
Figure 6. Synchronized NF-κB dynamics translates into functionally different dynamical patterns of gene expression, each corresponding to distinct pathways.
(A) Clusters 1–6 were obtained by an unsupervised k-means-like clustering from the genome-wide transcription profiling of samples harvested in the experiment shown in Figure 5A. Line colours are indicative of the membership value of each gene (colour scale at the bottom). Three clusters contain genes with increasing expression (1–3) and three with decreasing expression (4–6). On the y-axis, standardized expression profile in arbitrary units (see Materials and methods). (B) Plots show single-gene mRNA traces (median: thick blue line; 85% and 15% intervals, thin blue lines). The time courses can also be fitted using our minimal mathematical model: shown is the median of the single-gene fits (thick red line) and the 85% and 15% intervals (thin red lines). Fittings were performed using the same parameters for the external signal (PS) and the dynamics (PNF-κB) as in Figure 5, but using different gene expression parameters PG for each gene. (C) Top five pathways of hierarchical level 2 and 3 in the Reactome database significantly enriched in each dynamical cluster. Dot sizes are proportional to the percentage of genes in the cluster belonging to that pathway. Dot colours identify the corresponding p-values (p-value < 0.05 is set as threshold). Scale bars on the right. (D, E) Heatmaps shows the degree of overlap at gene level (D) and pathway level (E) between each of the 6 clusters. Colour scale bar on the right. Figure supplements from 1 to 6 are provided.

Figure 6—figure supplement 1. Transcription in cells chronically stimulated with TNF-α. .

Figure 6—figure supplement 2. Mathematical validation of clustering. .

Figure 6—figure supplement 3. Cluster enrichment analysis for NF-κB targets in genes clustered and displayed in Figure 6. .

Figure 6—figure supplement 4. Cluster enrichment analysis for NF-κB targets in genes clustered and displayed in Figure 7 (constant stimulation).

Figure 6—figure supplement 5. Distribution of fitting distances.
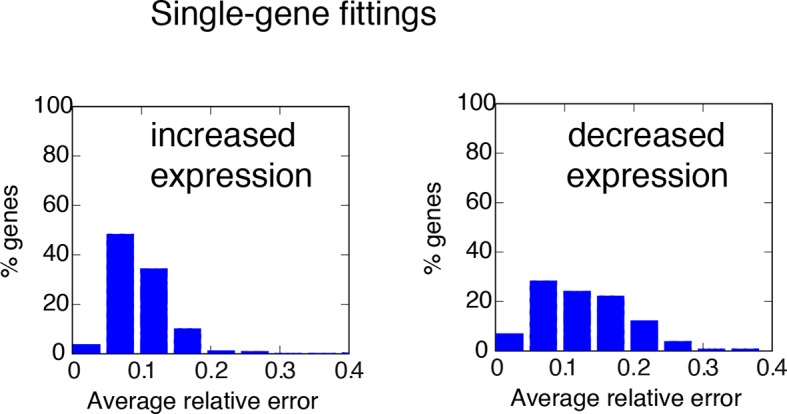
Figure 6—figure supplement 6. Degradation rates values are the key parameter to reproduce different gene expression patterns. .

Figure 6—figure supplement 7. Fitting of transcription data from the 180 min synchronization experiment with an alternative model of transcription.
