


Abstract
RNA cleavage by some endoribonucleases and self-cleaving ribozymes produces RNA fragments with 5′-hydroxyl (5′-OH) and 2′,3′-cyclic phosphate termini. To identify 5′-OH RNA fragments produced by these cleavage events, we exploited the unique ligation mechanism of Escherichia coli RtcB RNA ligase to attach an oligonucleotide linker to RNAs with 5′-OH termini, followed by steps for library construction and analysis by massively parallel DNA sequencing. We applied the method to RNA from budding yeast and captured known 5′-OH fragments produced by tRNA Splicing Endonuclease (SEN) during processing of intron-containing pre-tRNAs and by Ire1 cleavage of HAC1 mRNA following induction of the unfolded protein response (UPR). We identified numerous novel 5′-OH fragments derived from mRNAs: some 5′-OH mRNA fragments were derived from single, localized cleavages, while others were likely produced by multiple, distributed cleavages. Many 5′-OH fragments derived from mRNAs were produced upstream of codons for highly electrostatic peptides, suggesting that the fragments may be generated by co-translational mRNA decay. Several 5′-OH RNA fragments accumulated during the induction of the UPR, some of which share a common sequence motif that may direct cleavage of these mRNAs. This method enables specific capture of 5′-OH termini and complements existing methods for identifying RNAs with 2′,3′-cyclic phosphate termini.
INTRODUCTION
Many endoribonucleases including the tRNA splicing endonuclease (1), Ire1 (2) and the ribonucleases T2 (3) and L (4) create cleavage products with 5′-hydroxyl (5′-OH) and 2′,3′-cyclic phosphate termini, as do self-cleaving ribozymes like the hammerhead, HDV, VS, hairpin and twister ribozymes (5,6). In addition, accelerated RNA cleavage by intramolecular phosphoester transfer at structurally distorted phosphodiester bonds generates 5′-OH and 2′,3′-cyclic phosphate products (7). However, products of RNA cleavage with 5′-OH and 2′,3′-cyclic phosphate termini are not typically identified in global surveys of RNA populations, and thus their frequency and role in RNA metabolism remain unknown.
Methods for studying RNAs with specific termini can shed light on RNA transcription initiation, processing, and decay pathways. Several methods have been developed for the global study of mRNA 5′-termini, but most of these do not capture 5′-OH termini, or do so indirectly via a 5′-PO4 intermediate. In a recent example, 5′-OH termini were indirectly inferred from cloning of 5′-PO4 termini created upon phosphorylation of 5′-OH termini (8), but this method requires several enzymatic manipulations prior to the ligation step. Similarly, the Parallel Analysis of RNA Ends (PARE) method (9) begins with ligation of an RNA adapter to the 5′-PO4 terminus of an RNA substrate with T4 RNA ligase, and thus this method does not capture 5′-OH termini. Other methods compare signals among different enzymatic manipulations of 5′ mRNA cap structures to enable specific detection of mRNA 5′-termini (10), but these methods are unable to capture 5′-OH termini.
We previously developed a method to identify RNA fragments with 2′,3′-cyclic phosphate and 2′-PO4/3′-OH termini using the Arabidopsis thaliana tRNA ligase (11). Recent studies used this method to discover virus and host targets of ribonuclease L cleavage (4,12), and to identify the Gas2 mRNA as a novel target of the Ire1 endonuclease in the Schizosaccharomyces pombe unfolded protein response (UPR) (13). Fungal and plant tRNA ligases join RNAs with 5′-OH and 2′,3′-cyclic phosphate termini via 5′-PO4 and 5′-adenylate intermediates (14,15), and thus the method employing A. thaliana tRNA ligase does not distinguish between 5′-OH and 5′-PO4 RNA termini. In addition, libraries produced by this method typically contain high levels of ribosomal RNA fragments with cyclic-phosphate termini, which may mask signals from lower abundance fragments from mRNA. Therefore, we were motivated to develop a new method to capture the other products of RNA cleavage—5′-OH RNA fragments—that also allows mRNAs to be enriched via oligo-dT selection of their polyadenylate tails.
Escherichia coli RtcB RNA ligase catalyzes a unique chemistry involving the attack of a 3′-guanylylate intermediate by a 5′-OH nucleophile, yielding a 3′-5′ phosphodiester bond and GMP (16). As noted previously (17,18), RtcB RNA ligase therefore represents a unique reagent for direct capture of 5′-OH RNAs. Here, we developed a method using the E. coli RtcB RNA ligase to globally identify 5′-OH RNA fragments in complex RNA populations and applied the method to study 5′-OH RNA fragments in budding yeast.
MATERIALS AND METHODS
Expression and purification of E. coli RtcB RNA ligase
The E. coli RtcB gene was PCR amplified, recombined into pDONR221 (Gateway BP, Invitrogen) and Sanger sequenced to confirm lack of mutations in the open reading frame. The RtcB ORF was recombined into pET-53-DEST (creating an N-terminal 6xHis tag; Gateway LR, Invitrogen) and transformed into BL21 (DE3) RIPL cells (Stratagene). The pET-53-RtcB expression construct is available from Addgene (Plasmid 51282). For RtcB protein expression, cells were grown from a single colony in 1-l terrific broth with selective antibiotics (50 μg/ml ampicillin and 34 μg/ml chloramphenicol) at 37°C to an OD600 of 0.6 and then chilled on ice for 30 min. Ethanol was added to 2%, expression was induced with 0.1 mM isopropylthiogalactoside (IPTG). and cultures were grown 16 h at 17°C. Cells were harvested by centrifugation and lysed for 1 h at 4°C in Buffer A (50 mM Tris pH 7.4, 250 mM NaCl, 10% sucrose) with 0.2 mg/ml lysozyme. Triton X-100 was added to a final concentration of 0.1%, and the cells were sonicated briefly to reduce viscosity. Following clarification by centrifugation at 10 000 × g, the supernatant was incubated for 1 h at 4°C with 3 ml of pre-washed Nickel-NTA beads (Qiagen). The beads were washed with 25 ml of Buffer A, then loaded onto a column and washed twice with 25 ml Buffer E (50 mM Tris–HCl (pH 7.4), 10% glycerol, 250 mM NaCl) containing 15 mM imidazole. The column was eluted with 3 ml each of Buffer E with 50, 150 and 300 mM imidazole. Peak fractions were identified by SDS-PAGE and were pooled and diluted to 50 mM NaCl with Buffer D (50 mM Tris–HCl (pH 7.4), 10% glycerol, 5 mM dithiothreitol (DTT), 1 mM ethylenediaminetetraacetic acid (EDTA)). RtcB protein was further purified on a 1 ml HiTrap Heparin HP column (GE Healthcare Life Sciences), washed with Buffer D containing 50 mM NaCl, and eluted with 3 ml each of Buffer D containing 150, 300 and 500 mM NaCl. RtcB protein eluted predominantly in the 150–300 mM NaCl fractions, with a typical yield of 2 mg/l culture. Peak fractions were pooled, quantitated by Bradford assay, snap frozen in liquid nitrogen and stored at −80°C.
RtcB ligation assay
Oligonucleotides used in this study are listed in Table 1. Ligation assays were used to confirm recombinant RtcB function. A linker oligonucleotide with a 3′ phosphate was incubated at equimolar concentration (200 pM) with another oligonucleotide with a 5′-OH terminus in the presence of 1.25 μM RtcB in buffer (1 mM GTP, 50 mM MOPS pH 7.4, 2 mM MnCl2) for 1 h at 37°C. The reaction was quenched with stop dye (95% formamide, 5 mM EDTA pH 8.0, 0.01% xylene cyanol and bromophenol blue), denatured at 70°C for 3 min, and electrophoresed on 10% polyacrylamide gels containing 7 M urea and 1X 89 mM Tris, 89 mM boric acid and 2 mM EDTA (TBE) and stained with SYBR Gold (Invitrogen) prior to visualization.
Table 1. Oligonucleotides.
| Name | Sequence |
|---|---|
| RNA ligation assay | |
| M13-linker | 5′ Amino – rGrUrUrUrUrCrCrCrArGrUrCrArCrGrArC – 3′ phosphate |
| RPS31-5OH | 5′ OH – rArArGrArArGrArGrArArArGrArArGrArArGrGrUrCrUrArCrArCrCrArCrC 3′ Amino |
| 5OH library | |
| 5OH-Linker | 5′ desthiobiotin – ACACTCTTTCCCTACACGACGCTCTTCCGATCTrNrNrNrNrNrNrNrN 3′ phosphate |
| 5OH-RT-amino | 5′ Amino – GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNNNNN – 3′ |
| Validation primers | |
| M13-F | 5′ GTTTTCCCAGTCACGAC |
| RPS31-F | 5′ CCCTGGAAGTTGAATCTTCTG |
| RPS31-R | 5′ GCGTTAACCTTGTAGACGGAAT |
Yeast strains and culturing
Yeast strains used in this study are listed in Table 2. Single colonies were inoculated in YEPD and incubated at 30°C overnight with rotation. Cultures were diluted to an OD600 of 0.1 in YEPD. In untreated conditions, yeast cells were harvested in mid-log phase. To induce the UPR, yeast grown to mid-log phase were treated for 2 h with tunicamycin (final concentration of 2.5 μg/ml, Sigma-Aldrich). Cells were harvested by centrifugation and total RNA was isolated by hot acid phenol extraction and treated with TURBO DNase (Ambion) to remove genomic DNA.
Table 2. Yeast strains.
| Strain | Genotype |
|---|---|
| XRN1 (BY4742) | MATα his3Δ1leu2Δ0lys2Δ0ura3Δ0 |
| xrn1Δ | MATα his3Δ1leu2Δ0lys2Δ0 ura3Δ0 xrn1Δ::hygMX |
5′-Hydroxyl RNA library construction and sequencing
Total RNA from budding yeast (15 μg) was enriched for a polyadenylated mRNA fraction using Oligo-dT magnetic beads (Ambion). PolyA-enriched RNA was incubated with 1.1 μM RtcB in buffer (1 mM GTP, 50 mM MOPS pH 7.4, 2 mM MnCl2), 20 units RNase Inhibitor (Enzymatics) and 10 μM linker (‘5OH-Linker’ in Table 1) at 37°C for 1 h. Following phenol/chloroform extraction and ethanol precipitation, RNA was fragmented with Ambion Fragmentation Reagent for 15 min at 70°C, and the reaction was quenched with the addition of EDTA. Samples were denatured in stop dye (95% formamide, 0.01% xylene cyanol / bromophenol blue), heated to 65°C for 5 min and separated on a 10% acrylamide TBE–urea gel. Following staining with SYBR Gold (Invitrogen), the gel was visualized by blue light transillumination and a gel slice containing RNA above 80 nt was excised and pulverized. RNA was eluted from the gel by 2 h incubation at 40°C in 0.3 M sodium acetate, pH 5.2, 1 mM EDTA, pH 8.0 and precipitated with 2.5 volumes of ethanol. cDNA was prepared using an Illumina-compatible primer with a 6-basepair degenerate region (oligonucleotide 5OH-RT-amino) and Protoscript II RT (NEB), and was purified using Agencourt RNAClean XP Beads (Beckman Coulter). Second strand synthesis was performed with RNase H, E. coli DNA Polymerase, and E. coli ligase (Enzymatics). Double-stranded DNA products were purified using magnetic Streptavidin beads (Invitrogen), eluted with 25 mM biotin in elution buffer (Omega Bio-Tek) and PCR amplified with Illumina TruSeq primers and Phusion DNA polymerase. PCR reactions were purified with SPRI Size Select Beads (Beckman Coulter) to yield an average product size of 350 bp. Indexed libraries were quantified by Qubit (Invitrogen), mixed to a final concentration of 1–10 nM and sequenced on an Illumina MiSeq in a 50 cycle run. Raw and processed sequencing data are available at NCBI GEO under accession number GSE61527.
Analysis of DNA sequencing data
Sequences in FASTQ format were preprocessed to remove molecular indexes using umitools (https://github.com/brwnj/umitools). Processed reads were aligned to the S. cerevisiae genome (sacCer1) with Bowtie (19) and visualized in the UCSC Genome Browser (20). Coverage signals in bedGraph format were generated, normalized to UMI-corrected Counts Per Million reads mapped (CPM), and analyzed with BEDTools (21) and custom scripts. Studies of mRNA cleavage focused on the collection of 5′-OH fragments in the upper decile of the CPM distribution (i.e. the most abundant 10%) that were reproducible in replicate samples prepared from two independent cultures. Significant discrete 5′-OH signals within mRNAs were identified as previously described (22). Briefly, CPM values for each mRNA were fit to a negative binomial distribution, and P values were computed for each site. P values were adjusted using the Benjamini-Hochberg false discovery rate (FDR) (<0.05). Clustered hits were determined by identifying regions of high 5′-OH signal density using MACS (23) then selecting regions longer than 100 bp and discarding regions included in the list of mRNAs with single fragments. Additional workflow and analysis scripts are available at https://github.com/hesselberthlab/5OH. Processed data can be visualized on the UCSC Genome Browser in the track hub at https://github.com/hesselberthlab/trackhub.
Validation of 5′-hydroxyl RNA fragments by RtcB ligation and RT-PCR
5′-OH RNA fragments were verified by RtcB-mediated ligation of an oligonucleotide linker, followed by reverse transcription and gene-specific PCR. Total RNA (5 μg) was ligated to an RNA oligonucleotide with a 5′-amino group and a 3′-phosphate (M13-Linker, Table1 ) with RtcB. Reactions were phenol-chloroform extracted, ethanol precipitated, and reverse transcribed with Superscript III RT (Invitrogen) and gene-specific primers. cDNA was PCR amplified using primer pairs that hybridize to full length mRNA or the linker-ligated 5′-OH RNA fragment. PCR products were resolved on 10% TBE non-denaturing 29:1 acrylamide gels and stained with SYBR Gold.
RESULTS
Development of a method for capture of 5′-hydroxyl RNAs
To demonstrate the utility of RtcB RNA ligase as a reagent for global capture of 5′-OH RNA, we first asked whether RtcB could ligate unrelated oligonucleotide substrates—a prerequisite for global capture of 5′-OH molecules. We overexpressed and purified E. coli RtcB ligase (Figure 1A) and used the enzyme to perform ligations with oligoribonucleotide substrates with 2′-OH/3′-PO4 and 5′-OH termini with minimal predicted base pairing (Figure 1B). We found that RtcB could ligate these substrates in a bimolecular ligation with ∼25% efficiency, confirming previous studies of RtcB intermolecular ligation (24) and suggesting that RtcB could be used in global profiling of cellular RNAs with 5′-OH termini.
Figure 1.
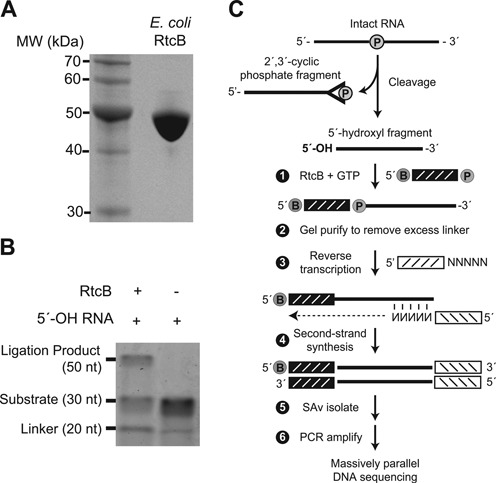
(A) SDS-PAGE analysis of E. coli RtcB protein. Recombinant RtcB protein (10 μg) was electrophoresed through a 10% SDS-PAGE gel and stained with Coomassie blue. (B) RtcB-mediated bimolecular ligation of RNA substrates with 3′-PO4 and 5′-OH termini was analyzed by polyacrylamide gel electrophoresis. The ligation product at 50 nt was dependent on RtcB, with an efficiency of ∼25%. (C) Schematic of 5′-hydroxyl (5′-OH) cloning. RNA cleavage generates two fragments with 2′,3′-cyclic phosphate and 5′-OH termini. An adaptor with a 5′-desthiobiotin (B) and 3′-phosphate (denoted by P) is ligated to the 5′-OH RNA fragment with E. coli RtcB ligase. This adaptor has eight randomized positions at its 3′ end which serve as a molecular index to uniquely identify individual ligation events upon sequencing (25). Following ligation, excess adaptor is removed by gel purification. A primer with a 3′ random hexamer region is used for reverse transcription to create cDNA, followed by second-strand synthesis to generated double-stranded DNA products. After streptavidin (SAv) isolation, DNA fragments are PCR amplified and analyzed by Illumina sequencing.
We next established a general protocol to capture 5′-OH RNA from complex RNA populations using the RtcB ligase (Figure 1C). We began by chemically fragmenting total RNA from budding yeast, producing many RNA fragments with 5′-OH and 2′,3′-cyclic phosphate termini. RNA fragments with 5′-OH termini were ligated to an oligonucleotide with 5′-desthiobiotin and 3′-PO4 groups using E. coli RtcB ligase. This oligonucleotide linker contains an 8-base random sequence at its 3′ end that serves as a molecular index during sequence analysis, enabling quantitation of unique ligation events and estimation of relative amounts of 5′-OH fragments in the initial RNA population (25). After gel purification to remove excess unligated linker, RNA was reverse transcribed with a second oligonucleotide primer containing an Illumina primer binding site and six randomized bases at the 3′ end to facilitate random priming to RNA templates. Double-stranded RNA:DNA hybrids were converted to double-stranded DNA by second-strand synthesis, and double-stranded DNA fragments containing the desthiobiotin linker were purified on immobilized streptavidin and eluted with free biotin. Purified double-stranded DNA fragments were PCR amplified with primers that incorporate library-specific indexes and flow cell sequences compatible with Illumina sequencing. Analysis of these libraries by gel electrophoresis yielded a broad distribution of double stranded DNA products from 120 bp to 1500 bp with a median size of 350 bp. These products were dependent on RtcB ligation and reverse transcription (data not shown).
Global analysis of 5′-hydroxyl RNAs in budding yeast
We used the cloning protocol (as described in ‘Materials and Methods’ section) to identify 5′-OH RNA fragments in the budding yeast S. cerevisiae. We prepared libraries from oligo-dT enriched yeast total RNA, in order to increase signals that might be present on polyadenylated mRNA fragments. These libraries were sequenced on the Illumina platform to identify 5′-OH RNA fragments. The libraries had an average of 2.7 million reads, with an average of 23% of the reads aligning uniquely to the yeast genome and an additional 30% of reads aligning multiple times. We examined the distribution of uniquely aligning reads within existing RNA annotations, and found that in polyA-enriched samples, 21% mapped to mRNA regions, 0.4% mapped to abundant non-coding RNA species (tRNA, snoRNA, and snRNA), and 65% of the reads were derived from ribosomal RNA. The remaining 13.6% of reads mapped to other regions including retrotransposon-derived and intergenic regions. Using these sequences, we also assessed whether RtcB exhibited a bias for specific sequences at the ligation junction. We found that the base composition of each position downstream of the ligation site (i.e. the 5′-OH terminus) mirrored budding yeast A/T content (62%), suggesting that RtcB does not have strong preference for 5′-OH RNAs with specific sequences. The 8 nt molecular index in ligated adaptors (Figure 1C and 5OH-Linker, Table 1) allowed us to examine base composition upstream of the ligation site, and we found that RtcB preferred adaptors with an adenosine at their 3′ terminus, which was present in 42% of sequences, while the remaining 7 positions had more random composition (data not shown). Because the molecular index is trimmed from reads prior to alignment, this ligation bias has no effect on sequence alignment, but does limit the diversity of possible molecular indexes.
To validate the protocol, we analyzed the capture of 5′-OH termini generated in vitro and in vivo. Ribosomal RNAs have 5′-PO4 termini produced by endonucleolytic (26) and exonucleolytic processing (27). These ends should be underrepresented in 5′-OH libraries, but we would expect to recover these termini in 5′-OH libraries prepared from RNA samples that had been dephosphorylated in vitro (Figure 2A). Therefore, we compared libraries prepared from total RNA samples that were treated or not with recombinant shrimp alkaline phosphatase (rSAP) to convert 5′-PO4 RNAs in the samples to 5′-OH groups. An average of 2574 reads aligned to the 5′ end of 5.8S rRNA in the dephosphorylated sample compared to an average of 358 5′-OH fragments aligned to the same position in the untreated samples, (Figure 2B), representing a 7-fold enrichment of reads at the 5′ end of this RNA upon phosphatase treatment and confirming that the method captures 5′-OH RNAs and discriminates against 5′-PO4 RNAs.
Figure 2.
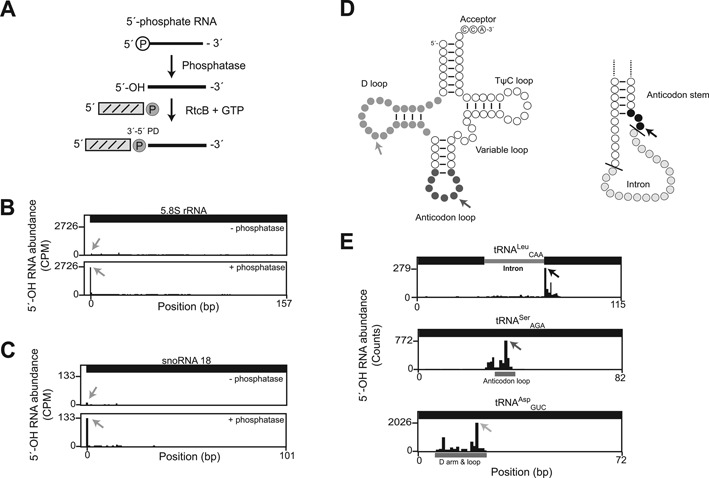
(A) Schematic of RtcB-mediated ligation to a 5′-OH RNA substrate produced by phosphatase-mediated removal of a 5′-PO4 group. (B) Abundance of 5′-OH fragments (per-site, UMI-corrected Counts Per Million reads mapped, CPM) mapped to 5.8S rRNA (157 nt). In an untreated sample (top), 5′-OH signal was uniformly low across the body of the 5.8S rRNA. 5′-OH RNA fragments cloned from total RNA treated with shrimp alkaline phosphatase (bottom) mapped to the 5′-end of 5.8S rRNA (gray arrows), indicating conversion of the 5′-PO4 to a 5′-OH group. (C) Abundance of 5′-OH fragments (CPM) in the snR18 locus show that the 5′-PO4 of snR18, produced by Rnt1-mediated processing of the EFB1 pre-mRNA (29), is specifically captured upon phosphatase treatment and 5′-OH cloning (gray arrows). (D) Schematics of tRNAs. On the left, a mature tRNA with the CCA terminator and the D, anticodon, variable, and TΨC loops. On the right, a portion of an intron-containing pre-tRNA with cleavage sites recognized by tRNA Splicing Endonuclease (SEN) represented by black lines. Sites within tRNAs that were captured abundantly by 5′-OH cloning are shaded. Arrows indicate the most abundant fragment in each tRNA shown in (E). (E) Examples of tRNA fragments captured by 5′-OH cloning in rRNA-depleted total RNA. Fragments from tRNALeuCAA mapped to the 5′-terminus of the 3′ exon (top panel). Anti-codon loop cleavages were found in tRNASerAGA (middle panel). tRNAAspGUC was the most abundant species captured by 5′-OH cloning and primarily showed D-arm and loop cleavage (bottom panel, Supplementary Table S1). Arrows indicate the most abundant fragment in each tRNA and correspond to the arrows in (D). Key regions of the tRNA are indicated by solid bars labeled as Intron, Anticodon loop, and D arm and loop.
The levels of ribosomal RNA fragments found in the poly(A)-enriched library suggested that oligo-dT capture of polyadenylated mRNA is not sufficient to deplete ribosomal RNA signals from libraries, and that other strategies for ribosomal RNA depletion might be used to further reduce signals from ribosomal RNA. We prepared libraries from an RNA sample following treatment with a commercial strategy to remove ribosomal RNA by hybridization to antisense probes (28). We found that signals from ribosomal RNAs were ∼10-fold less abundant in these libraries compared to libraries prepared from oligo-dT selected RNA, but also unexpectedly found that the commercial kit contained a phosphatase activity that converted the 5′-PO4 termini of ribosomal RNAs to 5′-OH groups, leading to their capture by 5′-OH cloning independent of additional phosphatase treatment (data not shown).
Similar to rRNA, some small nucleolar RNAs (snoRNAs) are produced from endonucleolytic and exonucleolytic processing of precursor RNA molecules and have 5′-monophosphate termini (29). Dephosphorylation of the 5′-PO4 termini of these snoRNAs would produce 5′-OH termini, and we found abundant signals for these in 5′-OH libraries following rSAP treatment. For example, the 5′-end of the box C/D snoRNA U18, which is produced by Rnt1 and Rat1 processing of the EFB1 pre-mRNA (30), was 6-fold more abundant in the dephosphorylated sample compared to the untreated sample (Figure 2C). Overall we identified six snoRNAs (snR18, 24, 55, 57, 61, 64) with 5′-PO4 termini that were captured by 5′-OH cloning after in vitro phosphatase treatment.
Cleavage of pre-tRNA molecules by the tRNA splicing endonuclease (SEN) creates intron and 3′-exon products with 5′-OH groups (1) (Figure 2D). Yeast pre-tRNA molecules range from 71 to 133 nt and processed tRNAs from 71 to 87 nt. Sequences in our libraries have an effective read length of 42 nt, and thus our libraries can be used to quantify cleavages within ∼30–40 nt of the 5′-end of processed tRNA molecules, depending on variable loop length. We analyzed signals from tRNAs in the 5′-OH library prepared from rRNA-depleted total RNA. The presence of 5′-OH fragments corresponding to the 5′-terminus of the 3′-exon of the intron-containing tRNALeuCAA (Figure 2E, top panel) confirms that this method can capture known 5′-OH termini generated in vivo. We also identified products of cleavage in the anticodon loop of tRNASerAGA (Figure 2E, middle panel), consistent with 2′,3′-cyclic phosphate capture of anticodon loop cleavage (11). Finally, we identified high levels of 5′-OH fragments that mapped to the D arm and loop of 28 tRNA species (Figure 2E, bottom panel; Supplementary Table S1). We compared the 5′-OH fragments in tRNAs to a previous study of 5′-PO4 RNA fragments (22), and found that 5′-PO4 signals were localized mainly to the 5′-ends of tRNA, indicating that the 5′-OH fragments represent a distinct pattern of tRNA cleavages (data not shown). We did not identify the 5′-OH intron products of SEN cleavage, presumably because they are phosphorylated by Trl1 and degraded by Xrn1 (31).
mRNA decay events identified with 5′-hydroxyl RNA capture
In order to focus on 5′-OH fragments present in mRNA, we collected sequence reads from libraries prepared from poly(A)-enriched RNA that mapped uniquely within annotated mRNA transcripts (32), and found 710 mRNAs that had significant signals in two replicate samples (35% overlap among mRNAs). We examined the distribution of 5′-OH fragments within these mRNAs (Figure 3A), and found that 76.7% of 5′-OH fragments mapped within open reading frames, 9.9% were in 5′ untranslated regions (UTRs) and 13.4% were in 3′ UTRs. We found 81 mRNAs with 5′-OH fragments that mapped exclusively to the 5′ and 3′ UTRs, and 551 mRNAs with 5′-OH fragments exclusively in their open reading frames. The remaining 78 mRNAs contained fragments distributed throughout the mRNA. Many 5′-OH fragments were biased toward the 3′-end of mRNAs, possibly reflecting a bias imposed by poly(A) purification prior to 5′-OH cloning (Figure 3A).
Figure 3.
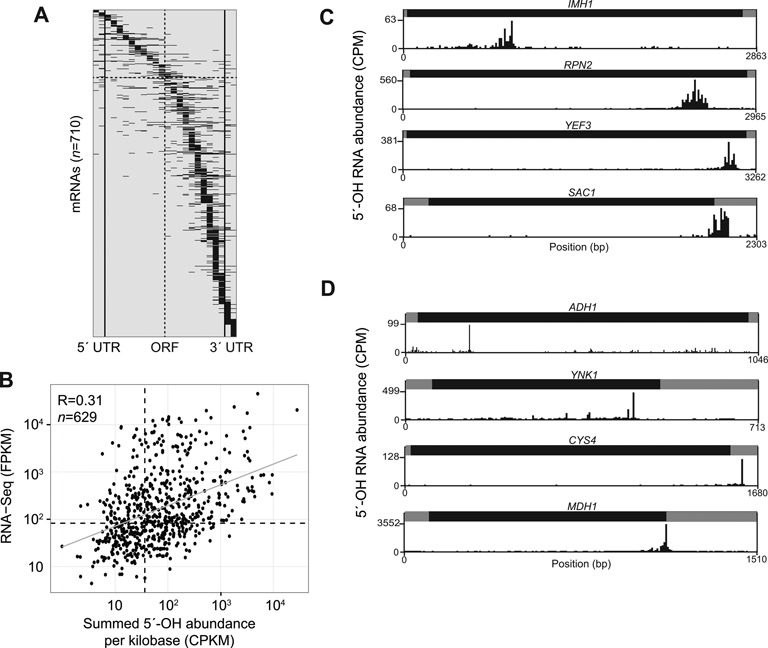
(A) 5′-OH fragments are found in numerous mRNAs. Each of the 710 mRNAs with a 5′-OH signal in the upper decile of replicates from a wild-type strain was divided into proportionally sized bins for the 5′ and 3′ UTR regions (two bins each, left and right of the solid vertical lines), and the open reading frame (ORF; 20 bins). 5′-OH fragment abundance (CPM) in each bin was summed, and mRNAs were sorted by their maximum bin value from 5′ to 3′. Each mRNA is an entry on the y-axis. A total of 11 mRNAs have signal exclusively within their 5′ UTRs (top) and 69 mRNAs have signal exclusively in their 3′ UTRs (bottom). The gray centerline marks the middle of the ORF region; 5′-OH signal was predominantly in the 5′ portion of 143 mRNAs (above horizontal dashed line), while remaining mRNAs had signals mainly localized toward their 3′ ends. (B) Comparison of signals in mRNAs in 5′-OH and RNA-seq libraries. There was a significant correlation (Pearson R2 = 0.31; P < 10−16) between 5′-OH signals (counts per million reads per kilobase; CPKM) and RNA-seq (FPKM) (33) for 629 mRNAs (a subset of the mRNAs in A). Dotted lines indicate median RNA-seq FPKM and 5′-OH CPKM values, and the linear regression is plotted as a gray line. (C) Plots of 5′-OH abundance (CPM) versus position (bp) for four representative mRNAs (of 187 total, Supplementary Table S2) with multiple, distributed 5′-OH fragments. All mRNAs are plotted from 5′ to 3′, with UTR regions in gray, and ORF regions in black. 5′-OH fragments in the IMH1, RPN2, and YEF3 mRNAs were predominantly localized to their coding regions, and 5′-OH fragments in the SAC1 mRNA were restricted to its 3′ UTR. (D) Three representative mRNAs (of 17 total, Supplementary Table S3) with single, predominant 5′-OH fragments. The ADH1 and YNK1 mRNAs each contained a single predominant 5′-OH fragment in their ORF regions, and the CYS4 mRNA contained a single 5′-OH fragment in its 3′ UTR. The MDH1 mRNA had a predominant 5′-OH fragment 8 nt downstream of its stop codon.
We identified a subset of mRNAs with 5′-OH signals that also were expressed under similar conditions as measured by a previous RNA-seq experiment (33). For each of these 629 mRNAs, we calculated a summary statistic by dividing the sum of its CPM values by the length of the mRNA in kilobases (Counts Per Kilobase per Million reads mapped; CPKM), and compared mRNA CPKM values to their expression levels in Reads Per Kilobase per Million mapped (RPKM) (33). There was a modest but significant correlation between levels of 5′-OH fragments and mRNA abundance (Pearson R2 = 0.31; P < 10−16), indicating that 5′-OH fragment abundance is partly a reflection of mRNA abundance (Figure 3B). We also evaluated whether these fragments were associated with intrinsic structural properties of RNAs. We compared 5′-OH fragment levels to previous measures of global RNA structures using Ribonuclease S1 and V1 mapping (34), and found minimal correlation between these data sets (Pearson R = −0.04; P < 0.048). This suggests that 5′-OH sites are uniformly distributed among RNA secondary structure elements (i.e., single and double-strand regions).
There were unique patterns of 5′-OH fragment distribution within mRNAs. We identified 187 mRNAs (Supplementary Table S2) with multiple fragments mapping to localized regions within the gene body, and these 5′-OH fragments clustered within a mean distance of 191 nt. For example, the IMH1, RPN2 and YEF3 mRNAs had multiple 5′-OH fragments within their coding regions, and the SAC1 mRNA had multiple fragments within its 3′ UTR (Figure 3C). The pattern of 5′-OH fragments in these mRNAs suggests that a localized process cleaves these specific mRNAs within a defined region, but cleavage occurs in different locations among several mRNA molecules.
We also found 17 mRNAs (Supplementary Table S3) that each had a single predominant 5′-OH fragment (FDR < 0.05). For example, the ADH1 and YNK1 mRNAs had single sites of cleavage in their open reading frames, CYS4 had a predominant cleavage in its 3′ UTR, and a predominant cleavage in the MDH1 mRNA is 8 nt downstream of its stop codon (Figure 3D). We also identified a prominent 5′-OH fragment in the RPS31 mRNA, which encodes an in-frame fusion of ubiquitin to the S31 protein of the ribosomal small subunit (35). The 5′-OH fragment mapped 3 nt downstream of the codons for the Arg-Gly-Gly (RGG) terminus of ubiquitin, suggesting that this fragment might be generated during translation and ubiquitin processing (36) (Figure 4A). However, another mRNA encoding a ubiquitin-ribosomal protein fusion, RPL40A, had significant levels of 5′-OH fragments located farther downstream of the codons encoding the ubiquitin C-terminus, suggesting that ubiquitin processing is not responsible for RPS31 mRNA cleavage (Figure 4B).
Figure 4.
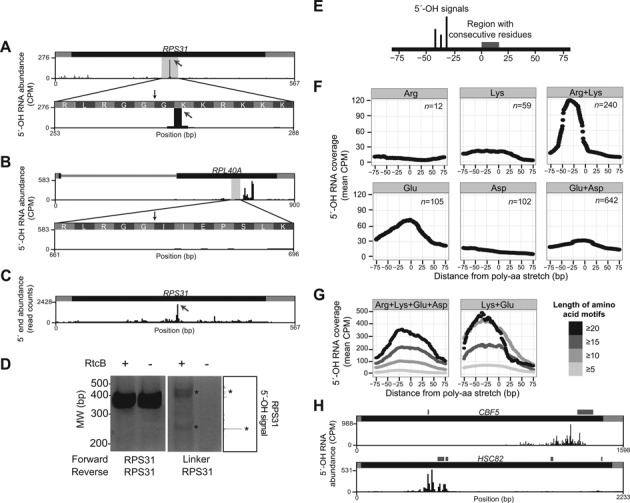
(A) A single predominant 5′-OH RNA fragment (gray arrow) maps to the RPS31 mRNA, encoding the ubiquitin-Rps31 fusion protein. The fragment maps to a position 3 nt downstream of the codons for Arg-Gly-Gly (RGG) residues (vertical black arrow), which signal ubiquitin removal by deubiquitylating enzymes (36). (B) Multiple, distributed 5′-OH signals are further downstream of RGG codons (vertical black arrow) in the RPL40A mRNA, which encodes the ubiquitin-Rpl40a fusion protein. (C) An abundant RNA fragment recovered in ribosome profiling experiments (38) maps to the same position in the RPS31 mRNA as the 5′-OH fragment in (A). This fragment was present in all samples, but was most abundant in ribosome profiling done in cells treated with the non-hydrolysable GTP analog GMP-PNP (shown). (D) The 5′-OH fragment in RPS31 was validated by ligation-mediated RT-PCR. 5′-OH fragments in total RNA were incubated with an RtcB linker (a synthetic oligonucleotide with a 3′-phosphate, M13-linker, Table) in the presence or absence of RtcB ligase. These reactions were divided and reverse transcribed with a primer specific for RPS31 that hybridizes downstream of the 5′-OH terminus. cDNA products of reverse transcription were PCR amplified with primers that hybridize to RPS31 up- and downstream of the 5′-OH terminus and analyzed by gel electrophoresis, yielding abundant fragments at the appropriate size (415 bp), independent of RtcB ligation (left panel). A second PCR was performed with a 5′ primer that hybridizes to the ligated linker, and analysis of these products showed ligation-dependent PCR products at the expected sizes (major 5′-OH fragment at 215 bp; minor products at ∼400 bp). A portion of (A) is displayed on the right for reference. (E) Schematic of 5′-OH signals with respect to regions encoding specific peptide regions in (F). (F) The mean 5′-OH fragment coverage in rRNA-depleted total RNA was calculated 75 nt upstream (negative) and downstream (positive) of the start position of all mRNA sequences encoding at least five consecutive basic or acidic residues, including Arg (n = 12), Lys (n = 59), a mixture of Lys and Arg (n = 280), Glu (n = 105), Asp (n = 102), and a mixture of Glu and Asp (n = 642). A peak of 5′-OH fragments is present ∼30 nt upstream of regions composed of Lys and Arg, but this signal is lower in regions encoding Lys or Arg alone (top panels). Coverage of 5′-OH fragments is elevated higher in regions encoding Glu than regions encoding Asp, or Glu and Asp (bottom panels). (G) The mean 5′-OH fragment coverage was calculated 75 nt upstream (negative) and downstream (positive) of the start position of all mRNA sequences encoding varying lengths of consecutive basic and acidic residues. 5′-OH fragments were enriched upstream of regions encoding polyelectrostatic (both basic and acidic) resides (Arg, Lys, Glu, Asp; left panel) and were highest upstream of regions encoding only Lys and Glu (right panel). In both panels, regions encoding longer consecutive stretches (≥20) of polyelectrostatic amino acids had higher 5′-OH signals than regions encoding shorter stretches (≥5). (H) Representative examples of mRNAs (CBF5 and HSC82) with 5′-OH fragments that map to positions upstream of sequences encoding polyelectrostatic amino acid motifs. Regions encoding polyelectrostatic amino acid stretches are indicated by gray boxes above the gene models.
In the RPS31 mRNA, the 7 codons following the Arg-Gly-Gly residues—and at the site of the 5′-OH fragment—are AAA and AAG codons that encode Lys and Arg residues, which are known to trigger co-translational mRNA cleavage (37). In addition, a similar fragment from the RPS31 mRNA was previously identified in ribosomal profiling experiments (38) (Figure 4C), suggesting that this fragment is associated with ribosomes. We validated the RPS31 mRNA fragment by RtcB-mediated linker ligation followed by reverse transcription and PCR, yielding product sizes consistent with 5′-OH cloning (Figure 4C). Based on this amplification, we estimate that these fragments are <1% of the abundance of full length RPS31 mRNA.
Given that the RPS31 5′-OH fragment mapped upstream of a region of six consecutive basic residues, we analyzed the global relationship between 5′-OH fragments and nearby codon content. We first examined annotated open reading frames to identify sequences encoding amino acid sequences of at least five consecutive identical or related residues (e.g. five or more consecutive lysine residues, or a mix of five consecutive lysine and arginine residues), and determined the average coverage of 5′-OH fragments 75 nt upstream and 75 nt downstream from the start of these regions (Figure 4E). We analyzed 5′-OH fragments cloned from rRNA-depleted RNA, and found that 5′-OH fragments accumulated upstream of the 240 instances of consecutive basic residues composed of both lysine and arginine codons (Figure 4F). This relationship was more pronounced than the accumulation of 5′-OH fragments upstream of regions with consecutive lysine or arginine residues alone (Figure 4F). We also found high levels of 5′-OH fragments upstream of the 105 instances of consecutive glutamate residues, but this signal was diminished when we considered consecutive acidic residues composed of aspartate alone (n = 102) or regions composed of both glutamate and aspartate (n = 642) (Figure 4F). The highest levels of 5′-OH fragments were upstream of regions composed of a mixture of acidic and basic residues, particularly the combination of Lys and Glu (Figure 4G). The levels of 5′-OH fragments were higher when longer lengths of consecutive amino acids were considered (Figure 4G). We identified specific examples of mRNAs with 5′-OH fragments upstream of regions encoding consecutive acidic residues including CBF5 and HSC82 (Figure 4H). Analysis of other consecutive codons showed that they are not associated with 5′-OH fragment accumulation (data not shown). These findings suggest a relationship between the electrostatic properties of a nascent peptide and mRNA cleavage events that yield 5′-OH fragments, possibly via a co-translational process such as no-go mRNA decay (39).
The eukaryotic 5′→3′ exonuclease Xrn1 specifically recognizes 5′-PO4 RNA substrates, but has greatly reduced activity on 5′-OH RNA in vitro (40). Consistent with this activity, xrn1Δ budding yeast accumulate several types of 5-PO4 RNAs (22), and phosphorylation of the 5′-OH RNA products of tRNA splicing is required for their turnover by Xrn1 (31). Given the high specificity of Xrn1 for 5′-PO4 RNA substrates in vitro, it is possible that other 5′-OH RNA fragments could be subject to a similar turnover pathway with a requisite phosphorylation event prior to Xrn1-mediated turnover. We cloned 5′-OH RNA fragments from xrn1Δ cells and compared the levels of these RNA fragments to those from XRN1 cells. Fifty percent of the mRNAs identified in XRN1 cells were also identified in xrn1Δ cells (n = 507). The Pearson correlation between the strains was 0.54 compared to a correlation of 0.68 between biological replicates of the same strain, indicating that some 5′-OH RNAs may enter an Xrn1 degradation pathway.
Identification of endonucleolytic cleavages produced during the unfolded protein response
During the unfolded protein response (UPR), misfolded or aggregated proteins accumulate in the lumen of the endoplasmic reticulum and activate the kinase-endoribonuclease Ire1 (41). Activated Ire1 endonuclease cleaves an intron from the HAC1 pre-mRNA (42), and the cleaved exons are subsequently ligated by yeast Trl1 tRNA ligase (43). After ligation, the mature HAC1 mRNA is translated, and Hac1 protein traffics to the nucleus where it drives expression of dozens of stress response genes (43). We mapped 5′-OH RNA fragments in cells treated with the N-linked glycosylation inhibitor tunicamycin, which causes protein misfolding and UPR activation. The primary 5′-OH RNA fragments captured in the HAC1 mRNA mapped upstream of consecutive electrostatic residues (Figure 5A), consistent with the overall enrichment of 5′-OH signal upstream of these residues in mRNAs (Figure 4F). The canonical sites of Ire1 cleavage in the HAC1 pre-mRNA were also identified, and 5′-OH signals increased upon in vitro phosphatase treatment, consistent with their phosphorylation in vivo, presumably by Trl1 5′-kinase (43) (Figure 5B).
Figure 5.
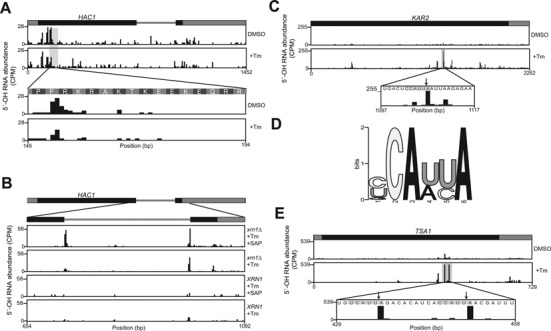
(A) The majority of 5′-OH fragments (CPM) in the HAC1 mRNA map upstream of codons for consecutive electrostatic amino acid residues. Their abundance was similar in vehicle-treated (DMSO, top) and tunicamycin-treated (bottom) cells. (B) Ire1 cleavage fragments in HAC1 are captured in 5′-OH libraries. Low levels of fragments resulting from intron cleavage were found in all conditions. The fragments were most abundant in the xrn1Δ strain treated with tunicamycin and subjected to in vitro phosphorylation (top panel). (C) 5′-OH fragment abundance (CPM) in KAR2 mRNA was examined in cells treated with vehicle (DMSO, top) and tunicamycin (middle). A 5′-OH fragment increased 17-fold upon tunicamycin treatment (middle) and mapped to the sequence 5′-CCAUU|A-3′ (bottom). (D) Consensus sequence motif in logo format derived from sequences flanking 5′-OH fragments (10 nt up- and downstream) in mRNAs that increased in abundance 5-fold or more upon tunicamycin treatment. A total of 10 instances of the motif were found among 23 mRNAs (Supplementary Table S4). (E) Two 5′-OH fragments in the TSA1 mRNA increased 11.4-fold and 8.7-fold upon tunicamycin treatment (top and middle panel) and mapped to the consensus motif in (D) (bottom panel, arrows).
We compared 5′-OH mRNA signals in untreated cells and cells treated with the tunicamycin, and found a 5′-OH fragment in the KAR2 mRNA (the yeast ortholog of BiP/HSP70) that was 17-fold more abundant in the presence of tunicamycin and occurred in the sequence 5′-CCAUU|A-3′ (Figure 5C). Initially, we attributed this signal to upregulation of KAR2 mRNA expression by the UPR transcriptional response (44), but we also identified six other ER-related mRNAs with 5′-OH fragments present in a similar sequence motif that increased 5-fold or more upon tunicamycin treatment (Figure 5D, Supplementary Table S4), including the UPR-induced thioredoxin peroxidase TSA1 mRNA (45), where 5′-OH fragments accumulated at two such motifs upon tunicamycin treatment (Figure 5E).
DISCUSSION
We developed a method to directly capture RNAs with 5′-OH termini using the unique substrate specificity of E. coli RtcB RNA ligase (16) (Figure 1). This method is highly selective for the capture of 5′-OH products (Figure 2B and C), and yields reproducible signals among several classes of cellular RNA. RNA fragments with 5′-OH termini have not been directly detected in other global studies, and the collection of 5′-OH RNA fragments reported here collectively comprise a previously uninterrogated RNA class. Future applications of this method could include synthetic 5′-OH RNAs spiked in at known concentrations to determine the lower limit of detection and provide a reference for absolute quantitation (46).
We identified three categories of tRNA fragments with 5′-OH termini: those derived from cleavage at the boundary of the intron and 3′ exon, cleavage in the anticodon loop, and cleavage in the D-arm/loop (Figure 2D and E). The first of these validates the method by demonstrating capture of a known 5′-OH terminus generated in vitro during the well-characterized processing of an intron-containing pre-tRNA by the tRNA Splicing Endonuclease (SEN) (1). A mechanism of anticodon loop cleavage is also known: Rny1 can cleave the anticodon loop during stress conditions (47), and related fragments have been previously captured by 2′,3′-cyclic phosphate cloning (11). However, the most abundant cleavages observed in 5′-OH cloning were in the D-arm and loop, which has not been previously described. This cleavage may represent a novel tRNA stress fragment or may be a consequence of mapping bias toward the 5′-end of a highly abundant species.
We identified a relationship between 5′-OH mRNA fragments and mRNA sequences that encoded consecutive basic peptide sequences (Figure 4A–F). In the RPS31 mRNA, a 5′-OH fragment accumulates upstream of codons encoding a polybasic peptide (Figure 4A). Production of the fragment does not appear to be related to ubiquitin processing, as another ubiquitin fusion RPL40A does not have 5′-OH fragments near the site of ubiquitin cleavage (Figure 4B). A similar fragment in the RPS31 mRNA was also identified in ribosomal profiling experiments done in budding yeast (38) (Figure 4C). These experiments predominantly recover RNA fragments that are protected from ribonuclease digestion by ribosomes, but do so without relying on a specific 5′ end chemistry (48). Additionally, several previous studies showed that nascent peptides with specific electrostatic properties cause ribosome pausing and trigger co-translational mRNA decay. In non-stop decay (NSD), translation of the polyadenylate tails of mRNAs that lack stop codons leads to a nascent polylysine peptide (49), which is proposed to interact with the negatively charged exit tunnel of the ribosome (50) to trigger ribosome stalling and mRNA decay. Similarly, internal polybasic peptides trigger co-translational mRNA decay, and this process is dependent on the non-canonical release factors Dom34 and Hbs1 (37). In addition, global measures of translation indicated that polybasic sequences cause ribosomal stalling, suggesting they interfere with ribosome elongation (51). The presence of this RPS31 5′-OH fragment in ribosome profiling data suggests that it may be associated with ribosomes and may be a product of a ribosome-directed decay process driven by the polybasic coding region.
Many studies have used synthetic reporter mRNAs to study the role of nascent peptides in determining decay of a translating mRNA (37,52–53). However, there are no known physiological mRNA substrates of no-go decay (NGD), and the endonuclease activity responsible for mRNA cleavage during NGD is not known (39). Detailed mapping of mRNA cleavage events during NGD showed that mRNA cleavage occurs around 20 nt upstream of a polybasic sequence (37), which is consistent with the distance between 5′-OH fragments and other polyelectrostatic sequences (Figure 4E–G). NGD decay intermediates are degraded by Xrn1 (52). However, insofar as Xrn1 requires 5′-PO4 substrates (40), the products of NGD that are degraded by Xrn1 could be generated by an endonuclease that creates 5′-OH RNA products that are phosphorylated by a cellular 5′-kinase activity (e.g., the tRNA ligase Trl1), promoting their decay by Xrn1— a scenario similar to 5′-kinase-mediated turnover of spliced tRNA introns by Xrn1 (31) and RegB mRNA cleavage products in bacteria (54). Thus, it is possible that many of the 5′-OH fragments we identified are products of NGD. Future studies will determine whether the production of 5′-OH fragments in mRNA sequences encoding highly electrostatic peptides (Figure 4E–H) is dependent on NGD factors such as Dom34 and Hbs1 (37,52).
We also identified 5′-OH fragments upstream of polyacidic and other polyelectrostatic sequences composed of a mixture of basic and acidic residues (Figure 4E–H). These signals were more prominent than any regions composed of any other single amino acid. Polyacidic stretches in the C-terminus of several human proteins are associated with ribosome frameshifting in these regions (55). However, it is not clear whether frameshifting is a consequence of the polyacidic nascent peptide, or whether it is a consequence of the propensity for out-of-frame polyglutamate codons to encode new Met initiation codons. Notably, the 5′-OH fragment we identified 8 nt downstream of the MDH1 stop codon (Figure 3D) is near the site of ribosomal stop codon read through in other fungi, which appends a peroxisomal targeting sequence (PTS) to the Mdh1 protein for its localization (56). If a similar phenomenon accounts for the MDH1 mRNA 5′-OH fragment in S. cerevisiae, it suggests that production of 5′-OH fragments is a general feature of mRNA decay at sites of non-canonical ribosome elongation (i.e. frameshifting or read through).
A potential artifact of mRNA purification by oligo-dT enrichment is the isolation of A-rich mRNA sequences independent of their non-templated 3′-polyadenylate tails. In particular, some codons for electrostatic residues (Arg, Lys, Glu) are A-rich, and might be preferentially isolated by oligo-dT enrichment. We found that 5′-OH fragments upstream of polylysine stretches (encoded by AAA and AAG codons) were indeed increased in polyA libraries relative to rRNA-depleted libraries, but 5′-OH fragment levels upstream of other mixed polyelectrostatic sequences were present at similar levels in polyA selected and rRNA-depleted libraries. Furthermore, in oligo-dT selected samples and rRNA-depleted samples, we detected no enrichment of 5′-OH signal upstream of polyasparagine residues, which are encoded by AAU and AAC codons and are abundant in yeast mRNAs (n = 138).
During activation of the unfolded protein response, the HAC1 pre-mRNA is cleaved by the kinase-endoribonuclease Ire1, producing 2′,3′-cyclic phosphate and 5′-OH RNA fragments (42) that are substrates for ligation by tRNA ligase (43). However, previous studies that identified 5′-PO4 mRNA fragments from dcp2Δ xrn1Δ yeast recovered HAC1 cleavage products at the sites of Ire1 cleavage (22). Using 5′-OH cloning, we recovered HAC1 mRNA cleavage products at their highest levels in xrn1Δ cells after their dephosphorylation in vitro (Figure 5B, top panel). The presence of these 5′-PO4 RNA fragments possibly indicates that the Trl1 5′-kinase step is rapid, but further HAC1 mRNA ligation by Trl1 is slow or often not completed. Corroborating this idea, fungal ligases have rapid 5′-kinase kinetics, but a slower ligation step (57). Similarly, the products of metazoan Regulated IRE1-Dependent Decay (RIDD) are degraded by Xrn1 (58), suggesting that a 5′-kinase activity phosphorylates 5′-OH products of Ire1 cleavage prior to their degradation by Xrn1.
Previous studies suggested that Hac1 is the sole target of Ire1 cleavage in budding yeast (59), but a more recent study suggested that Ire1 cleaves different substrates depending on its oligomeric state, and provided evidence that monomeric yeast Ire1 can cleave mRNAs other than HAC1 in vitro (60). We identified 23 mRNAs with 5′-OH fragments that increased more than 5-fold upon tunicamycin treatment, and found that seven of these fragments were present in the consensus motif 5′-CAUU|A-3′, suggesting that they are targeted by a site-specific endonuclease (Figure 5C–E, Supplementary Table S1). An obvious candidate for this endonuclease is Ire1, but this motif differs from the canonical Ire1 recognition motif for HAC1 mRNA in budding yeast (2), as well as motifs in other RIDD targets in fission yeast and metazoans (61). Further studies will determine whether these mRNAs are directly cleaved by Ire1, or are an indirect consequence of UPR activation.
The ability to globally map 5′-OH RNA fragments will enable new studies of 5′-kinase enzymes that phosphorylate 5′-OH substrates. For example, the human RNA 5′-kinase hClp1 was first identified as a component of the CF IIm sub-complex of cleavage and polyadenylation specificity factor (62), and has recently been implicated in quality control of RNA 5′-termini during transfer RNA processing (63). Mutations in hClp1 cause severe neurodevelopmental phenotypes, and reduce its 5′-kinase activity in vitro (64,65). Previous studies proposed that reduction in 5′-kinase activity causes accumulation of aberrant—and presumably 5′-OH—RNA fragments (66), but it is currently not known how accumulation of these molecules might lead to human neurodevelopmental defects or the spinal motor neuron deficits that develop in mice expressing kinase-dead Clp1 (66). Turnover of these molecules may be analogous to the turnover of tRNA introns in yeast, where phosphorylation of 5′-OH tRNA introns leads to their rapid 5′→3′ decay by Xrn1 (31). Application of the 5′-OH cloning method in cells harboring mutant hClp1 with reduced 5′-kinase activity might identify new 5′-OH RNA substrates of hClp1, providing insight into this class of neurodevelopmental disorders.
ACCESSION NUMBER
Data deposited in NCBI GEO under accession number GSE61527.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank R. Davis, S. Fields and S. Shuman for discussions and comments on the manuscript.
FUNDING
Damon Runyon-Rachleff Innovation award (in part); Research Scholar Grant from the American Cancer Society; National Institutes of Health Training [T32 GM008730 to S.P.]; Core facilities used were funded in part by the University of Colorado Cancer Center Grant [P30 CA046934]. Funding for open access charge: Internal University funds.
Conflict of interest statement. None declared.
REFERENCES
- 1.Trotta C.R., Miao F., Arn E.A., Stevens S.W., Ho C.K., Rauhut R., Abelson J.N. The yeast tRNA splicing endonuclease: a tetrameric enzyme with two active site subunits homologous to the archaeal tRNA endonucleases. Cell. 1997;89:849–858. doi: 10.1016/s0092-8674(00)80270-6. [DOI] [PubMed] [Google Scholar]
- 2.Sidrauski C., Walter P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell. 1997;90:1031–1039. doi: 10.1016/s0092-8674(00)80369-4. [DOI] [PubMed] [Google Scholar]
- 3.Luhtala N., Parker R. T2 Family ribonucleases: ancient enzymes with diverse roles. Trends Biochem. Sci. 2010;35:253–259. doi: 10.1016/j.tibs.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cooper D.A., Jha B.K., Silverman R.H., Hesselberth J.R., Barton D.J. Ribonuclease L and metal-ion-independent endoribonuclease cleavage sites in host and viral RNAs. Nucleic Acids Res. 2014;42:5202–5216. doi: 10.1093/nar/gku118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferré-D'Amaré A.R., Scott W.G. Small self-cleaving ribozymes. Cold Spring Harb. Perspect. Biol. 2010;2:a003574. doi: 10.1101/cshperspect.a003574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roth A., Weinberg Z., Chen A.G.Y., Kim P.B., Ames T.D., Breaker R.R. A widespread self-cleaving ribozyme class is revealed by bioinformatics. Nat. Chem. Biol. 2014;10:56–60. doi: 10.1038/nchembio.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soukup G.A., Breaker R.R. Relationship between internucleotide linkage geometry and the stability of RNA. RNA. 1999;5:1308–1325. doi: 10.1017/s1355838299990891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schifano J.M., Vvedenskaya I.O., Knoblauch J.G., Ouyang M., Nickels B.E., Woychik N.A. An RNA-seq method for defining endoribonuclease cleavage specificity identifies dual rRNA substrates for toxin MazF-mt3. Nat. Commun. 2014;5:3538. doi: 10.1038/ncomms4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.German M.A., Luo S., Schroth G., Meyers B.C., Green P.J. Construction of Parallel Analysis of RNA Ends (PARE) libraries for the study of cleaved miRNA targets and the RNA degradome. Nat. Protoc. 2009;4:356–362. doi: 10.1038/nprot.2009.8. [DOI] [PubMed] [Google Scholar]
- 10.Gu W., Lee H.-C., Chaves D., Youngman E.M., Pazour G.J., Conte D., Mello C.C. CapSeq and CIP-TAP identify Pol II start sites and reveal capped small RNAs as C. elegans piRNA precursors. Cell. 2012;151:1488–1500. doi: 10.1016/j.cell.2012.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schutz K., Hesselberth J.R., Fields S. Capture and sequence analysis of RNAs with terminal 2′, 3′-cyclic phosphates. RNA. 2010;16:621–631. doi: 10.1261/rna.1934910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cooper D.A., Banerjee S., Chakrabarti A., García-Sastre A., Hesselberth J.R., Silverman R.H., Barton D.J. Ribonuclease L targets distinct sites in influenza A virus RNAs. J. Virol. 2014;89:2764–2776. doi: 10.1128/JVI.02953-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kimmig P., Diaz M., Zheng J., Williams C.C., Lang A., Aragón T., Li H., Walter P. The unfolded protein response in fission yeast modulates stability of select mRNAs to maintain protein homeostasis. Elife. 2012;1:e00048. doi: 10.7554/eLife.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greer C.L., Peebles C.L., Gegenheimer P., Abelson J. Mechanism of action of a yeast RNA ligase in tRNA splicing. Cell. 1983;32:537–546. doi: 10.1016/0092-8674(83)90473-7. [DOI] [PubMed] [Google Scholar]
- 15.Phizicky E.M., Schwartz R.C., Abelson J. Saccharomyces cerevisiae tRNA ligase. Purification of the protein and isolation of the structural gene. J. Biol. Chem. 1986;261:2978–2986. [PubMed] [Google Scholar]
- 16.Tanaka N., Chakravarty A.K., Maughan B., Shuman S. Novel mechanism of RNA repair by RtcB via sequential 2′, 3′-cyclic phosphodiesterase and 3′-phosphate/5′-hydroxyl ligation reactions. J. Biol. Chem. 2011;286:43134–43143. doi: 10.1074/jbc.M111.302133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Desai K.K., Raines R.T. tRNA ligase catalyzes the GTP-dependent ligation of RNA with 3′-phosphate and 5′-hydroxyl termini. Biochemistry. 2012;51:1333–1335. doi: 10.1021/bi201921a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tanaka N., Meineke B., Shuman S. RtcB, a novel RNA ligase, can catalyze tRNA splicing and HAC1 mRNA splicing in vivo. J. Biol. Chem. 2011;286:30253–30257. doi: 10.1074/jbc.C111.274597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karolchik D., Barber G.P., Casper J., Clawson H., Cline M.S., Diekhans M., Dreszer T.R., Fujita P.A., Guruvadoo L., Haeussler M., et al. The UCSC Genome Browser database: 2014 update. Nucleic Acids Res. 2014;42:D764–D770. doi: 10.1093/nar/gkt1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harigaya Y., Parker R. Global analysis of mRNA decay intermediates in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 2012;109:11764–11769. doi: 10.1073/pnas.1119741109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nussbaum C., Myers R.M., Brown M., Li W., et al. Model-based Analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Das U., Chakravarty A.K., Remus B.S., Shuman S. Rewriting the rules for end joining via enzymatic splicing of DNA 3′-PO4 and 5′-OH ends. Proc. Natl. Acad. Sci. U.S.A. 2013;110:20437–20442. doi: 10.1073/pnas.1314289110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kivioja T., Vähärautio A., Karlsson K., Bonke M., Enge M., Linnarsson S., Taipale J. Counting absolute numbers of molecules using unique molecular identifiers. Nat. Methods. 2012;9:72–74. doi: 10.1038/nmeth.1778. [DOI] [PubMed] [Google Scholar]
- 26.Venema J., Tollervey D. Two distinct recognition signals define the site of endonucleolytic cleavage at the 5′-end of yeast 18S rRNA. EMBO J. 1995;14:4883–4892. doi: 10.1002/j.1460-2075.1995.tb00169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geerlings T.H., Vos J.C., Raué H.A. The final step in the formation of 25S rRNA in Saccharomyces cerevisiae is performed by 5′–>3′ exonucleases. RNA. 2000;6:1698–1703. doi: 10.1017/s1355838200001540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sigurgeirsson B., Emanuelsson O., Lundeberg J. Analysis of stranded information using an automated procedure for strand specific RNA sequencing. BMC Genomics. 2014;15:631. doi: 10.1186/1471-2164-15-631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petfalski E., Tollervey D. Processing of the precursors to small nucleolar RNAs and rRNAs requires common components. Mol. Cell. Biol. 1998;18:1181–1189. doi: 10.1128/mcb.18.3.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giorgi C., Fatica A., Nagel R., Bozzoni I. Release of U18 snoRNA from its host intron requires interaction of Nop1p with the Rnt1p endonuclease. EMBO J. 2001;20:6856–6865. doi: 10.1093/emboj/20.23.6856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu J., Hopper A.K. Healing for destruction: tRNA intron degradation in yeast is a two-step cytoplasmic process catalyzed by tRNA ligase Rlg1 and 5‘-to-3′ exonuclease Xrn1. Genes Dev. 2014;28:1556–1561. doi: 10.1101/gad.244673.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagalakshmi U., Wang Z., Waern K., Shou C., Raha D., Gerstein M., Snyder M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levin J.Z., Yassour M., Adiconis X., Nusbaum C., Thompson D.A., Friedman N., Gnirke A., Regev A. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat. Chem. Biol. 2010;7:1–10. doi: 10.1038/nmeth.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kertesz M., Wan Y., Mazor E., Rinn J.L., Nutter R.C., Chang H.Y., Segal E. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010;467:103–107. doi: 10.1038/nature09322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Finley D., Bartel B., Varshavsky A. The tails of ubiquitin precursors are ribosomal proteins whose fusion to ubiquitin facilitates ribosome biogenesis. Nature. 1989;338:394–401. doi: 10.1038/338394a0. [DOI] [PubMed] [Google Scholar]
- 36.Lacombe T., García-Gómez J.J., de la Cruz J., Roser D., Hurt E., Linder P., Kressler D. Linear ubiquitin fusion to Rps31 and its subsequent cleavage are required for the efficient production and functional integrity of 40S ribosomal subunits. Mol. Microbiol. 2009;72:69–84. doi: 10.1111/j.1365-2958.2009.06622.x. [DOI] [PubMed] [Google Scholar]
- 37.Tsuboi T., Kuroha K., Kudo K., Makino S., Inoue E., Kashima I., Inada T. Dom34:hbs1 plays a general role in quality-control systems by dissociation of a stalled ribosome at the 3′ end of aberrant mRNA. Mol. Cell. 2012;46:518–529. doi: 10.1016/j.molcel.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 38.Guydosh N.R., Green R. Dom34 rescues ribosomes in 3′ untranslated regions. Cell. 2014;156:950–962. doi: 10.1016/j.cell.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shoemaker C.J., Green R. Translation drives mRNA quality control. Nat. Struct. Mol. Biol. 2012;19:594–601. doi: 10.1038/nsmb.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stevens A. 5′-exoribonuclease 1: Xrn1. Methods Enzymol. 2001;342:251–259. doi: 10.1016/s0076-6879(01)42549-3. [DOI] [PubMed] [Google Scholar]
- 41.Ron D., Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Publishing Group. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 42.Gonzalez T.N., Sidrauski C., Dörfler S., Walter P. Mechanism of non-spliceosomal mRNA splicing in the unfolded protein response pathway. EMBO J. 1999;18:3119–3132. doi: 10.1093/emboj/18.11.3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sidrauski C., Cox J.S., Walter P. tRNA ligase is required for regulated mRNA splicing in the unfolded protein response. Cell. 1996;87:405–413. doi: 10.1016/s0092-8674(00)81361-6. [DOI] [PubMed] [Google Scholar]
- 44.Normington K., Kohno K., Kozutsumi Y., Gething M.J., Sambrook J. S. cerevisiae encodes an essential protein homologous in sequence and function to mammalian BiP. Cell. 1989;57:1223–1236. doi: 10.1016/0092-8674(89)90059-7. [DOI] [PubMed] [Google Scholar]
- 45.Kimata Y., Ishiwata-Kimata Y., Yamada S., Kohno K. Yeast unfolded protein response pathway regulates expression of genes for anti-oxidative stress and for cell surface proteins. Genes Cells. 2006;11:59–69. doi: 10.1111/j.1365-2443.2005.00921.x. [DOI] [PubMed] [Google Scholar]
- 46.Jiang L., Schlesinger F., Davis C.A., Zhang Y., Li R., Salit M., Gingeras T.R., Oliver B. Synthetic spike-in standards for RNA-seq experiments. Genome Res. 2011;21:1543–1551. doi: 10.1101/gr.121095.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thompson D.M., Parker R. The RNase Rny1p cleaves tRNAs and promotes cell death during oxidative stress in Saccharomyces cerevisiae. J. Cell Biol. 2009;185:43–50. doi: 10.1083/jcb.200811119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ingolia N.T., Brar G.A., Rouskin S., McGeachy A.M., Weissman J.S. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 2012;7:1534–1550. doi: 10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ito-Harashima S., Kuroha K., Tatematsu T., Inada T. Translation of the poly(A) tail plays crucial roles in nonstop mRNA surveillance via translation repression and protein destabilization by proteasome in yeast. Genes Dev. 2007;21:519–524. doi: 10.1101/gad.1490207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lu J., Deutsch C. Electrostatics in the ribosomal tunnel modulate chain elongation rates. J. Mol. Biol. 2008;384:73–86. doi: 10.1016/j.jmb.2008.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brandman O., Stewart-Ornstein J., Wong D., Larson A., Williams C.C., Li G.-W., Zhou S., King D., Shen P.S., Weibezahn J., et al. A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell. 2012;151:1042–1054. doi: 10.1016/j.cell.2012.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Doma M.K., Parker R. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature. 2006;440:561–564. doi: 10.1038/nature04530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dimitrova L.N., Kuroha K., Tatematsu T., Inada T. Nascent peptide-dependent translation arrest leads to Not4p-mediated protein degradation by the proteasome. J. Biol. Chem. 2009;284:10343–10352. doi: 10.1074/jbc.M808840200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Durand S., Richard G., Bontems F., Uzan M. Bacteriophage T4 polynucleotide kinase triggers degradation of mRNAs. Proceedings of the National Academy of Sciences of the U.S.A. 2012;109:7073–7078. doi: 10.1073/pnas.1119802109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gould P.S., Dyer N.P., Croft W., Ott S., Easton A.J. Cellular mRNAs access second ORFs using a novel amino acid sequence-dependent coupled translation termination-reinitiation mechanism. RNA. 2014;20:373–381. doi: 10.1261/rna.041574.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stiebler A.C., Freitag J., Schink K.O., Stehlik T., Tillmann B.A.M., Ast J., Bölker M. Ribosomal readthrough at a short UGA stop codon context triggers dual localization of metabolic enzymes in fungi and animals. PLoS Genet. 2014;10:e1004685. doi: 10.1371/journal.pgen.1004685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Remus B.S., Shuman S. Distinctive kinetics and substrate specificities of plant and fungal tRNA ligases. RNA. 2014;20:462–473. doi: 10.1261/rna.043752.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hollien J., Weissman J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–107. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- 59.Niwa M., Patil C.K., DeRisi J., Walter P. Genome-scale approaches for discovering novel nonconventional splicing substrates of the Ire1 nuclease. Genome Biol. 2005;6:R3. doi: 10.1186/gb-2004-6-1-r3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tam A.B., Koong A.C., Niwa M. Ire1 has distinct catalytic mechanisms for XBP1/HAC1 splicing and RIDD. Cell Rep. 2014;9:850–858. doi: 10.1016/j.celrep.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maurel M., Chevet E., Tavernier J., Gerlo S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 2014;39:245–254. doi: 10.1016/j.tibs.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 62.de Vries H., Rüegsegger U., Hübner W., Friedlein A., Langen H., Keller W. Human pre-mRNA cleavage factor II(m) contains homologs of yeast proteins and bridges two other cleavage factors. EMBO J. 2000;19:5895–5904. doi: 10.1093/emboj/19.21.5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weitzer S., Martinez J. The human RNA kinase hClp1 is active on 3′ transfer RNA exons and short interfering RNAs. Nature. 2007;447:222–226. doi: 10.1038/nature05777. [DOI] [PubMed] [Google Scholar]
- 64.Schaffer A.E., Eggens V.R.C., Caglayan A.O., Reuter M.S., Scott E., Coufal N.G., Silhavy J.L., Xue Y., Kayserili H., Yasuno K., et al. CLP1 founder mutation links tRNA splicing and maturation to cerebellar development and neurodegeneration. Cell. 2014;157:651–663. doi: 10.1016/j.cell.2014.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Karaca E., Weitzer S., Pehlivan D., Shiraishi H., Gogakos T., Hanada T., Jhangiani S.N., Wiszniewski W., Withers M., Campbell I.M., et al. Human CLP1 mutations alter tRNA biogenesis, affecting both peripheral and central nervous system function. Cell. 2014;157:636–650. doi: 10.1016/j.cell.2014.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hanada T., Weitzer S., Mair B., Bernreuther C., Wainger B.J., Ichida J., Hanada R., Orthofer M., Cronin S.J., Komnenovic V., et al. CLP1 links tRNA metabolism to progressive motor-neuron loss. Nature. 2013;495:474–480. doi: 10.1038/nature11923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.