

Abstract
AIM: To explore a rapid and easy sequencing method for hepatitis C virus (HCV) genome and establish a new sequencing method in China.
METHODS: Polymerase chain reaction (PCR) was combined with a DNA sequencing technique. PCR products were purified by agarose gel electrophoresis, polyacrylamide gel electrophoresis (PAGE) and polyethylene glycol (PEG) respectively. Then, in the presence of a 5’ labeling PCR primer, purified PCR products were directly sequenced. By this method, HCV NS5b cDNA from two HCV infected individuals (HC-42 and HC-49) were sequenced.
RESULTS: PCR directed sequencing worked best using PCR amplified DNA purified by electrophoresis as a sequencing template. When sequencing a large number of templates, the purification step can be bypassed by using a lower concentration of dNTPs (40 μmol of each dNTP) and primers (10 pmol of each primer) in the first stage of PCR. The aliquot of the first stage of PCR mixture was then directly used for amplification of chain terminated products but the sequencing ladders generated were of low intensity. Polyethylene glycol (PEG) could not remove nonspecific products of PCR, which affected the sequencing result to a certain extent and generated a background in sequencing ladders. Compared with the reported HCVJ and HC-C2, a new three nucleotide deletion was found in HC-42.
CONCLUSION: PCR directed sequencing is a rapid, simple and effective method, especially for sequencing large samples. A three nucleotide deletion was first reported.
Keywords: Hepatitis C virus DNA, Viral DNA, Complementary polymerase chain reaction, Sequence analysis, DNA mutation
INTRODUCTION
Hepatitis C virus (HCV) is a major causative agent of post transfusion non A and non B hepatitis and contains a positive stranded RNA molecule. Sequence analysis of the HCV genome plays an important role in the research on viral features, pathogenesis of chronic hepatitis C and discovery of a new genotype/subgenotype of HCV[1]. Traditional sequencing needs cloning which is time consuming and usually takes a lot of work. Recently, a novel DNA sequencing method, direct sequencing of polymerase chain reaction (PCR) amplified DNA, was described[2]. As a sequencing template, PCR amplified DNA was directly sequenced by a thermostable DNA polymerase from Thermus flavus in this method. In order to explore the possibility of sequencing HCV cDNA by this method and establish a new sequencing method in China, we purified and sequenced PCR amplified HCV NS5b cDNA. In the present study, this technique was optimized in ways so that it could be directly applicable to sequence a 376 bp fragment from patients with chronic hepatitis C. PCR direct sequencing was found to be easy, rapid and bypasses construction of the HCV cDNA clones. We believe that PCR direct sequencing is a practical method that would work well in research on the HCV genome and its variation.
MATERIALS AND METHODS
Serum samples
Two sera were collected from 2 patients with chronic HCV infection. All of them were positive for anti HCV (2nd generation EIA, Institute of Hepatology, Beijing Medical University), containing HCV RNA detectable by the nested PCR method with two pairs of external and internal primers deduced from the 5’non coding region[3].
Primers for PCR and sequencing
Primer no.51 (5’-GAGTTCCTGGTGAATACCTG-3’; nt 8189 to 8208, sense) and no.52 (5’-GATGTTATCAGCTCCAGGTC-3’; nt 8661 to 8680, antisense) were external primers. Primers no.53 (5’-CACCCGCTGTTTTGACTC-3’; nt 8246 to 8263, sense) and no.54 (5’-CCTAGTCATAGCCTCGGTC-3’; nt 8603 to 8621, antisense) were used as PCR primers as well as sequencing primers. All four primers were deduced from the putative nonstructural gene (NS) 5 b of the HCV-J isolate[4] and HC C2 isolate[5].
Preparation of nucleic acid, cDNA synthesis and PCR
RNA from 50 μL serum was extracted by the acid guanidium phenol/chloroform method. RNA obtained was denatured at 70 °C for 1 minute, chilled quickly on ice and used as a template for cDNA synthesis. Reverse transcription was carried out at 43 °C for 60 min in 10 μL of Tris HCl buffer (50 mmol/L, pH8.4) containing 8 mmol/L MgCl2, 30 mmol/L KCl, 1 mmol/L dithiothreitol, 50 pmol or 10 pmol primer no 52, 10 mmol/L each of the four deoxyribonucleoside triphosphate (dNTPs) (Promega Corporation), 10 units of RNase inhibitor (Promega Corporation) and 2 units avian myeloblastosis virus reverse transcriptase (Promega Corporation). The cDNA was amplified in two stages in a DNA thermal cycler (Perkin Elmer Cetus, United States). Both stages were carried out in a final volume of 50 μL containing 67 mmol/L Tris HCl, pH8.8, 1.5 mmol/L MgCl2, 1.5 units of Taq DNA polymerase (Promega Corporation), 2 or 10 mmol/L each of four dNTPs for the first stage, 10 mmol/L each of four dNTPs for the second stage, 10 or 50 pmol of each external primer for the first stage and 50 pmol of each internal primer for second stage). Both stages consisted of 35 cycles. In the first stage of PCR, each reaction cycle involved denaturation at 90 °C for 1 min, annealing of primers at 42 °C for 1.5 min and extension at 72 °C for 2 min. In the second stage of PCR, the program of the thermal cycle was similar to the first stage except that the annealing of primers was performed at 55 °C. Samples from the reaction mixture were subjected to electrophoresis on polyacrylamide gel (Promega Corporation) and DNA species on gel were stained with ethidium bromide for observation under ultraviolet light. The sizes of PCR products were estimated according to the migration pattern of pBR322 Hae III.
Purification of PCR products
Purification by electrophoresis in agarose gel PCR products were subjected to 2% agarose gel electrophoresis. The gel containing the positive band was cut off, wrapped up with PARA film (American National Can) and then stored at -70 °C for 30 min. After being eluted from the gel, the liquid was removed to a fresh tube, extracted by phenol/chloroform and precipitated by ethanol. At last, the precipitation was dissolved in ddH2O for sequencing.
Purification by PAG (polyacrylamide gel) electrophoresis PCR products were subjected to 6% PAG electrophoresis. The gel containing the positive band was cut off and immersed in the elution solution containing 0.5 mmol/L NH-4Ac and 1 mmol/L EDTA at 37 °C for 24 h. After this, the elution was treated as described above.
Purification by PEG (Polyethylene glycol) precipitation PCR products were purified according to the reported method[6].
Sequencing method[7]
The primer labeling Two internal primers were respectively used as sequencing primers in both directions. Ten pmol of sequencing primer, 50 μCi (r-32P) ATP (Ya Hui Company), 1 μL of 10 × kinase buffer (70 mmol/L Tris HCl, pH7.5, 10 mmol/L MgCl2, 5 mmol/L dithiothreitol) and H2O to a final reaction volume of 10 μL were mixed, then 5 units of T4 polynucleotide kinase was added. The mixture was incubated at 37 °C for 30 min.
Sequencing Two μL of each of the four dNTP/ddNTP (Boehringer Mannheim) extension termination mixtures were added to four tubes. In another fresh tube, purified PCR amplified DNA, 1.66 pmol of 5’-32P-labeled sequencing primer, 5 μL of 5 × sequencing buffer and 5 units of Taq polymerase were mixed in a total reaction volume of 16 μL. Then 4 μL of the above mixture was transferred to each of the four tubes containing the dNTP/ddNTP mixture. At last, the tubes were placed in a thermal cycler that was preheated to 95 °C. PCR involving 30 cycles was performed. Each cycle included denaturation at 95 °C for 45 s, primer annealing at 55 °C for 45 s and primer extension at 70 °C for 1 min. After completion of thermal cycling, 3 μL of stop solution (10 mmol/L NaOH, 95% formamide, 0.05% bromophenol blue and 0.05% xylene cyanol) were added to each reaction tube.
RESULTS
Amplification of HCV cDNA by reverse transcription nested PCR
HCV cDNA was detected in two sera samples from patients with chronic HCV infection. The sizes of PCR products estimated in electrophoresis gel were about 376 bp as predicted.
Effect of different methods recovering a template on sequencing
First stage PCR products with lower concentration dNTP (2 mmol/L each of four dNTPs) and primers (10 pmol of each external primer) and second stage PCR products were used as a sequencing template. PCR products purified by PEG precipitation, electrophoresis in agarose gel and PAG were sequenced respectively. The sequencing ladders are shown in Figure 1.
Figure 1.
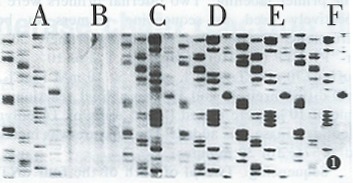
Effect of different methods recovering a template on sequencing. A: The first stage PCR products with lower concentration dNTPs and primers as a sequencing template; B: The second stage PCR products directly sequenced without purification; C: Sequencing of PCR amplified cDNA recovered by PEG; D: PCR products purified by agarose gel electrophoresis before sequencing; E:Sequencing of PCR products purified by PAG electrophoresis; F: Sequencing ladders of PCR products purified by agarose gel electrophoresis but without phenol/chloroform extraction.
Sequence analysis of HCV NS5b cDNA
By the method described above, HCV NS5B cDNA from two patients with chronic HCV infection (HC-42 and HC-C11) were sequenced. Compared with HCV-J, 91.84% and 92.63% of nucleotide’s homology, 91.84% and 95.79% with HC-C2, were found in HC-42 and HC-C11, respectively. A three nucleotide deletion occurred in HC-42 (Figure 2) and this deletion was reported for the first time.
Figure 2.
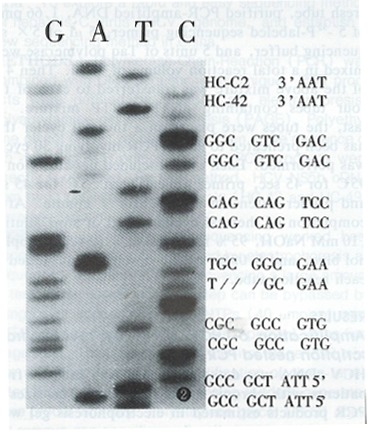
Deletion mutation in NS5B region of HC-42 and comparison with HC-C2. “/” denotes deletion.
DISCUSSION
The first consideration about PCR directed sequencing is the presence of unused dNTPs and primers in the PCR products. Typically, about 200 μmol of each dNTP are used in PCR reaction, whereas the sequencing reactions are routinely performed in the presence of around 10 μmol of each dNTP and a 10.100-fold higher concentration of appropriate ddNTP depending on the polymerase used. In fact, only about 10% of the dNTPs are consumed in PCR. Therefore, about 90% of the dNTPs added to the PCR mixture would remain unused at the end of amplification. Unless these are removed, the routine sequencing reaction would fail to generate sequence ladders because of negligible ddNTP terminations. The presence of unused primers is another problem since approximately 90% of the primers from the PCR amplification would remain unused, which would consume the polymerase as well as the ddNTP and dNTPs in the sequencing reaction[2,8]. Because of these factors, sequencing with unpurified second stage PCR products is unable to obtain sequencing ladders (Figure 1B). Non specific PCR products are also an important factor affecting sequencing. Since PEG could precipitate large molecular weight nucleotides and low molecular weight primers and dents would remain in the supernants, PEG was reported to be used to recover the PCR products[6]. However, PCR amplification produced expected DNA as well as some nonspecific products which were precipitated as expected DNA in PEG buffer. So PEG recovering PCR products could contain unremoved nonspecific products which would interfere with sequencing to some extent. With this as a sequencing template, sequencing ladders generated were not clear and had a dark background (Figure 1C). Purification by electrophoresis could remove unused primers and dNTPs. Moreover, this method could separate expected DNA fragments from nonspecific products. Because of this, sequencing with PCR products purified by electrophoresis worked best (Figure 1D and Figure 1E). Of course, extracting with phenol/chloroform and washing with 70% ethanol after electrophoresis were also needed (Figure 1F).
When sequencing a large number of templates, the purification step was heavy and complicated. Two strategies have been recommended to bypass this step[2]. In one strategy, lower concentrations of dNTPs (10-20 μmol of each dNTP) and primers (10 pmol of each primer) are used to amplify target DNA. As a result, the carryover of unused dNTPs and primers into the sequencing reactions is minimized. Alternatively, the target DNA is first amplified for 25 cycles and then an aliquot of the first PCR mixture is used for amplification of chain terminated products in the presence of a 5’ labeled sequencing primer positioned internally to the external primer and an appropriate ddNTP; this strategy is similar to semi nested PCR. Considering nested PCR is needed to detect HCV RNA, two strategies described above were combined in the present study. We used 40 μmol/L of each dNTPs and 10 pmol of each primer in the first stage PCR and the aliquot of the first PCR was then used as a sequencing template directly. By this method, the sequencing ladders could be read, although the overall quality of the sequence ladder was not as good as with purified second PCR products (Figure 1A).
Direct sequencing of PCR amplified DNA can lead to incorporation of errors in the DNA sequence due to Taq polymerase lacking proofreading 3’-5’exonuclease. Various estimates of misincorporation frequencies range from one error in 4000 nucleotides to one error in 400 nucleotides of the PCR amplified DNA[9]. Therefore, depending on the PCR conditions performed, it is possible that almost every molecule in a 1 kb size PCR amplified DNA could have one error[8]. The question is whether direct sequencing of the mutated PCR amplified DNA would result in the incorporation of errors in the DNA sequence generated. The answer to this question is that the copy number of starting DNA is in the order of 103-105 molecules in most PCR amplifications. Consequently, any errors incorporated in each cycle reaction are randomized and therefore any specific mutant sequences constitute only a minute fraction of the final product. Since the DNA sequence generated is a consensus sequence of millions of template DNA molecules, these errors would not be accounted for in the final sequence. In addition, using a lower concentration of dNTPs and primers in the first stage PCR experiment could also reduce the possibility of misincorporation. HC-42 sequences were sequenced from three times individual PCR products with three sequences the same, as with HC-C11. The above results suggested that sequences generated from PCR direct sequencing are reliable.
A three nucleotide deletion was reported for the first time in this paper. It was found that a deletion mutation in a HCV sequence often occurs at the NS5 region[10], as this deletion did in the present study. Since three nucleotides were deleted, the mutation did not result in significant codon changes. Further investigations and research are needed to elucidate the significance and epidemiology of this deletion.
The diversity of the HCV genome has been found to be related to the progression of liver disease[11]. Therefore, the variation of HCV awaits further studies to clarify the relationship so a rapid and easy sequencing technique is needed. PCR direct sequencing would be a very advantageous method to meet this need.
Footnotes
Original title: China National Journal of New Gastroenterology (1995-1997) renamed World Journal of Gastroenterology (1998-)
Supported by The China Medical Board of America (No.93-582)
S- Editor: Ma JY L- Editor: Ma JY E- Editor: Liu WX
References
- 1.Simmonds P. Variability of hepatitis C virus. Hepatology. 1995;21:570–583. doi: 10.1002/hep.1840210243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Du SC, Tao QM, Sun Y, Chen P, Xu XD, Liu JX. Detection of hepatitis C virus RNA with nested PCR (in Chinese with English abstract) Beijing Medical University. 1991;23:429–431. [Google Scholar]
- 3.Rao VB, Saunders NB. A rapid polymerase-chain-reaction-directed sequencing strategy using a thermostable DNA polymerase from Thermus flavus. Gene. 1992;113:17–23. doi: 10.1016/0378-1119(92)90665-c. [DOI] [PubMed] [Google Scholar]
- 4.Kato N, Hijikata M, Ootsuyama Y, Nakagawa M, Ohkoshi S, Sugimura T, Shimotohno K. Molecular cloning of the human hepatitis C virus genome from Japanese patients with non-A, non-B hepatitis. Proc Natl Acad Sci USA. 1990;87:9524–9528. doi: 10.1073/pnas.87.24.9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Y, Okamoto H, Tsuda F, Nagayama R, Tao QM, Mishiro S. Prevalence, genotypes, and an isolate (HC-C2) of hepatitis C virus in Chinese patients with liver disease. J Med Virol. 1993;40:254–260. doi: 10.1002/jmv.1890400316. [DOI] [PubMed] [Google Scholar]
- 6.Rosenthal A, Coutelle O, Craxton M. Large-scale production of DNA sequencing templates by microtitre format PCR. Nucleic Acids Res. 1993;21:173–174. doi: 10.1093/nar/21.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rao VB. Strategies for direct sequencing of PCR-amplified DNA. PCR Methods Appl. 1994;4:S15–S23. doi: 10.1101/gr.4.1.s15. [DOI] [PubMed] [Google Scholar]
- 8.Rao VB. Direct sequencing of polymerase chain reaction-amplified DNA. Anal Biochem. 1994;216:1–14. doi: 10.1006/abio.1994.1001. [DOI] [PubMed] [Google Scholar]
- 9.Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, Mullis KB, Erlich HA. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science. 1988;239:487–491. doi: 10.1126/science.2448875. [DOI] [PubMed] [Google Scholar]
- 10.Martell M, Esteban JI, Quer J, Genescà J, Weiner A, Esteban R, Guardia J, Gómez J. Hepatitis C virus (HCV) circulates as a population of different but closely related genomes: quasispecies nature of HCV genome distribution. J Virol. 1992;66:3225–3229. doi: 10.1128/jvi.66.5.3225-3229.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bukh J, Miller RH, Purcell RH. Genetic heterogeneity of hepatitis C virus: quasispecies and genotypes. Semin Liver Dis. 1995;15:41–63. doi: 10.1055/s-2007-1007262. [DOI] [PubMed] [Google Scholar]