
Fig. 4. MAF bias correction validation with spike-ins shows highly accurate clonal frequencies.
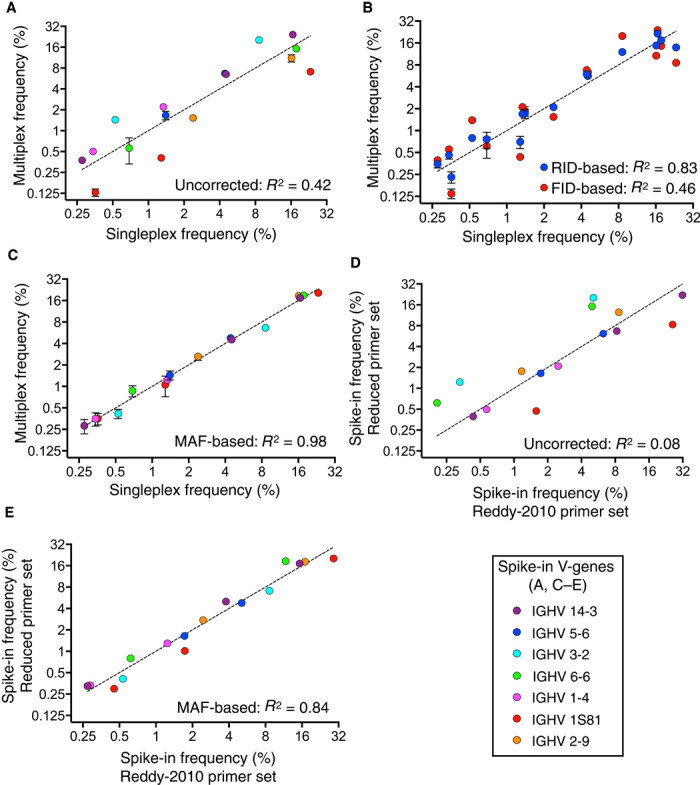
(A) Correlation of uncorrected spike-in clonal frequencies from multiplex PCR (with new reduced primer set) with singleplex PCR results in an R2 = 0.42. (B) Correlation of spike-in clonal frequencies based on FID counting or clonal frequencies based on RID counting with singleplex PCR. Data show that FID residuals are always larger than RID residuals. (C) Correlation of MAF bias corrected spike-in clonal frequencies from multiplex PCR with singleplex PCR results in a significantly improved R2 = 0.98. MAF bias–corrected counts were based on normalized RIDcount and MAF bias factor (see Fig. 2C). (D) Correlation of uncorrected spike-in clonal frequencies using two different multiplex PCR primer sets during library preparation results in an R2 = 0.08. (E) MAF-corrected spike-in clonal frequencies using two different multiplex PCR primer sets result in a significantly improved R2 = 0.84. The Ig-seq data sets used in this figure consisted of 1 × 106 preprocessed full-length antibody reads and were obtained from replicate library sample preparations (n = 3) from mouse splenic cDNA with synthetic spike-ins [for (A) to (C), data are presented as means ± SD and are from replicate data sets IM_1a, IM_1b, and IM_1c, see table S7; data sets Reddy-PS-Compare and TAK-PS-Compare were used for (D) and (E); see table S2]. Singleplex spike-in frequencies are mean values obtained from replicate libraries (n = 5) generated by singleplex PCR (see fig. S2 and table S1).