

Abstract
Objective:
To determine the frequency of amyotrophic lateral sclerosis (ALS) plateaus and reversals in the Pooled Resource Open-Access ALS Clinical Trials (PRO-ACT) database.
Methods:
We analyzed Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS) and ALSFRS–revised (ALSFRS-R) data from PRO-ACT participants. The frequencies of participants experiencing plateaus (periods where scores did not change) were calculated over 6-, 12-, and 18-month epochs. The percentage of participants ever experiencing reversals (periods where scores improved) of different lengths were also calculated and plotted.
Results:
Over 6 months, 25% of 3,132 participants did not decline. Over 12 months, 16% of 2,105 participants did not decline. Over 18 months, 7% of 1,218 participants did not decline. Small ALS reversals were also common, especially over shorter follow-up intervals; 14% of 1,343 participants had a 180-day interval where their ALSFRS-R slope was greater than zero. Fewer than 1% of participants ever experienced improvements of 4 or more ALSFRS-R points lasting at least 12 months.
Conclusion:
ALS plateaus and small reversals are common, especially over brief intervals. In light of these data, stable disease, especially for a short period of time, should not be interpreted as an ALS treatment effect. Large sustained ALS reversals, on the other hand, are rare, potentially important, and warrant further study.
It is widely appreciated that amyotrophic lateral sclerosis (ALS) progression can be variable. Less appreciated is the fact that progression can plateau or reverse with recovery of lost functions.1 ALS plateaus and reversals are thought to be so uncommon that they have been used as evidence of possible treatment benefit and justification for additional studies.2 In fact the frequencies of these have never been determined in a large population. Here, we elucidate these within the Pooled Resource Open-Access ALS Clinical Trials (PRO-ACT) database.3
METHODS
We analyzed the cohort in the PRO-ACT database.3 PRO-ACT contains data from 17 ALS clinical trials spanning more than 20 years.3 The most common measurements of progression in PRO-ACT (and ALS trials in general) are versions of the Amyotrophic Lateral Sclerosis Functional Rating Score (ALSFRS4,5). Five trials in PRO-ACT used the original ALSFRS,4 4 used the currently favored revised version (ALSFRS-R5), and 3 measured both. The plateau analysis described below was obtained using ALSFRS, which maximized available sample size. Reversal analyses were obtained using ALSFRS-R, which make them more comparable to contemporaneous studies. Additional PRO-ACT details including participant demographics and disease characteristics have been published previously.3
To examine longitudinal ALS progression in PRO-ACT, we generated histograms of ALSFRS changes at 6, 12, and 18 months of trial participation. Since studies in PRO-ACT gathered data on different schedules, we used windows to approximate these intervals. For example, the 6-month point was defined as the closest available time point to 180 days that occurred between 150 and 210 days.
To determine the potential for ALS reversals, we looked at rolling 180-day intervals with least 4 ALSFRS-R data points, applied a least-squares fit, and determined the slope, intercept, and 95% confidence interval. We reported the percentage of participants with slopes >0. We plotted the percentage of participants who ever experienced any periods of ALSFRS-R improvements of 1–4 points, and how long these lasted for intervals of up to 360 days. To ensure that positive or negative treatment effects did not influence findings, all analyses were repeated using only data from PRO-ACT participants assigned to placebo groups. We qualitatively reviewed individual curves for participants experiencing ALS reversals. We compared available demographics of participants who experienced reversals to participants who did not.
Standard protocol approvals, registrations, and patient consents.
The PRO-ACT database is institutional review board–approved. Participants in PRO-ACT were consented to participate in their original trials. All the data in PRO-ACT are de-identified; it was thus not possible to locate participants again and reconsent them for this study.
RESULTS
Figure 1 shows longitudinal ALSFRS changes. Over longer intervals, fewer participants were available for analysis. For the 6-month analysis, there were 3,132 participants available, and 25% did not decline. For the 12-month analysis, there were 2,105 participants available, and 16% did not decline. For the 18-month analysis, there were 1,218 participants available, and 7% did not decline. ALS plateaus, especially of short duration, are thus not uncommon.
Figure 1. Natural history of amyotrophic lateral sclerosis progression in PRO-ACT.

These histograms show the natural history of Pooled Resource Open-Access ALS Clinical Trials (PRO-ACT) participants' Amyotrophic Lateral Sclerosis Functional Rating Score (ALSFRS) changes at intervals of 6 months (A), 12 months (B), and 18 months (C).
In terms of potential ALS reversals, least-squares analysis showed that 14% of 1,343 participants had a 180-day interval where their ALSFRS-R slope was greater than zero. Plotting periods of ALSFRS-R improvements of 1–4 points vs the length of those periods out to 360 days confirmed that small ALS reversals are not uncommon, especially over shorter follow-up intervals (data not shown). Large sustained ALS reversals, however, are rare. Fewer than 1% of patients with ALS ever experienced reversals of 4 or more ALSFRS-R points lasting 12 months. Repeating these analyses in only placebo-treated participants showed no significant differences in the frequencies of ALS reversals. Thus, these are unlikely to be related to a previously undetected treatment effect.
Figure 2 presents selected PRO-ACT participants' ALSFRS-R scores over time. The region of the graph detected in the above analysis as an ALS reversal is highlighted. Some of these reversals (for example, participant A) are mainly the result of a single data point (in this case the last data point). Other reversals show a slow, steady improvement over time (for example, participant B). These are more interesting as discussed below.
Figure 2. Closer look at amyotrophic lateral sclerosis reversals.

These graphs show 2 individual Pooled Resource Open-Access ALS Clinical Trials participants' Amyotrophic Lateral Sclerosis Functional Rating Score–revised (ALSFRS-R) scores over time. The portion of the curve picked up as an amyotrophic lateral sclerosis reversal is highlighted. Some of these reversals appear to be the result of a single data point (participant A, last data point). Others include multiple incrementally improving data points, which are potentially more interesting (participant B).
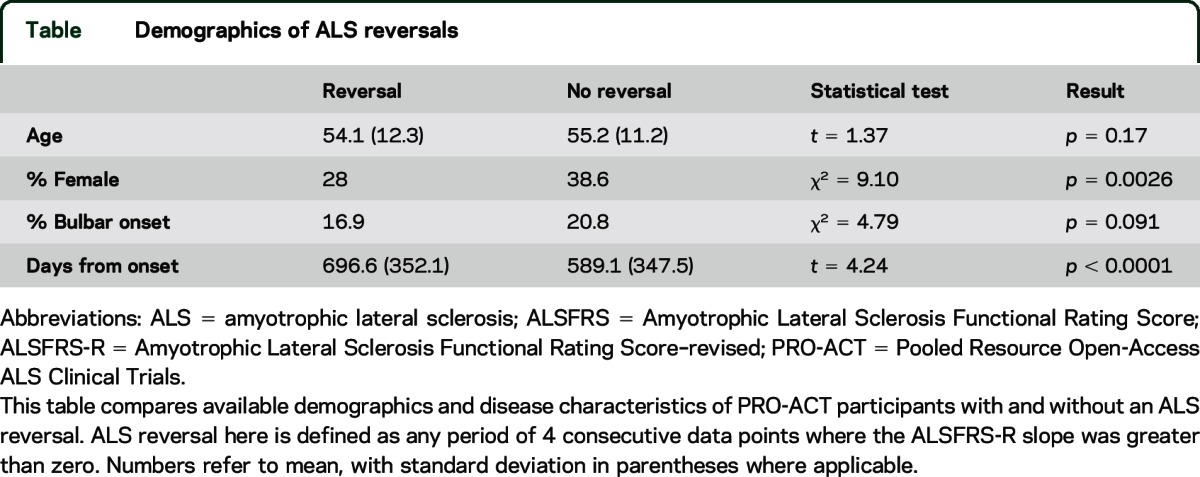
The table compares available demographics between participants with and without reversals (as defined by the least-squares method). Men and those with longer disease duration were more likely to experience reversals. The mean ALSFRS-R score at the start of a reversal was 37 (standard deviation 16.08); thus slow progressors (those with high ALSFRS-R score and long disease duration) appear more likely to experience a reversal. Age and onset site were not associated with reversals.
Table.
Demographics of ALS reversals
DISCUSSION
These data confirm previous case series, showing that ALS progression is variable, and extend our knowledge by showing that a high percentage of patients in a large database experience ALS plateaus. While brief plateaus are more common than longer ones, some of these last as long as 18 months. In light of this information, responder analyses that interpret stable disease over short intervals as evidence of treatment benefit must be met with skepticism.
These data also show that some patients with ALS experience a reversal of their disease. Small, brief reversals are not uncommon. Some of these are the result of a single data point being significantly different than the prior ones. These may indicate a measurement error, or the effect of a symptomatic treatment. Larger, sustained reversals are rare and more interesting. These may suggest an ALS mimic syndrome, an endogenous mechanism that is able to compensate for the disease, a deleterious exogenous environmental influence that was removed, or even an undocumented treatment that is working. Our leading hypotheses are that there may be an antibody-mediated ALS mimic syndrome, perhaps related to LRP4 antibodies,6 or a polymorphism that confers resistance to an ALS pathway allowing regeneration to occur at a faster rate than degeneration. Supporting the latter, there are polymorphisms associated with slow ALS progression.7 Genetic resistance allows some patients to unexpectedly control other fatal diseases such as HIV.8 To test these hypotheses, we will gather ALS reversals and measure their antibody levels and whole-exome sequencing. When we find reversals that are temporally correlated with an alternative therapy, we will try the same therapy in others to see if anyone else gets better.
There are limitations to our study. First, the demographics of participants in ALS trials (and thus those in PRO-ACT) differ from those of nonparticipating patients.9 It is thus not known how generalizable our results are. Second, the PRO-ACT database does not contain information about educational level, cognitive status, or the year when the original data points were collected3; it is thus not possible to use PRO-ACT to determine how these might influence disease progression. Third, while ALS reversals were more common in men and those with longer disease duration (and probably slower progression), it is difficult to know how much importance to ascribe to these findings. Our definitions of ALS reversals are somewhat arbitrary; thus comparison of demographics and disease characteristics between those with and without a reversal runs the risk of dichotomania.10 Finally, we measured ALS progression using the ALSFRS and ALSFRS-R. While these are the most commonly used measures of disease progression in ALS clinics and in current trials, they are unlikely to be perfectly sensitive to motor neuron loss. More sensitive ALS biomarkers are being sought and will eventually further illuminate the course of ALS progression. Until these arrive, the ALSFRS and ALSFRS-R remain useful, but we urge caution in interpreting plateaus or small reversals in these as evidence of treatment effects.
GLOSSARY
- ALS
amyotrophic lateral sclerosis
- ALSFRS
Amyotrophic Lateral Sclerosis Functional Rating Score
- ALSFRS-R
Amyotrophic Lateral Sclerosis Functional Rating Score–revised
- PRO-ACT
Pooled Resource Open-Access ALS Clinical Trials
AUTHOR CONTRIBUTIONS
R.S. Bedlack: study concept and design, writing. T. Vaughan: analysis and interpretation, critical revision of the manuscript for important intellectual content. P. Wicks: study concept and design, critical revision of the manuscript for important intellectual content. J. Heywood: study concept and design, critical revision of the manuscript for important intellectual content. E. Sinani: analysis and interpretation, critical revision of the manuscript for important intellectual content. R. Selsov: analysis and interpretation, critical revision of the manuscript for important intellectual content. E.A. Macklin: analysis and interpretation, critical revision of the manuscript for important intellectual content. D. Schoenfeld: analysis and interpretation, critical revision of the manuscript for important intellectual content. M. Cudkowicz: study concept and design, critical revision of the manuscript for important intellectual content. A. Sherman: analysis and interpretation, critical revision of the manuscript for important intellectual content.
STUDY FUNDING
Supported by the LVH ALS Foundation. The PatientsLikeMe Research Team has received research funding (including conference support and consulting fees) from Abbvie, Accorda, Actelion, Amgen, AstraZeneca, Avanir, Biogen, Boehringer Ingelheim, Genentech, Genzyme, Janssen, Johnson & Johnson, Merck, Novartis, Sanofi, and UCB. The PatientsLikeMe R&D team has received research grant funding from Kaiser Permanente, the Robert Wood Johnson Foundation, Sage Bionetworks, The AKU Society, and the University of Maryland.
DISCLOSURE
R. Bedlack has research grants from the ALS Association, Motor Neurone Disease Association, and Cytokinetics; serves on the DSMB for Brainstorm Cell; and is a paid consultant for the ALS Association and Neuraltus Pharmaceuticals. T. Vaughan is an employee of PatientsLikeMe and holds stock in this company. P. Wicks is an employee of PatientsLikeMe and holds stock in this company. J. Heywood is an employee of PatientsLikeMe and holds stock in this company. E. Sinani and R. Selsov report no disclosures relevant to the manuscript. E. Macklin serves on DSMBs for Acorda Therapeutics, Lantheus Medical Imaging, and Shire Human Genetic Therapies and receives research support from the Adolph Coors Foundation, ALS Association, ALS Therapy Alliance, ALS Therapy Development Institute, Autism Speaks, Biotie Therapies, Muscular Dystrophy Association, and Michael J. Fox Foundation. D. Schoenfeld reports no disclosures relevant to the manuscript. M. Cudkowicz consults for Biogen, Cytokinetics, Neuraltus, and Genetech. A. Sherman receives research support from the ALS Association, Muscular Dystrophy Association, American Academy of Neurology, ALS Finding a Cure Foundation, and Prize4Life. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Tucker T, Layzer R, Miller R, Chad D. Subacute reversible motor neuron disease. Neurology 1991;41:1541–1544. [DOI] [PubMed] [Google Scholar]
- 2.Immunosuppression in Amyotrophic Lateral Sclerosis (ALS) (NIPALS2013). Available at: https://clinicaltrials.gov/ct2/show/NCT01884571?term=als+immunosuppression&rank=1. Accessed May 6, 2015.
- 3.Atassi N, Berry J, Shui A, et al. The PRO-ACT database: design, initial analyses, and predictive features. Neurology 2014;83:1719–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cedarbaum J, Stambler N. Performance of the Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS) in multicenter clinical trials. J Neurol Sci 1997;152:S1–S9. [DOI] [PubMed] [Google Scholar]
- 5.Cedarbaum J, Stambler N, Malta E, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. J Neurol Sci 1999;169:13–21. [DOI] [PubMed] [Google Scholar]
- 6.Tzartos J, Zisimopoulou P, Rentzos M, et al. LRP4 antibodies in serum and CSF from amyotrophic lateral sclerosis patients. Ann Clin Transl Neurol 2014;1:80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Hoecke A, Schoonaert L, Lemmens R, et al. EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models and in humans. Nat Med 2012;18:1418–1422. [DOI] [PubMed] [Google Scholar]
- 8.Samson M, Libert F, Doranz B, et al. Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996;382:722–725. [DOI] [PubMed] [Google Scholar]
- 9.Chio A, Canosa A, Gallo S, et al. ALS clinical trials: do enrolled patients accurately represent the ALS population? Neurology 2011;77:1432–1437. [DOI] [PubMed] [Google Scholar]
- 10.Methods appraisal in psychiatry. Available at: http://www.methodsappraisal.com/education-dichotomania/. Accessed August 12, 2015.