
Large, multi-megabase sized regions of the human genome remain incomplete due to highly repetitive regions of the human genome, mapping to centromere/heterochromatin assigned gaps, and including sequences that remain missing from subtelomeric regions in the acrocentric short arms. As shown in (A), these regions are marked in the genome by gaps or space holders to indicate regions that are enriched for long arrays of tandemly repeated DNA. Often the edges of these gaps provide some representation of the sequences across the entirety of the array (shown as red if included in the assembly and shaded red if inferred to be present in the gap region). Sequence reads from the entire region are expected to be present in high-throughput, whole-genome datasets. When mapping to a partial reference, these reads find their best alignments on the regions represented in the assembly. As a result, a large number of reads (representing the multi-megabase arrays) align with high read depth, resulting in false positive sites in the genome. To account for these mapping errors we have designed mapping targets, collectively called a ‘sponge database’ with the various distribution of DNA families shown in (B) for the collection of 1.5 million remaining unassembled reads from the HuRef genome.