

ABSTRACT
Hepatitis C virus (HCV) infection is a global health problem, with millions of chronically infected individuals at risk for cirrhosis and hepatocellular carcinoma. HCV vaccine development is vital in the effort toward disease control and eradication, an undertaking aided by an increased understanding of the mechanisms of resistance to broadly neutralizing antibodies (bNAbs). In this study, we identified HCV codons that vary deep in a phylogenetic tree of HCV sequences and showed that a polymorphism at one of these positions renders Bole1a, a computationally derived, ancestral genotype 1a HCV strain, resistant to neutralization by both polyclonal-HCV-infected plasma and multiple broadly neutralizing monoclonal antibodies with unique binding epitopes. This bNAb resistance mutation reduces replicative fitness, which may explain the persistence of both neutralization-sensitive and neutralization-resistant variants in circulating viral strains. This work identifies an important determinant of bNAb resistance in an ancestral, representative HCV genome, which may inform HCV vaccine development.
IMPORTANCE Worldwide, more than 170 million people are infected with hepatitis C virus (HCV), the leading cause of hepatocellular carcinoma and liver transplantation in the United States. Despite recent significant advances in HCV treatment, a vaccine is needed. Control of the HCV pandemic with drug treatment alone is likely to fail due to limited access to treatment, reinfections in high-risk individuals, and the potential for resistance to direct-acting antivirals (DAAs). Broadly neutralizing antibodies (bNAbs) block infection by diverse HCV variants and therefore serve as a useful guide for vaccine development, but our understanding of resistance to bNAbs is incomplete. In this report, we identify a viral polymorphism conferring resistance to neutralization by both polyclonal plasma and broadly neutralizing monoclonal antibodies, which may inform HCV vaccine development.
INTRODUCTION
Hepatitis C virus (HCV) vaccine development has been complicated by the extraordinary genetic diversity of the virus and rapid viral evolution in infected individuals (1–7). The HCV genome is replicated by an error-prone NS5B polymerase (8), and past studies have demonstrated that cytotoxic T lymphocytes (CTL) and neutralizing antibodies (NAbs) against HCV exert selective pressure that results in selection of CTL and NAb escape mutations in the virus (9–15). While viral escape mutations allow for continued proliferation in the presence of CTL and NAbs, some of these mutations also carry a fitness cost, reducing the replication capacity of resistant viral variants (9–11, 16, 17).
Many NAbs are HCV strain specific, but broadly neutralizing human monoclonal antibodies (bNAbs) capable of neutralizing multiple diverse HCV variants have been isolated, proving that NAbs can also target relatively conserved regions of the envelope (E1 and E2) proteins (11, 18–30). Infusion of bNAbs is protective against infection in animal models of HCV (22, 31), and early high-titer bNAb responses to HCV are associated with viral clearance in humans (3, 10, 32–35). Unfortunately, resistance to bNAbs can also develop, and multiple studies have demonstrated that this resistance sometimes results from mutations distant from bNAb binding sites (11, 36–38). Since bNAbs may serve as a guide for HCV vaccine development, a more comprehensive understanding of resistance to bNAbs is essential.
Previously, our group generated a computationally derived, representative subtype 1a HCV genome known as Bole1a using Bayesian phylogenetics, ancestral sequence reconstruction, and covariance analysis (39). We demonstrated that Bole1a is ancestral to most circulating genotype 1a HCV strains, that it is representative of widely circulating strains, and that the envelope genes are functional on lentiviral particles (39). This genome contains fewer CTL escape mutations than natural circulating strains, since phylogenetic reconstruction places the more recent, host-specific changes, like escape mutations in HLA-restricted CTL epitopes, near the tips of the tree, while Bole1a falls near the root (40). This was confirmed in a prior study demonstrating that Bole1a contains more intact CTL epitopes than circulating HCV strains (40).
In contrast to changes near the tips of the tree, changes that occur deeper in the tree, closer to the Bole1a sequence, may represent selection that is less host specific. We hypothesized that this could include changes that enhance viral replicative fitness or confer resistance to bNAbs. In generation of the Bole1a genome, our analysis predicted a single most likely ancestral amino acid at all positions across the genome, but at some positions, posterior probabilities of a single ancestral amino acid were relatively low, suggesting complex evolution at these positions deep in a phylogenetic tree of diverse genotype 1a sequences. We examined 3 of these positions in the genes encoding E1 and E2 to determine whether variation at these positions could be explained by acquisition of E1E2 bNAb resistance or by an increase in viral replicative fitness or both (41).
MATERIALS AND METHODS
Sources of monoclonal Abs [MAbs].
CBH-5 (23), HC84.22 and HC84.26 (18), and HC33.4 (25) were gifts from Steven Foung (Stanford University School of Medicine, Stanford, CA, USA). AR3A (22) and AR4A (21) were gifts from Mansun Law (The Scripps Research Institute, La Jolla, CA, USA).
Source of plasma.
Plasma samples were obtained from the Baltimore Before-and-After Acute Study of Hepatitis (BBAASH) (42) cohort (Andrea Cox, Johns Hopkins University School of Medicine). Samples from each of the 18 HCV-infected subjects who had previously shown at least 50% neutralization by 1:100 plasma dilution of at least 2 HCV pseudoparticles (HCVpp) in the 19 HCVpp panel previously described (33) were selected.
HCVpp neutralization assays.
HCVpp were generated by cotransfection of pNL4-3.Luc.R-E- plasmid and an expression plasmid containing the gene encoding Bole1a HCV E1E2 as described elsewhere (35, 43, 44). Virus-containing medium was collected at 48 and 72 h, pooled, and stored at −4°C. For infectivity and neutralization testing of HCVpp, 8,000 Hep3B cells (American Type Culture Collection) per well were plated in flat-bottom 96-well tissue culture plates and incubated overnight at 37°C. The following day, HCVpp were mixed with either MAb (10 μg/ml) or heat-inactivated plasma (1:100 dilution) and then incubated at 37°C for 1 h. The medium was removed from the cells and replaced with 50 μl of HCVpp mixture. The plates were placed in a CO2 incubator at 37°C for 5 h, after which the HCVpp were removed and replaced with 100 μl of phenol-free Hep3B media and incubated for 72 h at 37°C. The medium was removed from the cells, 50 μl of 1× cell culture lysis reagent (Promega) was added and left to incubate for >5 min, and then 45 μl from each well was transferred to a white, low-luminescence 96-well plate (Berthold) and luciferase activity measured in relative light units (RLUs) in a Berthold Luminometer (Berthold Technologies Centro LB960). Infection by pseudoparticles was measured in the presence of MAb/test plasma (HCVppRLUtest) or nonspecific IgG/HCV-negative normal human plasma (HCVppRLUcontrol) at the same dilution. The percentage of neutralization was calculated as follows: 100% × [1 − HCVppRLUtest/HCVppRLUcontrol)]. Each sample was tested in duplicate. A mock pseudoparticle (no envelope) was used as a negative control. Neutralization was tested only for HCVpp with infectivity at least 10× greater than typical mock pseudoparticle values.
Generation of site-directed mutants.
E1E2 site-directed mutants were generated using a QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies).
HCV NS5A immunostaining.
Human hepatoma Huh7.5.1 cells (a gift from Jake Liang, NIH, Bethesda, MD, USA) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 1% sodium pyruvate, and 1% l-glutamate. At 48 h after infection, cells were fixed with 4% formaldehyde for 20 min and then stained for HCV NS5A using a mixture containing primary anti-NS5A 9E10 antibody (a gift from Charles Rice, The Rockefeller University, New York City, NY, USA) at a 1:10,000 dilution in phosphate-buffered saline (PBS), 3% bovine serum albumin, and 0.3% Triton X-100 for 1 h at room temperature. Cells were washed twice with PBS and stained using a mixture of Alexa Dylight 488-conjugated goat anti-mouse IgG secondary antibody (Life Technologies) at a 1:500 dilution in PBS, 3% bovine serum albumin, and 0.3% Triton X-100 for 1 h at room temperature. Cells were washed twice in PBS and then stored covered in 100 μl PBS at 4°C.
HCVcc neutralization assays.
Huh7.5.1 cells were plated at 10,000 cells per well onto 96-well plates. On the following day, antibodies were serially diluted 2.5-fold in growth medium starting from 50 μg/ml MAb. Controls were performed with a similar dilution series of nonspecific IgG. Viral supernatants diluted to fall in a linear range of infectivity were added to antibody dilutions in duplicate for 1 h at 37°C. The medium was removed from cells, and the antibody-virus mix was added onto cells and incubated overnight. The antibody-virus mix was replaced with 100 μl fresh medium the following day. After 48 h, the medium was removed and cells were fixed and stained. Images were acquired, and spot-forming units (SFU) were counted in the presence of MAb (HCVccSpotstest) or nonspecific IgG (HCVccSpotscontrol) using AID iSpot Reader Spectrum 219 operating AID ELISpot Reader version 7.0. Percent neutralization was calculated as 100% × [1 − (HCVccSpotstest/HCVccSpotscontrol)]. HCVcc neutralization assays using human plasma were performed similarly using 1:100 dilutions of test plasma and HCV-negative normal human plasma.
HCV E1E2 ELISA.
MAb or plasma binding to E1E2 was quantitated using an enzyme-linked immunosorbent assay (ELISA) as previously described (11). 293T cells were transfected with E1E2 expression constructs. At 48 h posttransfection, cell lysates were harvested. Plates were coated with 500 ng Galanthus nivalis lectin (Sigma-Aldrich) and blocked with phosphate-buffered saline containing 0.5% Tween 20, 1% nonfat dry milk, and 1% goat serum. E1E2 cell lysates were added. MAb or plasma was assayed in duplicate 2.5-fold serial dilutions, starting at 10 μg/ml or a 1:100 dilution, respectively. Binding was detected using horseradish peroxidase (HRP)-conjugated anti-human IgG secondary antibody (BD Pharmingen catalog no. 555788). For ELISA performed under denaturing conditions, lysates of cells were diluted in denaturing buffer (Tris-buffered saline with 10% fetal bovine serum, 1.0% sodium dodecyl sulfate, and 50 mM dithiothreitol) and then boiled for 5 min. Lysates were cooled on ice and then added to G. nivalis lectin-coated plates. The assay was completed as described above.
HCV load quantitation.
HCV RNA levels in infection supernatants were quantified using a process of RNA extraction and utilization of commercial real-time reagents (Abbot HCV Real Time assay) migrated onto a research-based real-time PCR platform (Roche 480 LightCycler).
Generation of HCVcc chimeras.
To introduce an AfeI restriction site, HCVcc chimera H77/JFH1 (45), a gift from Jens Bukh (Copenhagen University Hospital, Copenhagen, Denmark), was amplified in three sections, using primers insert_R_new (CACCAGCTGATATAGCGTTTGTAATATGGCGACAGAGTC), insert_F_new (GGATTCCGATCTACCAGCGCTTTGGAGAACCTCGTAATACTCAATGCAGCATCCCTGGCC), back_mid_F (CGGAATATGACCTGGAGCTAATAACATCC), backbone_outer_R (GGTGACATGGTAAAGCCCCG), Back-mid_R (CCAGGTCATATTCCGGTCTGG), and Backbone_outer_F (CTGTCGCCATATTACAAACGC).
This omitted nucleotides 916 to 2579. Amplified sections were reassembled using In-Fusion cloning (Clontech). This backbone was digested with enzyme AfeI (New England BioLabs). E1E2 insertions were amplified from library plasmids using primers HCVcc_E1E2_1R (GAGGTTCTCCAAAGCCGCCTCCGC) and HCVcc E1E2_1F (TGTGCCCGCTTCAGCCTACCAAG). The insertions were cloned into the digested HCVcc backbone using In-Fusion cloning.
To make HCVcc RNA, 2 μg plasmid DNA was linearized using XbaI (New England BioLabs). Linear DNA was used for in vitro RNA transcription with a T7 MEGAscript kit (Ambion). RNA cleanup was performed using a RNeasy minikit (Qiagen) and quantified using NanoDrop. RNA products were then stored at −80°C.
To transfect RNA, Nucelofector kit T was used (Amaxa). A 5-μg volume of RNA was transfected into 1.8e6 Huh7.5.1 cells and plated in a 6-cm-diameter plate. Transfection supernatants were collected 4 to 6 days later, once cells had reached confluence, and stored at −80°C for titer determinations by HCV NS5A immunostaining. High-titer transfection supernatants were used to infect Huh7.5.1 cells, and infection supernatants were collected; then, the titers of the infection supernatants were measured, and the infection supernatants were stored at −80°C for use in infectivity and neutralization experiments.
Western blotting.
HCVpp supernatants were concentrated and purified by ultracentrifugation through a 20% sucrose cushion (123,000 × g, 2 h, 4°C) with pellets resuspended in TNE buffer (50 mm Tris-HCl [pH 7.4], 100 mm NaCl, 0.1 mm EDTA). HCVpp and E1E2 lysates used in ELISAs were diluted and then denatured and run on a 4% to 12% Bis-Tris gel. After transfer, blots were probed with HC33.1.53 (25) (a gift from Steven Foung, Stanford University School of Medicine, Stanford, CA) and binding was detected with HRP-conjugated anti-human IgG secondary antibody (BD Pharmingen catalog no. 555788). HCVpp Western blots were also probed with mouse anti-HIV1 p24 (ab9044; Abcam) followed by HRP-conjugated anti-mouse IgG secondary antibody (ab97265; Abcam).
Statistical analyses.
Percent neutralization values were grouped by individual mutations, and a one-way analysis of variance (ANOVA) with a Tukey test was performed to compare the levels of significance of changes in neutralization mediated by different amino acid mutations at the same position (see Fig. 2). For each pair of R424S ELISAs, a two-way ANOVA was performed to determine the significance of the change in binding between E1E2 lysates.
FIG 2.
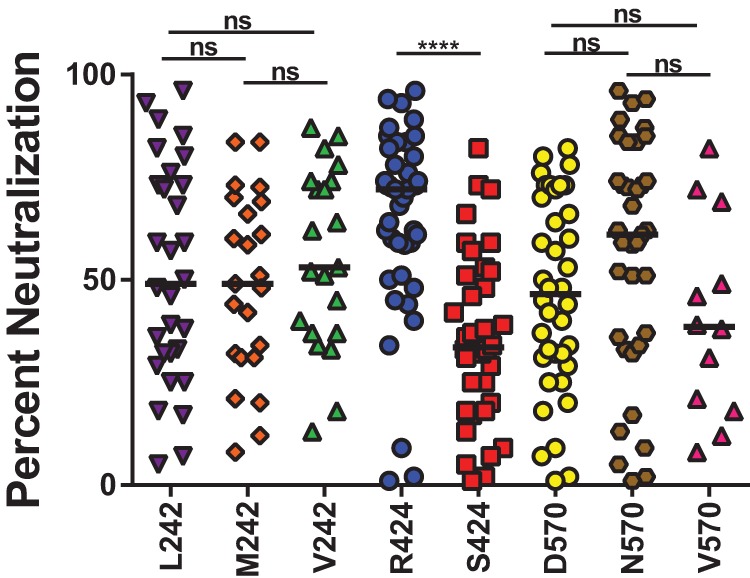
Mutation of arginine (R) 424 to serine (S) confers resistance to neutralization by plasma. A panel of 13 Bole1a E1E2 constructs encoding different combinations of amino acids at codons 242, 424, and 570 was generated using site-directed mutagenesis. Each Bole1a E1E2 variant construct was used to generate HCVpp. HCVpp were tested for neutralization by the use of 5 to 10 HCV-infected plasma samples in duplicate, and reported values represent the results of an average of 1 to 4 independent experiments. HCVpp with the same amino acid polymorphism at 242, 424, or 570 were grouped for analysis. Horizontal lines indicate median percent neutralization. Significance was tested using one-way ANOVA with a Tukey test for multiple comparisons (****, P < 0.0001; ns, not significant). Samples with percent neutralization values of <0 are not shown.
RESULTS
Some polyprotein positions show evidence of deep phylogenetic variation.
Through the use of Bayesian phylogenetics, ancestral sequence reconstruction, and covariance analysis, the most likely ancestral genotype 1a amino acid was predicted at each position across the HCV polyprotein, generating the Bole1a genome (39). For the majority of polyprotein positions, the most likely ancestral amino acid could be determined with very high posterior probability ranging from 0.99 to 1, but for 94 of the 3,012 codons analyzed, the posterior probability of the most likely ancestral codon was less than 0.99 (Fig. 1A), suggesting complex evolution at these positions deep in the phylogenetic tree. We selected three of these positions for further analysis. At position 242, which falls in HCV E1, leucine (L) was the most likely ancestral amino acid. At position 424, which falls near the CD81 binding site of E2, arginine (R) was the most likely ancestral amino acid. At position 570, which falls in the intergenotypic variable region (igVR) (46) near the carboxy terminus of E2, asparagine (N) was most likely. We also determined the most common amino acids at these positions in circulating viral strains (Fig. 1B). Circulating strains showed significant variability at the 242 and 570 positions, with 4 and 9 different amino acids observed at each position, respectively, in a reference alignment of 390 genotype 1a sequences. In contrast, the 424 codon is highly constrained, with either R or S observed in 387 of 390 circulating strains in the reference alignment.
FIG 1.

Codons 242, 424, and 570 show evidence of deep phylogenetic variation, and evolution at the 424 position is constrained. (A) Using a reference alignment of 390 genotype 1a HCV genomes and Bayesian phylogenetics, the identity of the most likely ancestral amino acid at each position across the HCV genome was calculated, generating the Bole1a genome (39). The histogram depicts the distribution of posterior probabilities with which a single most likely ancestral amino acid could be predicted at each of 3,012 codons. The posterior probabilities of leucine (L) at 242, arginine (R) at 424, and aspartic acid (D) at 570 are indicated. (B) Frequency of each observed amino acid at positions 242, 424, and 570 in a reference alignment of 390 genotype 1a sequences. Letters are standard IUPAC amino acid abbreviations.
Mutation of arginine 424 to serine confers resistance to neutralization by plasma.
HCV pseudoparticles (HCVpp) with Bole1a E1E2_L242, E1E2_R424, or E1E2_N570 or Bole1a E1E2 with other naturally occurring amino acids at these positions were generated by cotransfection of E1E2 expression plasmids with an HIV pNL4-3.Luc.R-E- reporter construct. The amino acid polymorphisms to be tested were chosen based on the predicted ancestral sequence, the most common amino acids in circulating strains, and some additional less frequently observed amino acids. To efficiently measure the effect of mutations at each of the three positions in multiple experiments, 13 HCVpp were generated with each mutation alone or in combination with mutations at one or both of the other two positions. These HCVpp were tested for sensitivity to neutralization by HCV-infected plasma samples, and all variants with the same amino acid at position 242, 424, or 570 were grouped for analysis (Fig. 2). Mutation of L242 to valine (V) or methionine (M) and mutation of N570 to valine (V) or aspartic acid (D) did not have a significant effect on neutralization sensitivity, but mutation of arginine (R) 424 to serine (S) conferred a significant increase in resistance to neutralization by plasma. Since variation at positions 242 and 570 did not significantly affect Bola1a neutralization sensitivity, subsequent experiments focused on variation at the 424 position, with L242 and N570 held constant.
To confirm the resistance phenotype of S424, Bole1a_R424 and Bole1a_S424 HCVpp were tested for neutralization by an additional panel of 19 HCV-infected plasma samples (Fig. 3A). As in prior experiments, S424 was strongly associated with resistance to neutralization (median neutralization, 91% for Bole1a_R424 and 44% for Bole1a_S424, P = 2E−10). Strikingly, for every plasma sample tested, Bole1a_S424 was more neutralization resistant than Bole1a_R424, despite the fact that each plasma sample was from a different donor and each sample presumably contained polyclonal NAbs. This result was confirmed with replication-competent cell culture virus (HCVcc) expressing either Bole1a_R424 or Bole1a_S424 E1E2 (Fig. 3B). In addition, mutation of R424 to S in two natural E1E2 variants isolated from HCV-infected donors (1a53 and 1a79) also conferred resistance to neutralization by heterologous HCV-infected plasma, and mutation of S424 to R in a third naturally occurring E1E2 variant (H77) conferred increased sensitivity to neutralization (Fig. 3C). To investigate whether changes in the neutralization sensitivity observed in Bole1a_R424 and Bole1a_S424 might be influenced by incorporation of different amounts of E2 per particle, we performed a Western blot analysis using purified HCVpp (Fig. 3D). Both E2 and the p24 loading control of Bole1a_S424 HCVpp showed approximately 2-fold less protein than Bole1a_R424 HCVpp, indicating that the two HCVpp incorporate approximately equal amounts of E2 protein per particle.
FIG 3.

Mutation of arginine (R) 424 to serine (S) confers resistance to neutralization by plasma. (A) Bole1a_R424 and Bole1a_S424 HCVpp were tested for neutralization by the use of an additional panel of 19 HCV-infected plasma samples in duplicate. Each point represents the mean of two replicate values. Each line indicates neutralization by a unique plasma sample. P values were calculated using a paired two-tailed Student's t test. (B) Bole1a_R424 and Bole1a_S424 E1E2 sequences were cloned into full-length replication-competent HCV (HCVcc) and used to produce infectious supernatants. Viral supernatants were incubated with a 1:100 dilution of HCV-infected plasma for 1 h and then used to infect Huh7.5.1 cells in triplicate. Values shown are means of results from replicate wells. Each line indicates neutralization by a unique plasma sample. P values were calculated using a paired two-tailed Student's t test. (C) E1E2 variants 1a53, 1a79, and H77 were mutated via site-directed mutagenesis to produce 1a53_S424, 1a79_S424, and H77_R424. Wild-type and mutant E1E2 HCVpp were made from these constructs and were then tested in duplicate for neutralization sensitivity using the panel of 19 HCV-patient plasma samples. Each point represents the mean of two replicate values, and each line indicates neutralization by a unique plasma sample. P values were calculated using a paired two-tailed Student's t test. Percentage neutralization values of <0 are not shown. (D) Bole1a_R424 and Bole1a_S424 HCVpp were purified by ultracentrifugation through a sucrose cushion and then analyzed by Western blotting. Blots were probed with human anti-E2 (HC33.1.53) (25) and mouse anti-HIV1 p24.
Mutation of arginine 424 to serine confers resistance to binding by antibodies in plasma.
To elucidate the mechanism of variable neutralization sensitivity between Bole1a_R424 and Bole1a_S424 variants, the same panel of 19 HCV-infected plasma samples was used in E1E2 binding assays. Lysates of cells transfected with Bole1a_R424 or Bole1a_S424 E1E2 expression constructs were used as the target antigen in enzyme-linked immunosorbent assays (ELISAs) performed with HCV-infected plasma (Fig. 4A). Binding of plasma antibodies to the S424 E1E2 protein was significantly lower than binding to the R424 variant (median optical density [OD] values of 1.44 for Bole1a_R424 and 0.0009 for Bole1a_S424, P = 1E−05), suggesting that mutation of R424 to S confers neutralization resistance by reducing binding of NAbs. To confirm that the reduced binding to Bole1a_S424 E1E2 relative to Bole1a_R424 was not due to a difference in levels of expression of the two E1E2 proteins, we performed a Western blot analysis with serial dilutions of both E1E2 lysate preparations, confirming that the quantities of the proteins used in the ELISAs were similar (Fig. 4B).
FIG 4.
Mutation of arginine (R) 424 to serine (S) confers resistance to binding of plasma antibodies. (A) Lysates of cells transfected with Bole1a_R424 and Bole1_S424 E1E2 expression constructs were used in enzyme-linked immunosorbent assays (ELISAs) to assess E1E2 binding by IgG in the same panel of 19 HCV-infected plasma samples. Each of the 19 plasma samples was assayed in duplicate, and antibody binding was detected using HRP-conjugated anti-human IgG secondary antibody. Each point represents the mean of two replicate values, and each line indicates binding by IgG from a unique plasma sample. P values were calculated using a paired two-tailed Student's t test. OD450, optical density at 450 nm. (B) Lysates of E1E2-transfected cells used for ELISA measurements were analyzed by Western blotting in three dilutions to confirm that equal quantities of E1E2 were present. Blots were probed using anti-E2 antibody HC33.1.53 (25).
Mutation of arginine 424 to serine confers resistance to neutralization by a diverse array of broadly neutralizing MAbs.
To further investigate the surprising result that an R424S mutation conferred resistance to polyclonal sera from numerous donors, we measured the sensitivity of replication-competent cell culture virus (HCVcc) expressing Bole1a_R424 or Bole1a_S424 E1E2 to neutralization by a panel of some of the most broadly neutralizing human monoclonal antibodies (MAbs) described to date, including CBH-5 (23), HC84.22, HC84.26 (18), HC33.4 (25), AR3A (22) and AR4A (21). These MAbs were selected because they are broadly neutralizing and also because they bind to distinct epitopes across E2. CBH-5, AR3A, HC84.22, and HC84.26 bind to distinct epitopes at or near the CD81 binding site of E2, while HC33.4 binds near the amino terminus of E2, and AR4A binds to E1 and the carboxy terminus of E2 (Fig. 5A). Amino acid 424 is a known binding residue for AR3A as determined by alanine scanning mutagenesis (22), but it is not a known binding residue for any other MAbs in the panel. Despite the differences in E2 binding sites of these bNAbs, mutation of R424 to S conferred resistance to each antibody (Fig. 5B). As shown in Fig. 5C, the 50% inhibitory concentration (IC50) of each MAb against Bole1a_R424 HCVcc was less than 2 μg/ml, while the IC50 of the majority of MAbs against Bole1a_S424 HCVcc was 50 μg/ml or greater. Similar results were observed with HCVpp expressing Bole1a_R424 or S424 E1E2 and with HCVpp expressing R424 and S424 variants of a naturally occurring HCV strain, 1a53 (Fig. 5D and E). Interestingly, no difference in MAb sensitivity was observed between R424 and S424 variants of two other naturally occurring HCV strains, 1a79 and H77. Taken together, these results indicate that mutation of Bole1a_R424 to S confers resistance to bNAb neutralization, regardless of the antibody binding epitope.
FIG 5.

Mutation of arginine 424 to serine confers resistance to neutralization by a diverse array of broadly neutralizing monoclonal antibodies. (A) A panel of six broadly neutralizing monoclonal human antibodies (CBH-5, HC84.22, HC84.26, AR3A, AR4A, and HC33.4) was selected to assess relative levels of neutralization sensitivity of Bole1a_R424 and Bole1a_S424 variants. The E2 core structure published by Kong and colleagues (20) (Protein Data Bank accession no. 4MWF) is shown with colors modified. The front layer is cyan; the CD81-binding loop is blue; the central β-sandwich is red. The 424 position is indicated with an arrow. Known critical binding residues of AR3A and HC84.22 are indicated with purple and yellow spheres, respectively. Critical binding residues of HC33.4 and AR4A are not present in this structure, but approximate binding positions are indicated along with their known critical binding residues. CBH-5 and HC84.26 (not shown) share multiple binding residues with AR3A and HC84.22, respectively. (B) Bole1a_R424 and Bole1a_S424 E1E2 sequences were cloned into full-length replication-competent HCV (HCVcc) and used to produce infectious virus. Titers of viral supernatants were determined and used to infect target cells in duplicate in neutralization assays with serial dilutions of six different broadly neutralizing MAbs or control IgG. Values shown are means, and error bars indicate standard deviations of the results of comparisons between replicates. (C) IC50s calculated from the curves presented in panel B. For curves with only the highest antibody concentration producing more than 50% neutralization, the IC50 is reported as ∼50 μg/ml; for curves with maximum neutralization of less than 50%, the IC50 is reported as >50 μg/ml. (D) Bole1a_R424 and Bole1a_S424 HCVpp were tested for sensitivity to neutralization by a panel of six broadly neutralizing MAbs. Each HCVpp was incubated with 10 μg/ml MAb for 1 h prior to incubation with Hep3B target cells. Percent neutralization was calculated by comparing infection of HVCpp incubated with MAb to infection of HCVpp incubated with nonspecific IgG. Each construct was tested in duplicate. Values shown are means, and error bars indicate standard deviations. P values were calculated using a paired two-tailed Student's t test. Percentage neutralization values of <0 are not shown. (E) HCVpp 1a53, 1a53_S424, 1a79, 1a79_S424, H77, and H77_R424 were tested for neutralization sensitivity as described for Bole1a HCVpp in the panel D legend.
Mutation of arginine 424 to serine confers resistance to binding of broadly neutralizing monoclonal antibodies.
Binding of MAbs to Bole1a_R424 and Bole1a_S424 E1E2 was measured using an E1E2 ELISA. MAbs HC84.22, CBH-5, AR3A, and HC33.4 showed greater binding to the R424 E1E2 lysate than the S424 E1E2 lysate (Fig. 6A and B). There was no detectable difference in binding of AR4A to either variant, suggesting either that the Bole1a_S424 resistance to neutralization by AR4A is not mediated by a reduction in binding of the MAb or that the binding ELISA is not sensitive enough to detect changes in binding affinity sufficient to influence neutralization. Taken together, these results show a small but consistent reduction in binding of MAbs to S424 E1E2 relative to R424 E1E2, in agreement with the observed reduction in binding of plasma antibodies to the S424 E1E2 protein, suggesting that resistance to bNAb binding is a likely mechanism of the observed neutralization resistance of Bole1a_S424.
FIG 6.

Mutation of arginine 424 to serine confers resistance to binding of broadly neutralizing monoclonal antibodies. (A) Binding of MAbs AR3A, CBH-5, HC84.22, and AR4A to Bole1a_R424 and Bole1a_S424 E1E2 lysates was measured by ELISA. MAbs were assayed in duplicate with 2.5-fold serial dilutions, starting at 10 μg/ml, and binding was detected using HRP-conjugated anti-human IgG secondary antibody. Values shown represent the means of the results determined with two replicates, and error bars indicate standard deviations. P values were determined by two-way ANOVA (****, P < 0.0001). (B) To compare the binding results seen under native and denatured E1E2 conditions, Bole1a_R424 and Bole1a_S424 E1E2 lysates were diluted either in a denaturing buffer containing sodium dodecyl sulfate (SDS) or in phosphate-buffered saline (PBS). The denatured lysates were also boiled for 5 min and then cooled on ice. Both native and denatured lysates were then added to G. nivalis lectin-coated plates, and binding of serial dilutions of MAb HC33.4 was quantitated in duplicate. Data shown represent the means of the results of two independent experiments, and error bars indicate standard deviations. (C) Binding of MAbs HC84.22, HC33.4, CBH-5, and AR4A to 1a53, 1a53_S424, 1a79, 1a79_S424, H77, and H77_R424 E1E2 lysates was measured by ELISA as described in the panel A legend. P values were determined by two-way ANOVA (*, P < 0.05; ****, P < 0.0001).
Since mutation of R424 to S conferred resistance to binding of MAbs for which 424 is not a known binding residue, we tested whether the effect is due to an induced structural change in the E1E2 proteins. We were able to test this directly for one MAb, HC33.4, since it binds to both native and denatured protein. Binding of HC33.4 to Bole1a_R424 and Bole1a_S424 E1E2 protein lysates was measured with the E1E2 protein in either native or denatured states (Fig. 6B). Surprisingly, the difference in MAb HC33.4 binding to Bole1a_R424 and Bole1a_S424 E1E2 was significantly more pronounced with the protein in a denatured state, suggesting that induction of a structural shift may not be the mechanism by which mutation of R424 to S reduces binding of HC33.4. Instead, R424 could be a previously undetected HC33.4 binding residue.
We also measured binding of four MAbs to R424 and S424 variants of 1a53, 1a79, and H77 E1E2 proteins (Fig. 6C). Introduction of S424 into 1a53 E1E2 reduced binding of all four MAbs, which is consistent with the observed resistance to these MAbs conferred to 1a53 HCVpp by S424. For 1a79 and H77 E1E2, S424 conferred resistance to binding by some MAbs but not others, which is also consistent with minimal resistance to MAb neutralization conferred to 1a79 and H77 HCVpp by S424.
Bole1a_S424 carries a fitness cost.
Despite the broad neutralization resistance conferred by S424, both R424 and S424 variants are abundant in circulating virus populations, suggesting selection pressure favoring R424 acting in opposition to the bNAb selection pressure favoring S424. We examined the relative levels of fitness of Bole1a_R424 HCVcc and Bole1a_S424 HCVcc in the absence of antibody (Fig. 7). Bole1a_R424 HCVcc showed higher specific infectivity than Bole1a_S424 HCVcc (18.9 SFU/105 international units [IU] for Bole1a_R424 versus 5.4 SFU/105 IU for Bole1a_S424). Taken together, these results suggest that selection by bNAbs favors persistence of S424 variants, while greater replicative fitness favors persistence of R424 variants.
FIG 7.
Bole1a_S424 carries a fitness cost. Bole1a_R424 and Bole1a_S424 E1E2 sequences were cloned into full-length replication-competent HCV (HCVcc). RNA was in vitro synthesized and transfected into Huh7.5.1 cells, and culture supernatants were collected. Transfection supernatants were subsequently used to infect Huh7.5.1 cells to expand viral stocks. Huh7.5.1 cells were infected in duplicate with serial dilutions of Bole1a_R424 HCVcc and Bole1a_S424 HCVcc supernatants. After 48 h, infection was quantified as the number of spot-forming units per milliliter of supernatant. Viral RNA was extracted from the same infection supernatants, and the viral RNA load was quantitated using real-time PCR and an international unit (IU) viral load standard. Specific infectivity is expressed as the number of spot-forming units per IU. Values shown represent means of the results of two independent experiments, and error bars indicate ranges.
DISCUSSION
In this study, we identified a position in E2, codon 424, that varies deep in a phylogenetic tree of diverse genotype 1a sequences and yet is constrained to only two possible amino acids, arginine and serine, in circulating strains. Mutation of R424 to S at this position in Bole1a, a representative, ancestral HCV strain, confers extremely broad resistance to neutralization by both polyclonal HCV-infected sera and a diverse array of broadly neutralizing MAbs. We showed that this neutralization resistance is due to a reduction in binding of NAbs. We also showed that the neutralization-resistant Bole1a_S424 HCVcc variant has reduced replicative fitness relative to the R424 variant, providing an explanation for persistence of both R424 and S424 polymorphisms in circulating viral strains.
This work supports prior studies showing that HCV resistance to NAbs may arise from mutations distant from antibody binding sites (11, 36–38). The resistance conferred to Bole1a by S424 is exceptionally broad. This mutation conferred resistance to the polyclonal NAbs present in every plasma sample tested from dozens of unique donors, as well as a panel of bNAbs selected specifically because their binding epitopes are well characterized and distant from each other. This work provides evidence that selective pressure from commonly expressed bNAbs and competing pressure for high replicative fitness may explain some amino acid changes deep in phylogenetic trees of diverse HCV sequences. Analysis of phylogenetically ancestral evolution may be a useful method to identify bNAb resistance mutations that are immunologically relevant at a population level.
It is interesting that the R424S mutation in Bole1a reduces binding of most MAbs and neutralization of all MAbs given that 424 is a known binding residue only for MAb AR3A. R424 may be a relatively minor, previously undetected binding residue for these MAbs with an unexpectedly significant influence on neutralizing activity. Alternatively, this mutation could influence glycosylation of the protein, although this seems less likely as it does fall at the first or third position of an N-linked glycosylation consensus sequence. It is also possible that the mutation alters the structure of E2, given the known structurally flexibility of the E2 protein in the region of 424 (47, 48). We were able to show for MAb HC33.4 that the reduction in binding to Bole1a_S424 was present even when Bole1a_R424 and Bole1a_S424 E1E2 were denatured, making it less likely that the effect is mediated by a shift in structure. These binding experiments are technically challenging, however, and the possibility of mutation-mediated structural shifts remains intriguing and warrants further investigation.
This study showed consistent and broad neutralization resistance conferred to Bole1a by R424S. The magnitude of the change in resistance to polyclonal serum antibodies was not large, but it was similar to the resistance described in a prior study of HCV neutralization escape (37). The effect of R424S was most striking for neutralization of Bole1a HCVcc by broadly neutralizing monoclonal antibodies (Fig. 5C), as a more than 100-fold increase in resistance to some bNAbs was observed. It is particularly interesting that the resistance conferred by S424 varied between Bole1a and other E1E2 variants. Work to understand the influence of combinations of polymorphisms on the neutralization resistance of E1E2 is ongoing. A better understanding of polymorphisms modulating the neutralization sensitivity of representative genomes like Bole1a is critical as work continues to identify the most representative and immunogenic HCV variants for inclusion in HCV vaccines.
In conclusion, we have identified a neutralization resistance polymorphism using a unique strategy which identifies amino acid changes deep in a phylogenetic tree of diverse HCV sequences. This R424S polymorphism confers extremely broad resistance to neutralization by both polyclonal HCV-infected sera and a diverse array of broadly neutralizing MAbs with distinct binding epitopes. The neutralization-resistant S424 variant has reduced replicative fitness, which may explain the persistence of both S424 and the neutralization-sensitive R424 variant in the population. A more complete understanding of determinants of bNAb resistance in candidate vaccine antigens like Bole1a should help to guide HCV vaccine development.
ACKNOWLEDGMENTS
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
This study was supported by NIH grants 1K08 AI102761 (Justin R. Bailey), U19 AI088791 (Stuart C. Ray), and R37 DA013806 (Stuart C. Ray) and The Johns Hopkins University Center for AIDS Research grant 1P30AI094189 (Justin R. Bailey).
We thank the faculty members and staff of the Viral Hepatitis Center at Johns Hopkins University School of Medicine as well as participants in the BBAASH cohort. Thanks also to Jeffry Quinn for help measuring HCVcc loads.
REFERENCES
- 1.Dienstag JL. 1997. The natural history of chronic hepatitis C and what we should do about it. Gastroenterology 112:651–655. doi: 10.1053/gast.1997.v112.agast970651. [DOI] [PubMed] [Google Scholar]
- 2.Liu L, Fisher BE, Dowd KA, Astemborski J, Cox AL, Ray SC. 2010. Acceleration of hepatitis C virus envelope evolution in humans is consistent with progressive humoral immune selection during the transition from acute to chronic infection. J Virol 84:5067–5077. doi: 10.1128/JVI.02265-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu L, Fisher BE, Thomas DL, Cox AL, Ray SC. 2012. Spontaneous clearance of primary acute hepatitis C virus infection correlated with high initial viral RNA level and rapid HVR1 evolution. Hepatology 55:1684–1691. doi: 10.1002/hep.25575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simmonds P. 2004. Genetic diversity and evolution of hepatitis C virus—15 years on. J Gen Virol 85:3173–3188. doi: 10.1099/vir.0.80401-0. [DOI] [PubMed] [Google Scholar]
- 5.Villano SA, Vlahov D, Nelson KE, Cohn S, Thomas DL. 1999. Persistence of viremia and the importance of long-term follow-up after acute hepatitis C infection. Hepatology 29:908–914. doi: 10.1002/hep.510290311. [DOI] [PubMed] [Google Scholar]
- 6.Farci P, Bukh J, Purcell RH. 1997. The quasispecies of hepatitis C virus and the host immune response. Springer Semin Immunopathol 19:5–26. doi: 10.1007/BF00945022. [DOI] [PubMed] [Google Scholar]
- 7.Bukh J, Miller RH, Purcell RH. 1995. Genetic heterogeneity of hepatitis C virus: quasispecies and genotypes. Semin Liver Dis 15:41–63. doi: 10.1055/s-2007-1007262. [DOI] [PubMed] [Google Scholar]
- 8.Neumann AU, Lam NP, Dahari H, Gretch DR, Wiley TE, Layden TJ, Perelson AS. 1998. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science 282:103–107. doi: 10.1126/science.282.5386.103. [DOI] [PubMed] [Google Scholar]
- 9.Cox AL, Mosbruger T, Mao Q, Liu Z, Wang XH, Yang HC, Sidney J, Sette A, Pardoll D, Thomas DL, Ray SC. 2005. Cellular immune selection with hepatitis C virus persistence in humans. J Exp Med 201:1741–1752. doi: 10.1084/jem.20050121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dowd KA, Netski DM, Wang XH, Cox AL, Ray SC. 2009. Selection pressure from neutralizing antibodies drives sequence evolution during acute infection with hepatitis C virus. Gastroenterology 136:2377–2386. doi: 10.1053/j.gastro.2009.02.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keck ZY, Li SH, Xia J, von Hahn T, Balfe P, McKeating JA, Witteveldt J, Patel AH, Alter H, Rice CM, Foung SK. 2009. Mutations in hepatitis C virus E2 located outside the CD81 binding sites lead to escape from broadly neutralizing antibodies but compromise virus infectivity. J Virol 83:6149–6160. doi: 10.1128/JVI.00248-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Timm J, Lauer GM, Kavanagh DG, Sheridan I, Kim AY, Lucas M, Pillay T, Ouchi K, Reyor LL, Schulze zur Wiesch J, Gandhi RT, Chung RT, Bhardwaj N, Klenerman P, Walker BD, Allen TM. 2004. CD8 epitope escape and reversion in acute HCV infection. J Exp Med 200:1593–1604. doi: 10.1084/jem.20041006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang XH, Netski DM, Astemborski J, Mehta SH, Torbenson MS, Thomas DL, Ray SC. 2007. Progression of fibrosis during chronic hepatitis C is associated with rapid virus evolution. J Virol 81:6513–6522. doi: 10.1128/JVI.02276-06z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimizu YK, Hijikata M, Iwamoto A, Alter HJ, Purcell RH, Yoshikura H. 1994. Neutralizing antibodies against hepatitis C virus and the emergence of neutralization escape mutant viruses. J Virol 68:1494–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thimme R, Bukh J, Spangenberg HC, Wieland S, Pemberton J, Steiger C, Govindarajan S, Purcell RH, Chisari FV. 2002. Viral and immunological determinants of hepatitis C virus clearance, persistence, and disease. Proc Natl Acad Sci U S A 99:15661–15668. doi: 10.1073/pnas.202608299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuntzen T, Timm J, Berical A, Lewis-Ximenez LL, Jones A, Nolan B, Schulze zur Wiesch J, Li B, Schneidewind A, Kim AY, Chung RT, Lauer GM, Allen TM. 2007. Viral sequence evolution in acute hepatitis C virus infection. J Virol 81:11658–11668. doi: 10.1128/JVI.00995-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ray SC, Fanning L, Wang XH, Netski DM, Kenny-Walsh E, Thomas DL. 2005. Divergent and convergent evolution after a common-source outbreak of hepatitis C virus. J Exp Med 201:1753–1759. doi: 10.1084/jem.20050122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keck ZY, Xia J, Wang Y, Wang W, Krey T, Prentoe J, Carlsen T, Li AY, Patel AH, Lemon SM, Bukh J, Rey FA, Foung SK. 2012. Human monoclonal antibodies to a novel cluster of conformational epitopes on HCV E2 with resistance to neutralization escape in a genotype 2a isolate. PLoS Pathog 8:e1002653. doi: 10.1371/journal.ppat.1002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keck ZY, Saha A, Xia J, Wang Y, Lau P, Krey T, Rey FA, Foung SK. 2011. Mapping a region of hepatitis C virus E2 that is responsible for escape from neutralizing antibodies and a core CD81-binding region that does not tolerate neutralization escape mutations. J Virol 85:10451–10463. doi: 10.1128/JVI.05259-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kong L, Giang E, Robbins JB, Stanfield RL, Burton DR, Wilson IA, Law M. 2012. Structural basis of hepatitis C virus neutralization by broadly neutralizing antibody HCV1. Proc Natl Acad Sci U S A 109:9499–9504. doi: 10.1073/pnas.1202924109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giang E, Dorner M, Prentoe JC, Dreux M, Evans MJ, Bukh J, Rice CM, Ploss A, Burton DR, Law M. 2012. Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc Natl Acad Sci U S A 109:6205–6210. doi: 10.1073/pnas.1114927109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Law M, Maruyama T, Lewis J, Giang E, Tarr AW, Stamataki Z, Gastaminza P, Chisari FV, Jones IM, Fox RI, Ball JK, McKeating JA, Kneteman NM, Burton DR. 2008. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat Med 14:25–27. doi: 10.1038/nm1698. [DOI] [PubMed] [Google Scholar]
- 23.Hadlock KG, Lanford RE, Perkins S, Rowe J, Yang Q, Levy S, Pileri P, Abrignani S, Foung SK. 2000. Human monoclonal antibodies that inhibit binding of hepatitis C virus E2 protein to CD81 and recognize conserved conformational epitopes. J Virol 74:10407–10416. doi: 10.1128/JVI.74.22.10407-10416.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keck ZY, Olson O, Gal-Tanamy M, Xia J, Patel AH, Dreux M, Cosset FL, Lemon SM, Foung SK. 2008. A point mutation leading to hepatitis C virus escape from neutralization by a monoclonal antibody to a conserved conformational epitope. J Virol 82:6067–6072. doi: 10.1128/JVI.00252-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keck Z, Wang W, Wang Y, Lau P, Carlsen TH, Prentoe J, Xia J, Patel AH, Bukh J, Foung SK. 2013. Cooperativity in virus neutralization by human monoclonal antibodies to two adjacent regions located at the amino terminus of hepatitis C virus E2 glycoprotein. J Virol 87:37–51. doi: 10.1128/JVI.01941-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krey T, Meola A, Keck ZY, Damier-Piolle L, Foung SK, Rey FA. 2013. Structural basis of HCV neutralization by human monoclonal antibodies resistant to viral neutralization escape. PLoS Pathog 9:e1003364. doi: 10.1371/journal.ppat.1003364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johansson DX, Voisset C, Tarr AW, Aung M, Ball JK, Dubuisson J, Persson MA. 2007. Human combinatorial libraries yield rare antibodies that broadly neutralize hepatitis C virus. Proc Natl Acad Sci U S A 104:16269–16274. doi: 10.1073/pnas.0705522104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bartosch B, Bukh J, Meunier JC, Granier C, Engle RE, Blackwelder WC, Emerson SU, Cosset FL, Purcell RH. 2003. In vitro assay for neutralizing antibody to hepatitis C virus: evidence for broadly conserved neutralization epitopes. Proc Natl Acad Sci U S A 100:14199–14204. doi: 10.1073/pnas.2335981100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Broering TJ, Garrity KA, Boatright NK, Sloan SE, Sandor F, Thomas WD Jr, Szabo G, Finberg RW, Ambrosino DM, Babcock GJ. 2009. Identification and characterization of broadly neutralizing human monoclonal antibodies directed against the E2 envelope glycoprotein of hepatitis C virus. J Virol 83:12473–12482. doi: 10.1128/JVI.01138-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perotti M, Mancini N, Diotti RA, Tarr AW, Ball JK, Owsianka A, Adair R, Patel AH, Clementi M, Burioni R. 2008. Identification of a broadly cross-reacting and neutralizing human monoclonal antibody directed against the hepatitis C virus E2 protein. J Virol 82:1047–1052. doi: 10.1128/JVI.01986-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morin TJ, Broering TJ, Leav BA, Blair BM, Rowley KJ, Boucher EN, Wang Y, Cheslock PS, Knauber M, Olsen DB, Ludmerer SW, Szabo G, Finberg RW, Purcell RH, Lanford RE, Ambrosino DM, Molrine DC, Babcock GJ. 2012. Human monoclonal antibody HCV1 effectively prevents and treats HCV infection in chimpanzees. PLoS Pathog 8:e1002895. doi: 10.1371/journal.ppat.1002895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pestka JM, Zeisel MB, Blaser E, Schurmann P, Bartosch B, Cosset FL, Patel AH, Meisel H, Baumert J, Viazov S, Rispeter K, Blum HE, Roggendorf M, Baumert TF. 2007. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc Natl Acad Sci U S A 104:6025–6030. doi: 10.1073/pnas.0607026104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Osburn WO, Snider AE, Wells BL, Latanich R, Bailey JR, Thomas DL, Cox AL, Ray SC. 2014. Clearance of hepatitis C infection is associated with the early appearance of broad neutralizing antibody responses. Hepatology 59:2140–2151. doi: 10.1002/hep.27013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.von Hahn T, Yoon JC, Alter H, Rice CM, Rehermann B, Balfe P, McKeating JA. 2007. Hepatitis C virus continuously escapes from neutralizing antibody and T-cell responses during chronic infection in vivo. Gastroenterology 132:667–678. doi: 10.1053/j.gastro.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 35.Logvinoff C, Major ME, Oldach D, Heyward S, Talal A, Balfe P, Feinstone SM, Alter H, Rice CM, McKeating JA. 2004. Neutralizing antibody response during acute and chronic hepatitis C virus infection. Proc Natl Acad Sci U S A 101:10149–10154. doi: 10.1073/pnas.0403519101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carlsen TH, Pedersen J, Prentoe JC, Giang E, Keck ZY, Mikkelsen LS, Law M, Foung SK, Bukh J. 2014. Breadth of neutralization and synergy of clinically relevant human monoclonal antibodies against HCV genotypes 1a, 1b, 2a, 2b, 2c, and 3a. Hepatology 60:1551–1562. doi: 10.1002/hep.27298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fofana I, Fafi-Kremer S, Carolla P, Fauvelle C, Zahid MN, Turek M, Heydmann L, Cury K, Hayer J, Combet C, Cosset FL, Pietschmann T, Hiet MS, Bartenschlager R, Habersetzer F, Doffoel M, Keck ZY, Foung SK, Zeisel MB, Stoll-Keller F, Baumert TF. 2012. Mutations that alter use of hepatitis C virus cell entry factors mediate escape from neutralizing antibodies. Gastroenterology 143:223–233.e9. doi: 10.1053/j.gastro.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bailey JR, Wasilewski LN, Snider AE, El-Diwany R, Osburn WO, Keck Z, Foung SK, Ray SC. 2015. Naturally selected hepatitis C virus polymorphisms confer broad neutralizing antibody resistance. J Clin Invest 125:437–447. doi: 10.1172/JCI78794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Munshaw S, Bailey JR, Liu L, Osburn WO, Burke KP, Cox AL, Ray SC. 2012. Computational reconstruction of Bole1a, a representative synthetic hepatitis C virus subtype 1a genome. J Virol 86:5915–5921. doi: 10.1128/JVI.05959-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burke KP, Munshaw S, Osburn WO, Levine J, Liu L, Sidney J, Sette A, Ray SC, Cox AL. 2012. Immunogenicity and cross-reactivity of a representative ancestral sequence in hepatitis C virus infection. J Immunol 188:5177–5188. doi: 10.4049/jimmunol.1103008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zibbell JE, Iqbal K, Patel RC, Suryaprasad A, Sanders KJ, Moore-Moravian L, Serrecchia J, Blankenship S, Ward JW, Holtzman D, Centers for Disease Control and Prevention (CDC). 2015. Increases in hepatitis C virus infection related to injection drug use among persons aged ≤30 years—Kentucky, Tennessee, Virginia, and West Virginia, 2006–2012. MMWR Morb Mortal Wkly Rep 64:453–458. [PMC free article] [PubMed] [Google Scholar]
- 42.Cox AL, Netski DM, Mosbruger T, Sherman SG, Strathdee S, Ompad D, Vlahov D, Chien D, Shyamala V, Ray SC, Thomas DL. 2005. Prospective evaluation of community-acquired acute-phase hepatitis C virus infection. Clin Infect Dis 40:951–958. doi: 10.1086/428578. [DOI] [PubMed] [Google Scholar]
- 43.Hsu M, Zhang J, Flint M, Logvinoff C, Cheng-Mayer C, Rice CM, McKeating JA. 2003. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc Natl Acad Sci U S A 100:7271–7276. doi: 10.1073/pnas.0832180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bailey JR, Dowd KA, Snider AE, Osburn WO, Mehta SH, Kirk GD, Thomas DL, Ray SC. 2015. CD4+ T-cell-dependent reduction in hepatitis C virus-specific neutralizing antibody responses after coinfection with human immunodeficiency virus. J Infect Dis 212:914–923. doi: 10.1093/infdis/jiv139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scheel TK, Gottwein JM, Jensen TB, Prentoe JC, Hoegh AM, Alter HJ, Eugen-Olsen J, Bukh J. 2008. Development of JFH1-based cell culture systems for hepatitis C virus genotype 4a and evidence for cross-genotype neutralization. Proc Natl Acad Sci U S A 105:997–1002. doi: 10.1073/pnas.0711044105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McCaffrey K, Boo I, Poumbourios P, Drummer HE. 2007. Expression and characterization of a minimal hepatitis C virus glycoprotein E2 core domain that retains CD81 binding. J Virol 81:9584–9590. doi: 10.1128/JVI.02782-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deng L, Ma L, Virata-Theimer ML, Zhong L, Yan H, Zhao Z, Struble E, Feinstone S, Alter H, Zhang P. 2014. Discrete conformations of epitope II on the hepatitis C virus E2 protein for antibody-mediated neutralization and nonneutralization. Proc Natl Acad Sci U S A 111:10690–10695. doi: 10.1073/pnas.1411317111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meola A, Tarr AW, England P, Meredith LW, McClure CP, Foung SK, McKeating JA, Ball JK, Rey FA, Krey T. 2015. Structural flexibility of a conserved antigenic region in hepatitis C virus glycoprotein E2 recognized by broadly neutralizing antibodies. J Virol 89:2170–2181. doi: 10.1128/JVI.02190-14. [DOI] [PMC free article] [PubMed] [Google Scholar]