

Abstract
Cachexia and muscle wasting are well recognized as common and partly reversible features of chronic obstructive pulmonary disease (COPD), adversely affecting disease progression and prognosis. This argues for integration of weight and muscle maintenance in patient care. In this review, recent insights are presented in the diagnosis of muscle wasting in COPD, the pathophysiology of muscle wasting, and putative mechanisms involved in a disturbed energy balance as cachexia driver. We discuss the therapeutic implications of these new insights for optimizing and personalizing management of COPD‐induced cachexia.
Keywords: COPD, Muscle, Emphysema, Energy balance, Cachexia, Lung cancer
List of abbreviations
- p70S6K
ribosomal protein S6 kinase, 70kDa, polypeptide 1 (PRPS6KB1)
- S6
ribosomal protein S6 (RPS6)
- 4EBP1
eukaryotic translation initiation factor 4E binding protein 1 (EIF4EBP1)
- AEE
activity induced energy expenditure
- AKT
signalling through v‐akt murine thymoma viral oncogene
- AMPK
AMP‐activated protein kinase
- ASM
appendicular skeletal muscle mass
- ATROGIN1
F‐box protein 32 (FBXO32)
- BAT
brown adipose tissue
- BMD
bone mineral density
- BMI
body mass index
- BNIP3
BCL2/ adenovirus E1B 19kDa interacting protein 3
- COPD
chronic obstructive chronic disease
- CT
computed tomography
- DALY
disability‐adjusted life years
- DEXA
dual‐energy X‐ray absorptiometry
- DIT
diet‐induced energy expenditure
- FFM
fat‐free mass
- FFMI
fat‐free mass index
- FM
fat mass
- FOXO
forkhead box O transcription factor
- HIF1‐α
hypoxia inducible factor 1, alpha subunit
- IGF1
insulin‐like growth factor 1
- LC3
microtubule‐associated protein 1 light chain 3 alpha (MAP1LC3A)
- MSTN
myostatin
- MtDNA
mitochondrial DNA
- MTORC1
mechanistic target of rapamycin (serine/threonine kinase) complex 1
- MURF1
tripartite motif containing 63, E3 ubiquitin protein ligase (TRIM63)
- MYOD1
myogenic differentiation 1
- MYOG
myogenin
- NEDD4
neural precursor cell expressed, developmentally downregulated 4, E3 ubiquitin protein
- NF‐κB
nuclear factor kappa‐light‐chain‐enhancer of activated B cells
- NSCLC
non‐small cell lung cancer
- OXPHEN
loss of muscle oxidative phenotype
- P62
sequestosome 1 (SQSTM1)
- PI3K
phosphatidylinositol‐4,5‐bisphosphate 3‐kinase, catalytic subunit alpha (PIK3CA)
- PMA
pectoralis muscle area
- PPARG
peroxisome proliferator‐activated receptor gamma
- REE
resting energy expenditure
- TEE
total energy expenditure
- TUNEL
terminal deoxynucleotidyl transferase
- UCP
uncoupling proteins
- ULK1
unc‐51 like autophagy activating kinase 1
- UPS
ubiquitin proteasome system
- VEGF
endothelial growth factor
- WAT
white adipose tissue
Introduction
Chronic obstructive pulmonary disease (COPD) is one of the leading causes of death worldwide. It has been estimated that COPD‐related mortality rates will even increase in the coming decades. This increase is not only related to the prevalence of smoking, but also ageing and reduced mortality from other common causes of death play a role.1 Additionally, COPD is a major contributor to global disease burden, accounting for 43.3 million disability‐adjusted life years in 2010.2 The disease is characterized by persistent airflow obstruction, resulting from inflammation and remodelling of the airways, and may include development of emphysema. Furthermore, systemic disease manifestations and acute exacerbations influence disease burden and mortality risk.3 Extending the classical descriptions of the ‘pink puffer’ and ‘blue bloater’, recent unbiased statistical approaches4, 5 support the concept that body weight and body composition discriminate pulmonary phenotypes and are predictors of outcome. Extra‐pulmonary degenerative manifestations that may occur in COPD include osteoporosis6 and muscle wasting. The prevalence of muscle wasting is relatively high in COPD: 15–40% depending on definition and disease stage.7, 8 Importantly, muscle wasting not only contributes to diminished skeletal muscle function, reduced exercise capacity, and decreased health status,9, 10 but is also a determinant of mortality in COPD, independent of airflow obstruction.8, 11
Muscle wasting in COPD has been demonstrated by decreases in fat‐free mass (FFM) at whole body level, but also specifically at the level of the extremities.12 Whole body and trunk FFM reduction are more pronounced in the emphysematous phenotype, whereas reduced FFM in extremities is not different between the pulmonary phenotypes.13, 14 In addition, muscle wasting is apparent as a decrease in the size of individual muscle fibres, and this muscle fibre atrophy in COPD seems selective for type II fibres in peripheral muscle,15, 16 which is in line with other chronic diseases prone to cachexia such as chronic heart failure.17 Furthermore, a shift in muscle fibre composition from type I (oxidative) to type II (glycolytic), accompanied by a decrease in oxidative capacity, culminates in reduced muscle endurance.18 This not only contributes to reduced exercise capacity19 but may also affect muscle mass in COPD,20 because type I and II fibres display different responses to anabolic and catabolic signals.21, 22
While unintended weight loss was initially considered to be an indicator of inevitable and terminal progression of the disease process, there is now convincing evidence that it is an independent determinant of survival, arguing for weight maintenance in patient care. There are indications that the pathophysiology of unintended weight loss is different between clinically stable COPD and during acute flare‐ups of the disease. To date, data in acute exacerbations of COPD are, however, very limited. Therefore, lung cancer is used as a comparative acute pulmonary cachexia model.
A recent unbiased statistical approach suggests that not all COPD patients but only the emphysematous phenotype is prone to cachexia,4 although the informative value of available clinical studies is limited by a cross‐sectional study design. The last two decades have also yielded insight in the impairments of the processes governing muscle mass and identified putative triggers of muscle wasting in COPD. However, it remains unclear to what extent acute flare‐ups of COPD may accelerate chronic wasting of muscle mass and whether muscle wasting involves similar mechanisms as in other chronic diseases or in lung cancer cachexia. In this review, we present recent insights in the pathophysiology of muscle wasting in COPD and (putative) mechanisms involved in the pathophysiology of a disturbed energy balance as important driver of cachexia, which may lead to novel targets for clinical management of cachexia in COPD.
Recent developments in identifying muscle wasting in COPD
Incorporation of body composition into nutritional assessment has been a major step forward in understanding systemic COPD pathophysiology, since changes in weight and classification of body mass index (BMI) do not account for (hidden) body compositional shifts in fat mass (FM), FFM, and bone mineral density. In clinical research, bioelectrical impedance analysis is commonly used to identify cachexia. Traditionally, reference values for fat‐free mass index (FFMI) in COPD were developed based on age‐specific and gender‐specific 10th percentile values.8 These reference values were defined as abnormally low, based on well‐established adverse effects of low FFMI on physical performance and survival in normal to underweight COPD patients.7, 11 However, this might underestimate low muscle mass in the increasing proportion of overweight to obese COPD patients.23 The recent European Respiratory Society statement on nutritional assessment and therapy in COPD24 proposed dual‐energy X‐ray absorptiometry (DEXA) as the most appropriate method for body composition analysis in COPD, mostly because it combines screening for osteoporosis with assessment of FM and FFM at the regional level in addition to whole body level. Consequently, body composition assessed by DEXA also allows measurement of appendicular skeletal muscle mass (ASM), which has been demonstrated to be stronger related to physical functioning than total FFM.23 Moreover, we recently showed that this particularly applies to overweight to obese COPD patients.23, 25
Whereas low muscle mass is prevalent in ±15% of well‐functioning elderly in the general aged population,26 a higher prevalence can be expected in COPD as a reflection of accelerated ageing.25 Indeed, Van de Bool et al. recently identified low ASM in even 87% of Dutch COPD patients eligible for pulmonary rehabilitation, along with a high persisting prevalence across all BMI categories.23 The ASM‐wasted phenotype was not only associated with impaired strength but in men also with decreased endurance capacity. Coexistent abdominal obesity was identified in 78% of muscle‐wasted patients, which appeared to have a protective effect on physical functioning.23 Physiological alterations in terms of less hyperinflation and a larger inspiratory capacity in obese COPD patients contribute to certain advantages during physical activity.27 In addition, mortality rates in advanced COPD are the lowest among obese subjects.28 This prognostic advantage typically reflects the ‘obesity paradox’, because obesity, on the other hand, is also associated with increased risk of cardiovascular and metabolic diseases.
Although clinically useful estimates can be derived by DEXA, a more precise distinction between muscle mass, visceral and subcutaneous adipose tissue requires more advanced imaging technologies. This could be relevant in COPD as Van den Borst et al. and Furutate et al. recently reported a higher visceral adipose tissue in older‐age patients with COPD compared with age‐matched healthy controls, despite comparable subcutaneous adipose tissue and BMI.29, 30
McDonald et al.31 recently demonstrated that CT‐derived pectoralis muscle area (PMA) provides a more clinically relevant measure of COPD‐related outcomes in comparison with BMI, as lower PMA was associated with more severe expiratory airflow obstruction, lower quality of life, and impaired exercise capacity. Because gender differences have been documented in body composition and its functional implications,23 Diaz et al. explored gender differences in computed tomography (CT)‐derived PMA and observed lower PMA in women compared with men.32 CT scans are often used to exclude other underlying illnesses, and therefore, chest CT‐derived analysis of body composition may be an attractive diagnostic tool to combine screening for pulmonary and systemic pathology. However, it first needs to be properly validated against reference methods of whole body and regional body composition to allow use in clinical practice. Furthermore, due to the radiation exposure, use of CT scans for body composition assessment is only admissible when scans are already performed to screen for pulmonary pathology.
New insights in the pathophysiology of muscle wasting in chronic obstructive pulmonary disease
The loss of muscle mass and cross sectional area in COPD patients as determined by imaging techniques has been confirmed at the cellular level, that is, a reduction in muscle fibre cross‐sectional area.16 As reviewed by Langen et al. 33 and Remels et al.,20 triggers of muscle wasting include hypoxemia, oxidative stress, inflammation, impaired growth factor signalling, oral glucocorticoids, disuse, and malnutrition, some of which are influenced by smoking.34 Wasting of skeletal muscle is due to a net catabolic state, which may result from an imbalance in muscle protein synthesis and breakdown (protein turnover), as well as from an imbalance in myonuclear accretion and loss (myonuclear turnover).
Protein turnover
To get insight in the rate of (muscle) protein turnover in COPD, information on (muscle) protein synthesis and breakdown is required. Both increased and normal rates of whole body protein turnover have been reported in patients with COPD,35, 36 but the relative contribution of muscle versus other tissues to protein turnover is unknown. Rutten et al. observed an increase in myofibrillar protein breakdown in cachectic COPD patients compared with non‐cachectic patients and controls,35 but no data are available regarding muscle protein synthesis rate, except for a small study showing depressed muscle protein synthesis rates in malnourished patients with emphysema.37 Numerous studies, however, have addressed molecular regulation of anabolic and catabolic pathways in the quadriceps muscle of COPD patients, which provides some insight in altered muscle protein turnover in muscle‐wasted COPD patients.
Proteolytic signalling
Several environmental triggers can lead to catabolic signalling in the skeletal muscle, mediated by transcriptional regulators including nuclear factor kappa‐light‐chain‐enhancer of activated B cells (NF‐κB) and forkhead box O transcription factors (FOXOs). NF‐κB activity is increased in COPD patients compared with controls38, 39 and in cachectic COPD patients compared with non‐cachectic COPD patients.38, 40 Furthermore, limb muscle NF‐κB activity is increased in patients with lung cancer cachexia.41 FOXO mRNA and protein expression are increased in patients with COPD,38, 39, 42, 43, 44, 45 seemingly independent of body composition, although it is noticeable that in all studies, the patient group showed signs of emphysema. In the COPD patient group, FOXO‐1 protein expression was higher in limb muscles than in respiratory muscles, while this difference was not found in controls.46 Interestingly, the respiratory muscles of COPD patients show an opposite fibre type shift compared with limb muscles, that is, towards more type I fibres.47, 48 This will have implications for the expression levels of constituents of atrophy signalling pathways.22, 49 Increased catabolic signalling through FOXO and NF‐κB can induce gene expression of key factors in both the ubiquitin proteasome system (UPS)50, 51 and the autophagy lysosome pathway.33, 52
Ubiquitin proteasome‐mediated degradation
The ubiquitin 26S‐proteasome pathway consists of coordinated actions of the ubiquitin conjugating and ligating enzymes that link ubiquitin chains onto proteins to mark them for degradation by the proteasome.53, 54 These enzymes include tripartite motif containing 63, E3 ubiquitin protein ligase (TRIM63, referred to as MURF1), F‐box protein 32 (FBXO32, referred to as ATROGIN1), and neural precursor cell expressed, developmentally downregulated 4, E3 ubiquitin protein (NEDD4).
MURF1 limb and respiratory muscle mRNA and protein expression appear unaltered in COPD patients compared with controls,38, 40, 55, 56, 57 although one study reported increased MURF1 protein expression in the limb muscles of cachectic COPD patients.39 In COPD patients, MURF1 protein expression is relatively increased in limb muscle than in the respiratory muscle, while this difference was not found in controls.46 Furthermore, cachectic COPD patients show increased limb muscle mRNA expression of MURF1 compared with a control population.44 ATROGIN1 mRNA and protein expression are increased in limb muscles,38, 39, 40, 44, 56, 57 but unaltered in respiratory muscles of patients with COPD.55 Similarly, ATROGIN1 mRNA expression is increased in the limb muscles of smokers.58 Furthermore, Doucet et al. found that COPD patients display a higher ATROGIN1 protein expression in limb muscles than in respiratory muscles, while this difference was not found in controls.46 Additionally, limb muscle NEDD4 protein expression is increased in patients with COPD.57 Total poly‐ubiquitinated protein is increased in limb muscles of COPD patients compared with healthy controls39, 56 and in cachectic COPD patients compared with non‐cachectic COPD patients.38
Taken together, the majority of the literature suggests that wasting in COPD is accompanied by an increase in UPS activation. The increase in catabolic signalling in cachectic COPD patients is site specific. This may reflect disuse atrophy of the limb muscle with maintained or increased respiratory muscle activity, or it may result from an interaction between inactivity and other triggers of atrophy, such as smoking.
Autophagy‐lysosome‐mediated degradation
The autophagy‐lysosome pathway is a protein degradation pathway, which recently gained interest in the context of COPD‐associated muscle dysfunction. Upon activation, autophagosomes form and mature to subsequently fuse with lysosomes. The autophago‐lysosomes degrade the cargo and release amino‐acids for de novo protein synthesis or other metabolic fates.59
Signalling through v‐akt murine thymoma viral oncogene (AKT) regulates mechanistic target of rapamycin (serine/threonine kinase) complex 1 (MTORC1) activity and downstream of MTORC1, unc‐51 like autophagy activating kinase 1 (ULK1) activity, thereby regulating autophagy initiation.60, 61 Inhibitory MTORC1‐mediated ULK1 phosphorylation is decreased in limb muscles of COPD patients compared with controls,45 which may implicate an increase in autophagic flux induction. The increase of FOXO mRNA and protein expression in COPD patients may induce the transcription of autophagy‐related genes. However, it should be taken into account that FOXO transcriptional activity is also regulated by post‐translational modifications. Plant et al. found that the mRNA expression of autophagy‐related genes Beclin‐1 and microtubule‐associated protein 1 light chain 3 alpha (MAP1LC3A, referred to as LC3) is unaltered in the limb muscles of COPD patients compared with controls.57 Limb muscle sequestosome 1 (SQSTM1, referred to as P62) mRNA expression, however, is increased in COPD patients.45 Although the mRNA expression of autophagy‐related genes and the activation of ULK1 may give some insight in the level of autophagy initiation, it does not directly reflect the level of autophagic flux. In muscle biopsies, the conversion of LC3BI to LC3BII can be used as a measure for autophagic flux. Furthermore, P62 is used as a marker for autophagic flux, because it is broken down by the lysosome. In the limb muscles of COPD patients, the LC3BII/I ratio is increased,39, 45 pointing to an increase in the autophagic flux. In contrast, P62 protein expression is increased, pointing to a decrease in autophagic flux.45 However, it cannot be excluded that the increase in P62 protein expression is due to the increase in P62 transcription. The number of autophagosomes was found to be increased in the limb muscle of COPD patients,39, 45 which suggests an increase in autophagic flux. However, it is only possible to speculate on the level of autophagic flux in the limb muscles of patients with COPD based on the currently applied markers, as these incompletely cover autophagic flux, autophagy induction, and autophagic‐lysosomal degradation. Therefore, autophagic flux markers should be analysed coupled to the activity status of upstream regulators such as MTORC1 and AMP‐activated protein kinase (AMPK) in muscle biopsies. Moreover, besides its role in protein breakdown, autophagy also acts as a quality control mechanism for proteins and intracellular components.62, 63 Therefore, an impaired autophagic flux in COPD patients may have consequences for the integrity and function of intracellular components such as the nucleus and mitochondria.
It currently is unknown if the autophagic‐lysosome pathway activity is altered during acute exacerbations of COPD, because most studies were conducted in stable COPD patients. However, in lung cancer cachexia, LC3BII protein expression and BCL2/ adenovirus E1B 19kDa interacting protein 3 (BNIP3) mRNA expression are induced,41 pointing to an increase in autophagy. From this, autophagy induction in skeletal muscle might be anticipated during acute stages of COPD wasting.
Protein synthesis signalling
A major anabolic pathway is the insulin‐like growth factor 1 (IGF1)/phosphatidylinositol‐4,5‐bisphosphate 3‐kinase, catalytic subunit alpha (PIK3CA referred to as PI3K)/AKT pathway. Most studies found an increase in IGF1 mRNA expression in the limb muscle of COPD patients compared with controls,42, 43, 64 although Crul et al. found a decrease in IGF1 mRNA expression in stable COPD patients.65 Unfortunately, this study did not provide body composition data. Cachectic COPD patients seem to have a lower limb muscle IGF1 mRNA and protein expression than non‐cachectic COPD patients.40 Furthermore, during an acute exacerbation, muscle IGF1 mRNA expression is lower in COPD patients than in controls, although IGF1 protein expression remains unaltered.65
Even though IGF1 mRNA expression is increased in limb muscles of COPD patients, AKT activation remains unaltered.39, 45, 57 AKT activity is relatively increased in cachectic patients compared with non‐cachectic patients and healthy controls,40, 44 while the decrease in IGF1 mRNA expression in this group would generally implicate a decrease of IGF1/AKT signalling. Interestingly, an increase in AKT activation is also observed in patients with lung cancer‐related cachexia,41 suggesting it may be a common feature of pulmonary cachexia. The discrepancy in IGF1 mRNA expression and AKT activation suggests altered regulation at the IGF‐receptor or IGF‐receptor protein expression level.66
Signalling through AKT inhibits the upstream inhibitor of MTORC1, thereby inducing MTORC1 activation and subsequent phosphorylation of its downstream targets eukaryotic translation initiation factor 4E‐binding protein 1 (EIF4EBP1, also called 4EBP1) and ribosomal protein S6 kinase, 70kDa, polypeptide 1 (PRPS6KB1, also called p70S6K).67 The increased AKT activation in the limb muscle of cachectic patients compared with non‐cachectic COPD patients is paralleled by an increase in phosphorylation of the downstream targets 4E‐BP1 and p70S6K.44 P70S6K phosphorylation is unaltered in COPD patients compared with controls,44 while ribosomal protein S6 (RPS6 referred to as S6) phosphorylation was even decreased in COPD patients.45 Together, these studies show an increase in protein synthesis signalling in the limb muscles of cachectic COPD patients compared with non‐cachectic COPD patients, but no alteration in the general COPD population. In patients with lung cancer‐related cachexia, AKT activation is increased without concurrent activation of MTOR or its downstream targets.41 This may indicate that, although impaired AKT signalling is found in lung cancer cachexia, AKT signalling is largely intact in COPD‐induced muscle wasting. However, one limitation of these studies concerns the evaluation of the activation status of protein synthesis signalling solely in a basal state. Although the current data may suggest that the protein synthesis pathway is a promising target for the treatment of COPD‐induced muscle wasting, the integrity of the anabolic response should be further addressed.
Only limited data are available on anabolic signalling in respiratory muscles of COPD patients, and although the results also point to an increase in anabolic signalling, it remains unclear if this is different between cachectic and non‐cachectic COPD patients. Martinez‐Llorens et al. found an increase in IGF1 mRNA expression in the intercostal muscles of patients with COPD.68 Doucet et al. compared the ratio of quadriceps to diaphragm AKT activation in COPD patients with controls and found a lower ratio in COPD.20 This implicates that the AKT activation is relatively higher in the diaphragm than in the quadriceps. In line with this, the p70S6K phosphorylation is relatively higher in the diaphragm, while 4E‐BP1 phosphorylation is higher in the quadriceps.20
Interestingly, Tannerstedt et al. showed a difference in anabolic response between type‐I and type‐II muscle fibres, with increased AKT phosphorylation and downstream pathway activation in the type‐II fibres.69 In contrast, in COPD, the respiratory muscle with a shift towards more type‐I fibres displayed a larger AKT activation and downstream signalling than the limb muscles with a shift towards more type‐II fibres. Therefore, the shift in fibre type does not explain the variation in AKT phosphorylation and downstream signalling between the limb muscle and respiratory muscle. Other discriminating factors, such as the muscle activity level, may be more closely linked to differences in AKT phosphorylation.
Taken together, anabolic signalling is increased in the skeletal muscle of patients with COPD, with an even larger increase in the diaphragm than the limb muscles. One may speculate that the increased activation of AKT signalling in the respiratory muscles is an attempt to preserve respiratory function by compensating catabolic triggers, although it may also reflect intrinsic alterations in muscle fibre composition.
Myonuclear turnover
Besides the turnover of proteins, the turnover of myonuclei appears essential for muscle regeneration.70, 71, 72 Furthermore, although at a lower rate, myonuclear turnover might be indispensable for the maintenance of skeletal muscle mass. The regulated loss of a nucleus may involve apoptosis. Increased apoptosis, as determined by an increase in terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) staining and poly (ADP‐ribose) polymerase cleavage in the skeletal muscle of cachectic COPD patients has been reported.73 In the diaphragm of COPD patients with muscle wasting, elevated caspase‐3 levels indicated an increase in apoptosis.55 No difference in apoptosis measured by TUNEL staining or caspase‐3 was found in COPD patients with maintained muscle mass compared with controls.74 A possible alteration in apoptosis in the muscle of COPD patients remains inconclusive due to the lack of studies and the use of different markers. Furthermore, the possible role of apoptotic signalling in the skeletal muscle atrophy remains obscure,75 and multinucleated muscle fibres may utilize other mechanisms for selective myonuclear loss, such as autophagy.76
To replenish the myonuclear pool, satellite cells are essential.72 In contrast to muscle wasting in ageing,77, 78 the number of satellite cells per muscle fibre is unaltered in the limb and respiratory muscles of patients with COPD compared with controls.68, 79, 80, 81 Furthermore, no differences in satellite cell number have been reported between muscle‐wasted and non‐muscle‐wasted COPD patients.80
Upon activation, satellite cells proliferate, differentiate, and fuse with myofibres. Activation and proliferation of satellite cells in the limb muscles does not seem to differ between COPD patients and age‐matched controls based on number of satellite cells 24 h after a resistance‐exercise bout.81 However, molecular markers of satellite cell activation may be more sensitive than satellite cell number to quantify the satellite cell response.
In a basal condition, myogenic factor 5 mRNA expression is unaltered in the limb muscles of patients with COPD compared with controls.57 Myogenic differentiation 1 (MYOD1) mRNA expression40 and protein expression appear similar in the limb muscles of COPD patients and healthy controls.57 Furthermore, Myogenin (MYOG) mRNA expression is unaltered in COPD patients.57 However, in cachectic COPD patients, MYOD140 and MYOG38, 39 protein expression are reduced compared with controls, while no alteration in MYOG protein expression was found in the limb muscles of cachectic compared with non‐cachectic COPD patients.38 It should be considered that in different populations and disease stages, the course of the satellite cell response might be altered, which may have implications for the timing of the measurements.
A negative regulator of myogenesis is myostatin (MSTN).82 Limb muscle MSTN mRNA expression is increased in COPD compared with controls,57, 83 while no difference seems to be present between cachectic and non‐cachectic COPD patients.40 The increased MSTN mRNA expression in COPD patients may be partially explained by smoking status, as this increase is also found in smokers.58 MSTN protein expression is unaltered in the limb muscle of COPD patients, independent of the pulmonary phenotype,38, 39, 40 while the circulatory level of MSTN is increased in COPD compared with control subjects.84 It should be noted that Snijders et al. showed a delayed response in MSTN protein levels upon a single bout of resistance exercise in the elderly, while basal levels did not seem to differ.85 This implies that in the absence of a myogenic trigger, intrinsic alterations in satellite cell plasticity responses in muscle of COPD patients may be masked.
Centrally localized myonuclei in myofibres are considered derived from newly fused satellite cells prior to their final location peripherally in the myofibre against the sarcolemma. COPD patients with preserved muscle mass have higher amounts of central nuclei in the limb muscle than muscle‐wasted COPD patients and controls.80 This could be interpreted as an attempt to counteract atrophic signalling in COPD patients, which may be essential to preserve muscle mass. However, as there is only indirect evidence that central myonuclei reflect recent regenerative events, central nuclei could also reflect an increase in myonuclear turnover to compensate for increased loss of myonuclei.
To gain further insight in the regulation of myonuclear turnover and possible defects in COPD‐induced skeletal muscle wasting, it is essential to incorporate satellite cell activation stimuli and sensitive techniques to monitor myonuclear accretion and turnover in the study design. This will require further development of new techniques, in parallel with novel ex vivo and in vitro approaches to monitor myonuclear accretion and possibly myonuclear loss, and assessment of the role of alterations in myonuclear turnover in muscle atrophy.
Loss of muscle oxidative phenotype
Besides the importance of the muscle quantity for muscle function, the quality of the muscle should also be considered. This is highlighted by the finding that muscle mass‐specific muscle strength and endurance are reduced in patients with COPD.86, 87, 88 A well‐established qualitative alteration in the skeletal muscle of COPD patients is the loss of oxidative phenotype (OXPHEN) characterized by a muscle fibre type I to type II shift and a loss of oxidative capacity.20, 88, 89 The loss of OXPHEN is associated with increased oxidative stress,88, 90 which may render the muscle more susceptible to muscle atrophy.38 In addition, type II fibres are generally more susceptible to atrophy stimuli including, for example, inflammation21 and hypoxia.22 Therefore, the loss of OXPHEN in COPD may accelerate the loss of muscle mass, thereby linking muscle quality to muscle quantity. This is supported by the fact that non‐symptomatic smokers already exhibit reduced mitochondrial capacity and a similar fibre‐type shift.91 Although less extensively investigated, striking similarities are reported regarding muscle oxidative metabolism in chronic heart failure.92 As these patients also share other systemic features and lifestyle characteristics (e.g. muscle wasting and low physical activity level) comparative analyses between well‐phenotyped patients with COPD and chronic heart failure may provide more insight in common and disease‐specific denominators and mechanisms.
Therapeutic perspective
Because muscle wasting may result from alterations in the protein and myonuclear turnover, targeting key pathways in these processes will be required to combat muscle wasting.
Ubiquitin proteasome system activity is increased in the muscles of cachectic COPD patients, which implicates the atrogenes MURF1 and ATROGIN1 as targets to normalize UPS activity. This is supported by the finding that in a cell culture model and in a mouse model of muscle disuse, MURF1 inhibition and knockout, respectively, prevented muscle fibre atrophy.93, 94 Pharmacological inhibitors that target specific ubiquitin‐conjugating and deconjugating enzymes are being developed to treat cancer, neurodegenerative disorders, and autoimmune diseases95 but may also be highly relevant for the treatment of COPD‐induced muscle wasting. Furthermore, exercise training may attenuate MURF1 expression, as was observed in the skeletal muscle of chronic heart failure patients.50, 96 In contrast to exercise training, one bout of exercise leads to an increase in MURF1 expression, albeit blunted in COPD,97, 98 while the increase in proteolytic signalling is reduced by branched‐chain amino acid supplementation in a healthy population.97
Autophagy is disturbed in patients with COPD, although it remains unclear whether there is an increased induction of autophagy or an inhibition of autophagic‐lysosomal degradation. Low amino acid availability can activate autophagy by inhibition of MTOR.99 In line with this, branched‐chain amino acid supplementation leads to an inhibition of autophagy by activation of MTOR.100 Furthermore, overall low energy status, DNA damage, and hypoxia can inhibit MTOR through AMPK and hypoxia inducible factor 1, alpha subunit (HIF1‐α)101, 102 and thereby induce autophagy. Interestingly, exercise targets these factors, and exercise training results in elevated levels of basal autophagy.103 However, exercise leads to an increase in muscle mass and strength in COPD patients.98 Moreover, autophagy is required for muscle adaptations to training.103, 104 The counterintuitive effect of exercise on autophagy may therefore be more tightly linked to its function in quality control.
A more speculative thought is that autophagy may play a role in selective removal of damaged myonuclei. Besides causing mitochondrial DNA (mtDNA) damage, oxidative stress also causes nuclear DNA damage. COPD patients display elevated levels of oxidative stress, which may lead to increased DNA damage and requires increased removal of damaged nuclei. Although exercise induces oxidative stress, which is even accentuated in COPD patients,105 exercise also triggers myogenesis.106 In COPD, this myogenic response to exercise seems intact, although it is specifically impaired in cachectic COPD.40, 98 Exercise‐induced satellite cell activation is mediated by IGF signalling.107 In contrast, MSTN signalling inhibits satellite cell activation.108 Because MSTN expression is increased in COPD, pharmacological inhibition of MSTN might be beneficial to prevent COPD‐induced muscle wasting. This idea is supported by the finding that in mice with chronic kidney failure, pharmacological inhibition of MSTN blocks muscle atrophy,109 and that pharmacological inhibition of the ActII‐receptor, which mediates MSTN signalling, prevents glucocorticoid‐induced muscle atrophy.110 In vitro, glutamine reduced the tumour necrosis factor alpha‐dependent increase of MSTN.111 This implicates that availability of amino acids is important for normal satellite cell function in COPD and that restoration of normal amino acid levels may be required for muscle maintenance. Taken together, the dual function and differential regulation of UPS and autophagy in the maintenance of muscle mass and quality reflects a highly interactive signalling network that is regulated by several upstream pathways. The effect of specific modulation of UPS and autophagy mediators may therefore transcend catabolic signalling and may affect a range of other cellular processes, yielding it difficult to predict long‐term side effects. So far, exercise seems to be the only intervention that can target UPS and autophagy leading to improved quantity, as well as an improved quality of the muscle in COPD patients. One prerequisite is that COPD patients, and specifically cachectic COPD patients, have maintained responsiveness to exercise stimuli, which remains to be established. Exercise capacity in COPD may be limited by impaired pulmonary function, leading to incapability to supply a sufficiently strong exercise trigger to the muscles. In this case, pharmacological or nutritional activators of AMPK, sirtuin 1, and peroxisome proliferator‐activated receptors such as metformin, resveratrol, rosglitazone, and polyunsaturated fatty acids could be used as exercise mimetics and may help sensitize the muscle to a following exercise bout. Furthermore, anabolic steroids could be considered in the treatment of COPD‐induced muscle wasting, although a recent meta‐analysis showed that exercise capacity of COPD patients was not improved.112 It should also be considered that an appropriate nutritional status is necessary for the beneficial effects of exercise and that exercise (in particular, endurance type of exercise) in a malnourished state could even have detrimental effects by worsening the energy imbalance. Taken together, a multi‐modal approach may be required to combat COPD‐induced muscle wasting, in which exercise training is central. However, to establish such intervention, further research is crucial to determine whether the response to exercise is intact or if specific defects occur, in cachectic patients with COPD.
Putative mechanisms involved in a disturbed energy balance in COPD
Specific loss of muscle mass in weight‐stable COPD patients has been observed, which may reflect a tissue‐specific sensitivity to an overall catabolic state. A net catabolic state may also result from an imbalance in energy expenditure and energy availability (energy balance).
Increased energy expenditure
It is well established that components of whole body energy expenditure may be increased in patients with COPD.113 Total daily energy expenditure (TEE) is the sum of resting energy expenditure (REE), activity‐induced energy expenditure (AEE), and diet‐induced thermogenesis (DIT). Assessment of TEE requires sophisticated methodology including a respiration chamber114 or doubly labelled water to allow assessment of TEE in free living conditions.115, 116 Data on TEE in COPD are scarce and sometimes contradicting, which may be related to the use of different methodology or patient characteristics. Based on the available evidence, it seems that in particular the emphysematous phenotype is prone to increased TEE, as high values are more often observed in patients with low carbon monoxide diffusing capacity.117, 118, 119 This is in line with a positive effect of lung volume‐reduction surgery on body weight in emphysema, although surgical intervention might not only decrease energy requirements but also improve dietary intake by alleviating dyspnoea.120 More research is needed to assess putative differences in whole‐body energy metabolism and its components among COPD phenotypes under comparable standardized circumstances.
Numerous studies have shown that REE is raised.121, 122, 123 This is more prevalent in emphysema124, 125 during acute exacerbations,126 and appears inversely correlated with forced expiratory volume in 1 s when comparing different studies.118, 119, 122, 127 Highest values are found among weight‐losing patients.122 This is in contrast with non‐pathology‐induced malnutrition, where subjects with low BMI have lower REE due to hypometabolic adjustments.128 The same results are found for non‐small‐cell lung cancer (NSCLC), where REE is found to be unregulated in 74% of primary lung cancer patients.129, 130, 131 Hypermetabolism at rest was also found to be more pronounced in weight‐losing compared with weight‐stable lung cancer patients.132 Thus, increased REE is a consistent feature of chronic and more acute cachexia and seems to be more pronounced in the emphysematous subtype.
Activity‐induced energy expenditure is the most variable component of TEE, and it has been postulated that COPD patients reduce physical activity to compensate for dyspnoea severity or to anticipate to breathlessness. Indeed, lower physical activity levels are seen in COPD.133 Physical inactivity is associated with advanced disease stage,134, 135, 136 exacerbations,137, 138 and degree of emphysema.139 In addition, lung volume‐reduction surgery in patients with severe emphysema improved exercise performance due to reduced lung hyperinflation, less dyspnoea severity, and less cost of breathing.140 However, it did not cause augmentation of physical activity level, implying other factors play a role, including motivation or anxiety.141 There are several indications that when COPD patients perform physical activities, they require more energy. For example, Lahaije et al. found a higher daily activity‐related oxygen consumption assessed by a face mask measuring ventilatory and metabolic demand in COPD patients compared with healthy controls,142 while Vaes et al. found an increase in FFM adjusted oxygen consumption in COPDpatients compared with controls, although total oxygen consumption was not altered.143 Indirect evidence for altered AEE is a rise in plasma ammonia in COPD patients during low intensity walking, which is an indicator of muscle ATP depletion and metabolic stress.144 These collective data may indicate that COPD patients use oxygen less efficiently and exhibit an altered energy metabolism during physical activity. This is not surprising in view of the shift in lower limb muscle fibre type composition in COPD towards less oxidative fibres, which appears to be more pronounced in the emphysematous phenotype.18 The opposite shift in muscle fibre type of the diaphragm relative to the limb muscle47, 48 indicates an adaptation to chronic increase in work of breathing. Together with hyperinflation‐induced mechanical inefficiency, this muscle fibre type shift could contribute to enhanced oxygen cost of breathing, illustrated by the effects of lung volume‐reduction surgery145 and non‐invasive positive‐pressure ventilation therapy.146 For comparison, in lung cancer, physical activity level assessed by accelerometry is also reduced,147, 148 but no specific data about AEE.
Diet‐induced thermogenesis represents metabolic oxygen cost for processing of ingested nutrients. Green and colleagues125 indeed reported enhanced DIT in emphysematous COPD patients, but this was not confirmed by other authors,149, 150 independently of BMI.151 These differences may be due to different test meal composition and portion size. Although oxygen desaturation during meals was noticed in severe COPD patients,152 it is unknown whether this is DIT related or not. Therefore, the thermic effect of dietary intake remains unclear. Taken together, it indicates that energy requirements are increased in COPD, and there is certainly no adaptive reduced energy demand.
In addition to the hypermetabolic state, early clinical trials have shown that enhanced systemic inflammation is a contributing factor to elevated REE, both in COPD153 and in lung cancer,154 the source of which is yet unclear. Besides pulmonary inflammation,155 adipose tissue has also been suggested to contribute to a higher inflammatory gene expression in adipose tissue, as has been reported in malnourished patients with advanced COPD.156
Adipose tissue metabolism
In cachexia, muscle wasting is accompanied by loss of adipose tissue.14, 157 In fact, in cancer‐induced cachexia, adipose tissue is often one of the first affected organs, illustrated by decreasing fat cell volume and upregulation of fatty acid metabolism.158 Regarding COPD, low BMI11 and fat mass depletion14 particularly occur in those with advanced disease and in the emphysematous phenotype.
Schols et al. observed low leptin levels in the blood of patients with emphysema compared with chronic bronchitis in line with a lower BMI and fat mass.159 After adjustment for FM and oral corticosteroid use as possible confounders, leptin was associated with systemic inflammation, in particular in the emphysematous patients. More recently, Brusik et al. investigated serum levels and adipose tissue expression of leptin and adiponectin in patients with COPD and reported an association between decreased serum and tissue leptin levels, and decreased serum adiponectin and increased REE adjusted for body weight in underweight patients.160 In adipose tissue, two cell types can be distinguished: white adipose tissue (WAT) and brown adipose tissue (BAT). Brown adipose tissue is differentiated from WAT by the presence of cold and diet‐induced thermogenesis. Thermogenesis is facilitated by BAT‐specific uncoupling proteins (UCP) that dissipate the proton gradient in mitochondria in order to generate heat.161 High amounts of mitochondria and high vascularisation are responsible for the brown colour of BAT.162 Additionally, WAT can be converted in BAT, called WAT browning.163 BAT activation negatively correlates with BMI, as demonstrated by decreased BAT activation in obese subjects164 and during ageing.165 Cachexia on the other hand is characterized by fat mass depletion. This raises the question whether there is a role of BAT activity in the hypermetabolic state, as seen in pulmonary cachexia.
No studies are performed to determine BAT activity in COPD patients. With respect to lung cancer, results are conflicting and scarce. Despite negative results of BAT activation reported by some authors,166, 167 Shellock and colleagues provided evidence for BAT activation as a cachexia mediator. Autopsy reports of cachectic cancer patients revealed high incidence of BAT in this group compared with age‐matched controls.168 Furthermore, a correlation between BAT activity and neoplastic status has been suggested,169 although the authors also reported high amount of BAT activation in non‐malignancy subjects.
There is indirect evidence that BAT activation might be a potential cachexia driver in COPD as well. Hypoxia170 and hypermetabolism122 are hallmarks of COPD. In response to hypoxia, cells can produce vascular endothelial growth factor (VEGF) in order to restore oxygen supply.171 This has been established by Van Den Borst et al., who found an upregulation of the VEGF gene in adipose tissue in response to chronic hypoxia in mice. Congruently, adipose tissue showed a brown appearance. This browning of adipose tissue was established by increased expression of UCP1,172 which proposes a link between hypoxia‐induced VEGF activation and browning. Indeed, Sun et al. revealed upregulation of UCP‐1, the main characteristic of BAT, in a VEGF overexpressing mouse model.173 In addition, recently, increased thermogenesis and energy expenditure were found in mice with VEGF overexpression in BAT and WAT.174
Another hypoxia alignment occurs in the form of lactate. In peripheral muscle of COPD patients, increased glycolysis metabolism is observed,175 which in turn causes rising lactate levels.176, 177 Lactate is indeed increasingly released by adipocytes in a hypoxic environment,178 which in turn is able to control the expression of UCP‐1. The UCP‐1 regulation is independent of HIF‐1α and thereby also promotes WAT browning under normoxic circumstances.179
Another possible browning factor is beta‐adrenergic stimulation mediated by norepinephrine.180, 181 Emphysematous COPD patients indeed exhibit increased plasma norepinephrine levels,182 indicating a possible activation of the autonomic nervous system. However, Schiffelers et al. showed a blunted beta‐adrenoceptor‐mediated lipolytic and thermogenic response,183 suggesting desensitization. Additionally, in 10 lean healthy men, BAT activity in response to a systemic non‐selective beta‐agonist was not enhanced.184 In contrast, blocking the receptors by propranolol decreases BAT activity.185 Therefore, activation of brown fat through beta‐adrenergic stimulation remains disputable.
It can be concluded that there is some indirect evidence pointing towards a role of BAT in pulmonary cachexia, but this area requires more research to identify therapeutic potential.
Compromised dietary intake
In order to compensate for increased energy requirements in COPD, patients should be able to adapt their dietary intake. Systematic analyses of dietary intake in COPD patients are rare. In terms of caloric content, dietary intake was found to be normal compared with healthy controls, but inadequate for measured energy expenditure.118, 186, 187, 188 During severe acute exacerbations, the gap between energy intake and energy expenditure becomes even wider, which slowly decreases upon recovery.126, 189 To our knowledge, no human studies have systematically investigated the relation between dietary intake and disease severity or putative differences between emphysematous and non‐emphysematous patients. Advanced disease stages and acute exacerbations are often characterized by chronic or acute hypoxemia.170 It is well established that mice under chronic hypoxic circumstances experience weight loss, which is partly due to temporarily decreased dietary intake.172
Anorexia
It is acknowledged that apparently normal dietary intake in COPD patients may be insufficient to meet elevated energy requirements, but reduced food intake may also be caused by anorexia, that is, loss of appetite.190 A few underlying causes have been mentioned, including nicotine use,191 physical discomfort such as dyspnoea and increased breathing effort,192 depression, and anxiety,193 seen in COPD194 as well as in NSCLC.195, 196
Besides pulmonary and psychological symptoms, COPD patients often experience pain. In a Norwegian study, which controlled for age and gender, 45% of the COPD patients reported chronic pain, compared with 34% of the general population.197 Opioids are commonly used to combat pain in COPD.198 Side‐effects of opioids occur regularly, and opioids are able to cause gastrointestinal motility disorders,199 of which constipation is the most common.200 People suffering from constipation often present with anorexia,201 probably due to early satiety. Separate from use of pain medication, early satiety and abdominal bloating is highly prevalent in COPD.202
Chemosensory alterations
Food intake is regulated by taste and smell,203, 204 and chemosensory dysfunction could influence dietary intake. Nordén et al. showed that 21 out of 169 stable COPD patients reported taste changes, which contributed to a decreased energy intake.205 In addition, Dewan et al. compared 20 COPD subjects with long‐term oxygen therapy to 20 COPD patients without oxygen therapy and 20 healthy elderly controls. They found reduced smell and taste test scores among COPD patients compared with controls, independent of oxygen supply.206 Also, Wardwell and colleagues found that healthy elderly tended to be able to identify more different tastes correctly than COPD patients, although not statistically significant.207 Both authors did not report medication use, and therefore, the influence of treatment is unknown. Although data are scarce and methodological quality of the studies is limited, these data suggest that COPD or its treatment could modify taste and smell detection.
Food reward system
Fullness is regulated by gastrointestinal hormones, including leptin and ghrelin,208 and their secretion is affected by dietary intake and nutritional status. Clinically stable emphysematous COPD patients exhibit low leptin levels compared with the chronic bronchitis subtype.159 During acute flare ups, these plasma levels rise temporarily,189 as seen in NSCLC.209, 210 Likewise, enhanced plasma ghrelin levels are noticed in COPD211 and NSCLC212 and are related to cachectic status.
The peripheral hormonal satiety system closely interacts with the central nervous system in order to regulate food intake. Brain imaging studies have revealed reward‐specific brain regions related to food reward,213 and activation of these regions correlate with food rewarding.214 Different orexigenic and anorexigenic peptides and hormones can stimulate neurons in these specific cerebral regions.208, 215 For instance, leptin inhibits neurons, causing reduced food intake and increased energy expenditure.216 Ghrelin, considered to be a leptin counterpart, can induce food intake mediated by stimulation of neurons in this area.217
There is surprisingly no human study available that explored the role of central dysregulation in food reward in patients with COPD. In relation to lung cancer cachexia, only one study was performed identifying brain activity in anorectic and non‐anorectic patients while receiving pleasant and unpleasant food cues.218 In contrast to non‐anorectic patients, anorectic patients showed no brain activity differences in response to pleasant versus unpleasant pictures. This implies an overall blunted response in the perceptual and motivational system that could also be involved in COPD but requires further investigation.
Therapeutic perspective
The importance of nutritional status is not only emphasized by adverse effects on muscle function and exercise performance but also by detrimental effects of malnutrition on lung tissue. These effects have mostly been studied in animal models. Following the clinical phenotyping of the pink puffer and the blue bloater in the 1960s, Sahebjami et al. found reinforcement of pre‐existing emphysematous processes due to caloric food deprivation in rats,219 which was more pronounced in young rats.220 These deleterious effects could partly be reversed by refeeding.221 In contrast, Bishai et al. found no alveolar size changes in calorie‐restricted mice, although the lungs became stiffer and lung capacity was decreased.222 Supplementary evidence was provided by emphysema‐like changes present in anorexia nervosa patients, which underscores the impact of chronic malnutrition on alveoli.223 In addition to lung tissue, respiratory muscles also contribute to breathing. Weight loss does not spare the respiratory muscles, because weight loss is related to diminished diaphragm weight224 and decreased function225 in experimental models and in humans.
As proof of concept, Efthimiou et al. conducted a randomized controlled trial to investigate the effect of nutritional support on respiratory and peripheral muscle function in malnourished COPD patients. They reported improvement in respiratory muscle strength and hand grip strength, accompanied by less dyspnoea and enhanced distance in 6‐min walk test. Importantly, these effects diminished after quitting the dietary supplementation.226 The positive effects of dietary support on body weight was verified by Weekes et al., who found weight gain in the intervention group, whereas the control group continued to lose weight. Addition of dietary counselling to dietary support has been shown to maintain weight loss after cessation of intervention.227
Initially, the focus was primarily on caloric intake to balance energy requirements, but more recent proof of concept experiments also highlighted the importance of optimizing protein intake.228, 229 Low intake of other essential nutrients is identified, including vitamin D and calcium,230 which are also relevant in the context of osteoporosis as clustering comorbid condition.
One should keep in mind that dietary intake does not reflect actual availability of ingested micronutrients. There are indications that intestinal function is impaired in COPD, illustrated by splanchnic hypoperfusion and reduced intestinal permeability.231 Altered intestinal function translates into reduced splanchnic extraction of amino acids derived from nutritional intake,36, 232 but as a result, the amino acid uptake in the skeletal muscle of clinically stable COPD patients appears increased.233 Thus, the significance for clinical applications remains ambiguous.
Both dietary intake and nutrient availability are controlled by gastrointestinal hormones. By binding to the growth hormone secretagogue receptor, ghrelin can induce secretion of growth hormone.234 This leads to modulation of the growth hormone/IGF1 axis, which is an important anabolic pathway in human skeletal muscle.235 Furthermore, ghrelin can induce food intake, mediated by stimulation of specific neurons in the food reward centre.217 Due to its orexigenic property, ghrelin analogues have been proposed for clinical application in cachexia. One clinical trial with ghrelin analogues have been conducted in COPD patients. They reported improvements of ventilatory efficiency at peak exercise, reflected by increased peak oxygen uptake.236 However, it did not translate in improved 6‐min walk distance, and no data are available about body composition or food intake. Clinical trials in cancer cachexia,237 including lung cancer,238 demonstrate an enhanced lean body mass and quality of life. Hence, ghrelin analogues warrant further investigation in COPD. Besides dietary and pharmacological interventions, cognitive behavioural interventions are relatively underexplored in the management of cachexia in COPD. Although results from different functional neuroimaging studies are inconsistent and sometimes conflicting,239 there might be altered reactivity in the brain reward system in response to perceived food stimuli in people with altered eating patterns, including anorexia nervosa and obesity.240, 241 Therefore, cognitive behavioural therapy may serve as a treatment for patients with an eating disorder like anorexia nervosa.242 Recently, a randomized controlled trial was conducted in obese subjects, receiving behavioural therapy for 6 months in order to reduce weight. Analysis of functional magnetic resonance imaging revealed changes in reward system activity in the intervention group versus controls. Further research has to identify whether it is possible to enhance neuroplasticity in the food reward centre in order to increase successfulness for eating disorder treatment.243 This opens up new insights and therapeutic opportunities for suspected nutritional therapy‐resistant cachectic COPD patients, if disturbances in the central food reward system are indeed identified.
Conclusions
It is well established that the prevalence and related disease burden of cachexia is high in COPD and likely to increase in the near future given the high and increasing prevalence of the disease in an ageing population. Nevertheless, cachexia management is still poorly implemented in clinical practice. In 2014, the European Respiratory Society published a statement on nutritional assessment and therapy in COPD including a nutritional risk stratification diagram based on assessment of BMI, weight changes, and body composition, which could be useful in patient counselling.24 In order to increase overall survival and compress morbidity, a multi‐modal intervention approach is needed, which should target the discussed factors involved in cachexia (Figure 1). Such a multi‐modal intervention approach, encompassing exercise training and improvement of energy balance and nutrient availability, is currently feasible as supported by recent statements and meta‐analyses, possibly improved in the near future by targeted pharmacological interventions and cognitive behavioural therapy to sensitize patients to anabolic stimuli.
Figure 1.
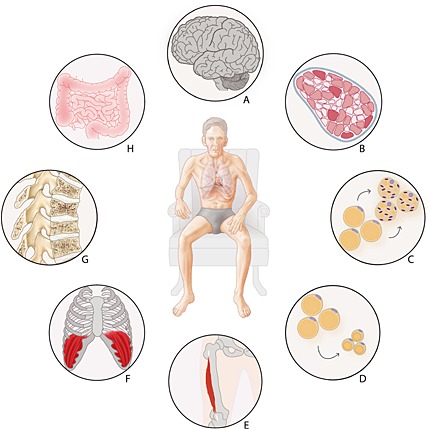
Pulmonary and extra‐pulmonary cross‐talk in COPD cachexia. (A) Altered brain responses to food stimuli; (B) muscle fibre type shifting and oxidative metabolism; (C) altered adipose tissue metabolism; (D) adipose tissue wasting; (E) limb muscle dysfunction; (F) respiratory muscle dysfunction; (G) osteoporosis; (H) altered gut integrity and reduced splanchnic extraction.
Conflict of interest
None.
Acknowledgements
The authors comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle (von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. J Cachexia Sarcopenia Muscle. 2010;1:7‐8).
Sanders, K. J. C. , Kneppers, A. E. M. , van de Bool, C. , Langen, R. C. J. , and Schols, A. M. W. J. (2016) Cachexia in chronic obstructive pulmonary disease: new insights and therapeutic perspective. Journal of Cachexia, Sarcopenia and Muscle, 7: 5–22. doi: 10.1002/jcsm.12062.
References
- 1. Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 2006;3:e442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Prince MJ, Wu F, Guo Y, Gutierrez Robledo LM, O'Donnell M, Sullivan R, et al. The burden of disease in older people and implications for health policy and practice. Lancet 2015;385:549–562. [DOI] [PubMed] [Google Scholar]
- 3. Vestbo J, Hurd SS, Agusti AG, Jones PW, Vogelmeier C, Anzueto A, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med 2013;187:347–365. [DOI] [PubMed] [Google Scholar]
- 4. Vanfleteren LE, Spruit MA, Groenen M, Gaffron S, van Empel VP, Bruijnzeel PL, et al. Clusters of comorbidities based on validated objective measurements and systemic inflammation in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2013;187:728–735. [DOI] [PubMed] [Google Scholar]
- 5. Burgel PR, Paillasseur JL, Peene B, Dusser D, Roche N, Coolen J, et al. Two distinct chronic obstructive pulmonary disease (COPD) phenotypes are associated with high risk of mortality. PLoS One 2012;7:e51048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Romme EA, Murchison JT, Phang KF, Jansen FH, Rutten EP, Wouters EF, et al. Bone attenuation on routine chest CT correlates with bone mineral density on DXA in patients with COPD. J Bone Miner Res 2012;27:2338–2343. [DOI] [PubMed] [Google Scholar]
- 7. Schols AM, Soeters PB, Dingemans AM, Mostert R, Frantzen PJ, Wouters EF. Prevalence and characteristics of nutritional depletion in patients with stable COPD eligible for pulmonary rehabilitation. Am Rev Respir Dis 1993;147:1151–1156. [DOI] [PubMed] [Google Scholar]
- 8. Vestbo J, Prescott E, Almdal T, Dahl M, Nordestgaard BG, Andersen T, et al. Body mass, fat‐free body mass, and prognosis in patients with chronic obstructive pulmonary disease from a random population sample: findings from the Copenhagen City Heart Study. Am J Respir Crit Care Med 2006;173:79–83. [DOI] [PubMed] [Google Scholar]
- 9. Mostert R, Goris A, Weling‐Scheepers C, Wouters EF, Schols AM. Tissue depletion and health related quality of life in patients with chronic obstructive pulmonary disease. Respir Med 2000;94:859–867. [DOI] [PubMed] [Google Scholar]
- 10. Engelen MP, Schols AM, Baken WC, Wesseling GJ, Wouters EF. Nutritional depletion in relation to respiratory and peripheral skeletal muscle function in out‐patients with COPD. Eur Respir J 1994;7:1793–1797. [DOI] [PubMed] [Google Scholar]
- 11. Schols AM, Broekhuizen R, Weling‐Scheepers CA, Wouters EF. Body composition and mortality in chronic obstructive pulmonary disease. Am J Clin Nutr 2005;82:53–59. [DOI] [PubMed] [Google Scholar]
- 12. Bernard S, LeBlanc P, Whittom F, Carrier G, Jobin J, Belleau R, et al. Peripheral muscle weakness in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998;158:629–634. [DOI] [PubMed] [Google Scholar]
- 13. Engelen MP, Schols AM, Does JD, Wouters EF. Skeletal muscle weakness is associated with wasting of extremity fat‐free mass but not with airflow obstruction in patients with chronic obstructive pulmonary disease. Am J Clin Nutr 2000;71:733–738. [DOI] [PubMed] [Google Scholar]
- 14. Engelen MP, Schols AM, Lamers RJ, Wouters EF. Different patterns of chronic tissue wasting among patients with chronic obstructive pulmonary disease. Clin Nutr 1999;18:275–280. [DOI] [PubMed] [Google Scholar]
- 15. Caron MA, Debigare R, Dekhuijzen PN, Maltais F. Comparative assessment of the quadriceps and the diaphragm in patients with COPD. J Appl Physiol (1985) 2009;107:952–961. [DOI] [PubMed] [Google Scholar]
- 16. Gosker HR, Engelen MP, van Mameren H, van Dijk PJ, van der Vusse GJ, Wouters EF, et al. Muscle fiber type IIX atrophy is involved in the loss of fat‐free mass in chronic obstructive pulmonary disease. Am J Clin Nutr 2002;76:113–119. [DOI] [PubMed] [Google Scholar]
- 17. Mancini DM, Coyle E, Coggan A, Beltz J, Ferraro N, Montain S, et al. Contribution of intrinsic skeletal muscle changes to 31P NMR skeletal muscle metabolic abnormalities in patients with chronic heart failure. Circulation 1989;80:1338–1346. [DOI] [PubMed] [Google Scholar]
- 18. Gosker HR, van Mameren H, van Dijk PJ, Engelen MP, van der Vusse GJ, Wouters EF, et al. Skeletal muscle fibre‐type shifting and metabolic profile in patients with chronic obstructive pulmonary disease. Eur Respir J 2002;19:617–625. [DOI] [PubMed] [Google Scholar]
- 19. Maltais F, LeBlanc P, Whittom F, Simard C, Marquis K, Belanger M, et al. Oxidative enzyme activities of the vastus lateralis muscle and the functional status in patients with COPD. Thorax 2000;55:848–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Remels AH, Gosker HR, Langen RC, Schols AM. The mechanisms of cachexia underlying muscle dysfunction in COPD. J Appl Physiol (1985) 2013;114:1253–62. [DOI] [PubMed] [Google Scholar]
- 21. Files DC, D'Alessio FR, Johnston LF, Kesari P, Aggarwal NR, Garibaldi BT, et al. A critical role for muscle ring finger‐1 in acute lung injury‐associated skeletal muscle wasting. Am J Respir Crit Care Med 2012;185:825–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. de Theije CC, Langen RC, Lamers WH, Gosker HR, Schols AM, Koehler SE. Differential sensitivity of oxidative and glycolytic muscles to hypoxia‐induced muscle atrophy. J Appl Physiol (1985) 2015;118:200–211. [DOI] [PubMed] [Google Scholar]
- 23. van de Bool C, Rutten EP, Franssen FM, Wouters EF, Schols AM. Antagonistic implications of sarcopenia and abdominal obesity on physical performance in COPD. Eur Respir J 2015. doi: 10.1183/09031936.00197314. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 24. Schols AM, Ferreira IM, Franssen FM, Gosker HR, Janssens W, Muscaritoli M, et al. Nutritional assessment and therapy in COPD: a European Respiratory Society statement. Eur Respir J 2014;44:1504–1520. [DOI] [PubMed] [Google Scholar]
- 25. Jones SE, Maddocks M, Kon SS, Canavan JL, Nolan CM, Clark AL, et al. Sarcopenia in COPD: prevalence, clinical correlates and response to pulmonary rehabilitation. Thorax 2015;70:213–218. [DOI] [PubMed] [Google Scholar]
- 26. Cherin P, Voronska E, Fraoucene N, de Jaeger C. Prevalence of sarcopenia among healthy ambulatory subjects: the sarcopenia begins from 45 years. Aging Clin Exp Res 2014;26:137–146. [DOI] [PubMed] [Google Scholar]
- 27. O'Donnell DE, Ciavaglia CE, Neder JA. When obesity and chronic obstructive pulmonary disease collide. Physiological and clinical consequences. Ann Am Thorac Soc 2014;11:635–644. [DOI] [PubMed] [Google Scholar]
- 28. Guenette JA, Jensen D, O'Donnell DE. Respiratory function and the obesity paradox. Curr Opin Clin Nutr Metab Care 2010;13:618–624. [DOI] [PubMed] [Google Scholar]
- 29. van den Borst B, Gosker HR, Koster A, Yu B, Kritchevsky SB, Liu Y, et al. The influence of abdominal visceral fat on inflammatory pathways and mortality risk in obstructive lung disease. Am J Clin Nutr 2012;96:516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Furutate R, Ishii T, Wakabayashi R, Motegi T, Yamada K, Gemma A, et al. Excessive visceral fat accumulation in advanced chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis 2011;6:423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McDonald ML, Diaz AA, Ross JC. San Jose Estepar R, Zhou L, Regan EA et al. Quantitative computed tomography measures of pectoralis muscle area and disease severity in chronic obstructive pulmonary disease. A cross‐sectional study. Ann Am Thorac Soc 2014;11:326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Diaz AA, Zhou L, Young TP, McDonald ML, Harmouche R, Ross JC, et al. Chest CT measures of muscle and adipose tissue in COPD: gender‐based differences in content and in relationships with blood biomarkers. Acad Radiol 2014;21:1255–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Langen RC, Gosker HR, Remels AH, Schols AM. Triggers and mechanisms of skeletal muscle wasting in chronic obstructive pulmonary disease. Int J Biochem Cell Biol 2013;45:2245–2256. [DOI] [PubMed] [Google Scholar]
- 34. Zuo L, He F, Sergakis GG, Koozehchian MS, Stimpfl JN, Rong Y, et al. Interrelated role of cigarette smoking, oxidative stress, and immune response in COPD and corresponding treatments. Am J Physiol Lung Cell Mol Physiol 2014;307:L205–L218. [DOI] [PubMed] [Google Scholar]
- 35. Rutten EP, Franssen FM, Engelen MP, Wouters EF, Deutz NE, Schols AM. Greater whole‐body myofibrillar protein breakdown in cachectic patients with chronic obstructive pulmonary disease. Am J Clin Nutr 2006;83:829–834. [DOI] [PubMed] [Google Scholar]
- 36. Engelen MP, Deutz NE, Wouters EF, Schols AM. Enhanced levels of whole‐body protein turnover in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2000;162:1488–1492. [DOI] [PubMed] [Google Scholar]
- 37. Morrison WL, Gibson JN, Scrimgeour C, Rennie MJ. Muscle wasting in emphysema. Clin Sci (Lond) 1988;75:415–420. [DOI] [PubMed] [Google Scholar]
- 38. Fermoselle C, Rabinovich R, Ausin P, Puig‐Vilanova E, Coronell C, Sanchez F, et al. Does oxidative stress modulate limb muscle atrophy in severe COPD patients? Eur Respir J 2012;40:851–862. [DOI] [PubMed] [Google Scholar]
- 39. Puig‐Vilanova E, Rodriguez DA, Lloreta J, Ausin P, Pascual‐Guardia S, Broquetas J, et al. Oxidative Stress, Redox Signaling Pathways, And Autophagy In Cachectic Muscles Of Male Patients With Advanced Copd And Lung Cancer. Free Radic Biol Med 2014;79:91–108. [DOI] [PubMed] [Google Scholar]
- 40. Vogiatzis I, Simoes DC, Stratakos G, Kourepini E, Terzis G, Manta P, et al. Effect of pulmonary rehabilitation on muscle remodelling in cachectic patients with COPD. Eur Respir J 2010;36:301–310. [DOI] [PubMed] [Google Scholar]
- 41. Op den Kamp CM, Langen RC, Snepvangers FJ, de Theije CC, Schellekens JM, Laugs F, et al. Nuclear transcription factor kappa B activation and protein turnover adaptations in skeletal muscle of patients with progressive stages of lung cancer cachexia. Am J Clin Nutr 2013;98:738–748. [DOI] [PubMed] [Google Scholar]
- 42. Debigare R, Cote CH, Maltais F. Ubiquitination and proteolysis in limb and respiratory muscles of patients with chronic obstructive pulmonary disease. Proc Am Thorac Soc 2010;7:84–90. [DOI] [PubMed] [Google Scholar]
- 43. Debigare R, Maltais F, Cote CH, Michaud A, Caron MA, Mofarrahi M, et al. Profiling of mRNA expression in quadriceps of patients with COPD and muscle wasting. COPD 2008;5:75–84. [DOI] [PubMed] [Google Scholar]
- 44. Doucet M, Russell AP, Leger B, Debigare R, Joanisse DR, Caron MA, et al. Muscle atrophy and hypertrophy signaling in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2007;176:261–269. [DOI] [PubMed] [Google Scholar]
- 45. Guo Y, Gosker HR, Schols AM, Kapchinsky S, Bourbeau J, Sandri M, et al. Autophagy in locomotor muscles of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2013;188:1313–1320. [DOI] [PubMed] [Google Scholar]
- 46. Doucet M, Dube A, Joanisse DR, Debigare R, Michaud A, Pare ME, et al. Atrophy and hypertrophy signalling of the quadriceps and diaphragm in COPD. Thorax 2010;65:963–970. [DOI] [PubMed] [Google Scholar]
- 47. Levine S, Gregory C, Nguyen T, Shrager J, Kaiser L, Rubinstein N, et al. Bioenergetic adaptation of individual human diaphragmatic myofibers to severe COPD. J Appl Physiol (1985) 2002;92:1205–1213. [DOI] [PubMed] [Google Scholar]
- 48. Levine S, Kaiser L, Leferovich J, Tikunov B. Cellular adaptations in the diaphragm in chronic obstructive pulmonary disease. N Engl J Med 1997;337:1799–1806. [DOI] [PubMed] [Google Scholar]
- 49. Mofarrahi M, Guo Y, Haspel JA, Choi AM, Davis EC, Gouspillou G, et al. Autophagic flux and oxidative capacity of skeletal muscles during acute starvation. Autophagy 2013;9:1604–1620. [DOI] [PubMed] [Google Scholar]
- 50. Gielen S, Sandri M, Kozarez I, Kratzsch J, Teupser D, Thiery J, et al. Exercise training attenuates MuRF‐1 expression in the skeletal muscle of patients with chronic heart failure independent of age: the randomized Leipzig Exercise Intervention in Chronic Heart Failure and Aging catabolism study. Circulation 2012;125:2716–2727. [DOI] [PubMed] [Google Scholar]
- 51. Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. Foxo transcription factors induce the atrophy‐related ubiquitin ligase atrogin‐1 and cause skeletal muscle atrophy. Cell 2004;117:399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 2007;6:458–471. [DOI] [PubMed] [Google Scholar]
- 53. Glickman MH, Ciechanover A. The ubiquitin‐proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 2002;82:373–428. [DOI] [PubMed] [Google Scholar]
- 54. Pickart CM. Back to the future with ubiquitin. Cell 2004;116:181–190. [DOI] [PubMed] [Google Scholar]
- 55. Ottenheijm CA, Heunks LM, Li YP, Jin B, Minnaard R, van Hees HW, et al. Activation of the ubiquitin‐proteasome pathway in the diaphragm in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2006;174:997–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lemire BB, Debigare R, Dube A, Theriault ME, Cote CH, Maltais F. MAPK signaling in the quadriceps of patients with chronic obstructive pulmonary disease. J Appl Physiol (1985) 2012;113:159–166. [DOI] [PubMed] [Google Scholar]
- 57. Plant PJ, Brooks D, Faughnan M, Bayley T, Bain J, Singer L, et al. Cellular markers of muscle atrophy in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 2010;42:461–471. [DOI] [PubMed] [Google Scholar]
- 58. Petersen AM, Magkos F, Atherton P, Selby A, Smith K, Rennie MJ, et al. Smoking impairs muscle protein synthesis and increases the expression of myostatin and MAFbx in muscle. Am J Physiol Endocrinol Metab 2007;293:E843–E848. [DOI] [PubMed] [Google Scholar]
- 59. Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol 2009;335:1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 2010;22:132–139. [DOI] [PubMed] [Google Scholar]
- 61. Mammucari C, Schiaffino S, Sandri M. Downstream of Akt: FoxO3 and mTOR in the regulation of autophagy in skeletal muscle. Autophagy 2008;4:524–526. [DOI] [PubMed] [Google Scholar]
- 62. Mai S, Muster B, Bereiter‐Hahn J, Jendrach M. Autophagy proteins LC3B, ATG5 and ATG12 participate in quality control after mitochondrial damage and influence lifespan. Autophagy 2012;8:47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rello‐Varona S, Lissa D, Shen S, Niso‐Santano M, Senovilla L, Marino G, et al. Autophagic removal of micronuclei. Cell Cycle 2012;11:170–176. [DOI] [PubMed] [Google Scholar]
- 64. Lewis A, Riddoch‐Contreras J, Natanek SA, Donaldson A, Man WD, Moxham J, et al. Downregulation of the serum response factor/miR‐1 axis in the quadriceps of patients with COPD. Thorax 2012;67:26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Crul T, Spruit MA, Gayan‐Ramirez G, Quarck R, Gosselink R, Troosters T, et al. Markers of inflammation and disuse in vastus lateralis of chronic obstructive pulmonary disease patients. Eur J Clin Invest 2007;37:897–904. [DOI] [PubMed] [Google Scholar]
- 66. Adams GR. Invited Review: Autocrine/paracrine IGF‐I and skeletal muscle adaptation. J Appl Physiol (1985) 2002;93:1159–67. [DOI] [PubMed] [Google Scholar]
- 67. Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev 2004;18:1926–1945. [DOI] [PubMed] [Google Scholar]
- 68. Martinez‐Llorens J, Casadevall C, Lloreta J, Orozco‐Levi M, Barreiro E, Broquetas J, et al. Activation of satellite cells in the intercostal muscles of patients with chronic obstructive pulmonary disease. Arch Bronconeumol 2008;44:239–244. [PubMed] [Google Scholar]
- 69. Tannerstedt J, Apro W, Blomstrand E. Maximal lengthening contractions induce different signaling responses in the type I and type II fibers of human skeletal muscle. J Appl Physiol (1985) 2009;106:1412–1418. [DOI] [PubMed] [Google Scholar]
- 70. Lepper C, Partridge TA, Fan CM. An absolute requirement for Pax7‐positive satellite cells in acute injury‐induced skeletal muscle regeneration. Development 2011;138:3639–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Relaix F, Zammit PS. Satellite cells are essential for skeletal muscle regeneration: the cell on the edge returns centre stage. Development 2012;139:2845–2856. [DOI] [PubMed] [Google Scholar]
- 72. Adams GR, Caiozzo VJ, Haddad F, Baldwin KM. Cellular and molecular responses to increased skeletal muscle loading after irradiation. Am J Physiol Cell Physiol 2002;283:C1182–C1195. [DOI] [PubMed] [Google Scholar]
- 73. Agusti AG, Sauleda J, Miralles C, Gomez C, Togores B, Sala E, et al. Skeletal muscle apoptosis and weight loss in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2002;166:485–489. [DOI] [PubMed] [Google Scholar]
- 74. Gosker HR, Kubat B, Schaart G, van der Vusse GJ, Wouters EF, Schols AM. Myopathological features in skeletal muscle of patients with chronic obstructive pulmonary disease. Eur Respir J 2003;22:280–285. [DOI] [PubMed] [Google Scholar]
- 75. Dupont‐Versteegden EE. Apoptosis in skeletal muscle and its relevance to atrophy. World J Gastroenterol 2006;12:7463–7466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mijaljica D, Devenish RJ. Nucleophagy at a glance. J Cell Sci 2013;126:4325–4330. [DOI] [PubMed] [Google Scholar]
- 77. Verdijk LB, Snijders T, Drost M, Delhaas T, Kadi F, van Loon LJ. Satellite cells in human skeletal muscle; from birth to old age. Age (Dordr) 2014;36:545–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Snijders T, Verdijk LB, van Loon LJ. The impact of sarcopenia and exercise training on skeletal muscle satellite cells. Ageing Res Rev 2009;8:328–338. [DOI] [PubMed] [Google Scholar]
- 79. Eliason G, Abdel‐Halim S, Arvidsson B, Kadi F, Piehl‐Aulin K. Physical performance and muscular characteristics in different stages of COPD. Scand J Med Sci Sports 2009;19:865–870. [DOI] [PubMed] [Google Scholar]
- 80. Theriault ME, Pare ME, Maltais F, Debigare R. Satellite cells senescence in limb muscle of severe patients with COPD. PLoS One 2012;7:e39124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Menon MK, Houchen L, Singh SJ, Morgan MD, Bradding P, Steiner MC. Inflammatory and satellite cells in the quadriceps of patients with COPD and response to resistance training. Chest 2012;142:1134–1142. [DOI] [PubMed] [Google Scholar]
- 82. Rodriguez J, Vernus B, Chelh I, Cassar‐Malek I, Gabillard JC, Hadj Sassi A, et al. Myostatin and the skeletal muscle atrophy and hypertrophy signaling pathways. Cell Mol Life Sci 2014;71:4361–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Man WD, Natanek SA, Riddoch‐Contreras J, Lewis A, Marsh GS, Kemp PR, et al. Quadriceps myostatin expression in COPD. Eur Respir J 2010;36:686–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ju CR, Chen RC. Serum myostatin levels and skeletal muscle wasting in chronic obstructive pulmonary disease. Respir Med 2012;106:102–108. [DOI] [PubMed] [Google Scholar]
- 85. Snijders T, Verdijk LB, Smeets JS, McKay BR, Senden JM, Hartgens F, et al. The skeletal muscle satellite cell response to a single bout of resistance‐type exercise is delayed with aging in men. Age (Dordr) 2014;36:e9699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Malaguti C, Nery LE, Dal Corso S, Napolis L, De Fuccio MB, Castro M, et al. Scaling skeletal muscle function to mass in patients with moderate‐to‐severe COPD. Eur J Appl Physiol 2006;98:482–488. [DOI] [PubMed] [Google Scholar]
- 87. Malaguti C, Napolis LM, Villaca D, Neder JA, Nery LE, Dal CS. Relationship between peripheral muscle structure and function in patients with chronic obstructive pulmonary disease with different nutritional status. J Strength Cond Res 2011;25:1795–1803. [DOI] [PubMed] [Google Scholar]
- 88. van den Borst B, Slot IG, Hellwig VA, Vosse BA, Kelders MC, Barreiro E, et al. Loss of quadriceps muscle oxidative phenotype and decreased endurance in patients with mild‐to‐moderate COPD. J Appl Physiol (1985) 2013;114:1319–1328. [DOI] [PubMed] [Google Scholar]
- 89. Gosker HR, Zeegers MP, Wouters EF, Schols AM. Muscle fibre type shifting in the vastus lateralis of patients with COPD is associated with disease severity: a systematic review and meta‐analysis. Thorax 2007;62:944–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Barreiro E, de la Puente B, Minguella J, Corominas JM, Serrano S, Hussain SN, et al. Oxidative stress and respiratory muscle dysfunction in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2005;171:1116–1124. [DOI] [PubMed] [Google Scholar]
- 91. Degens H, Gayan‐Ramirez G, van Hees HW. Smoking‐induced skeletal muscle dysfunction: from evidence to mechanisms. Am J Respir Crit Care Med 2015;191:620–625. [DOI] [PubMed] [Google Scholar]
- 92. Gosker HR, Wouters EF, van der Vusse GJ, Schols AM. Skeletal muscle dysfunction in chronic obstructive pulmonary disease and chronic heart failure: underlying mechanisms and therapy perspectives. Am J Clin Nutr 2000;71:1033–1047. [DOI] [PubMed] [Google Scholar]
- 93. Labeit S, Kohl CH, Witt CC, Labeit D, Jung J, Granzier H. Modulation of muscle atrophy, fatigue and MLC phosphorylation by MuRF1 as indicated by hindlimb suspension studies on MuRF1‐KO mice. J Biomed Biotechnol 2010;2010:e693741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Eddins MJ, Marblestone JG, Suresh Kumar KG, Leach CA, Sterner DE, Mattern MR, et al. Targeting the ubiquitin E3 ligase MuRF1 to inhibit muscle atrophy. Cell Biochem Biophys 2011;60:113–118. [DOI] [PubMed] [Google Scholar]
- 95. Edelmann MJ, Nicholson B, Kessler BM. Pharmacological targets in the ubiquitin system offer new ways of treating cancer, neurodegenerative disorders and infectious diseases. Expert Rev Mol Med 2011;13:e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Gosker HR, Lencer NH, Franssen FM, van der Vusse GJ, Wouters EF, Schols AM. Striking similarities in systemic factors contributing to decreased exercise capacity in patients with severe chronic heart failure or COPD. Chest 2003;123:1416–1424. [DOI] [PubMed] [Google Scholar]
- 97. Borgenvik M, Apro W, Blomstrand E. Intake of branched‐chain amino acids influences the levels of MAFbx mRNA and MuRF‐1 total protein in resting and exercising human muscle. Am J Physiol Endocrinol Metab 2012;302:E510–E521. [DOI] [PubMed] [Google Scholar]
- 98. Constantin D, Menon MK, Houchen‐Wolloff L, Morgan MD, Singh SJ, Greenhaff P, et al. Skeletal muscle molecular responses to resistance training and dietary supplementation in COPD. Thorax 2013;68:625–633. [DOI] [PubMed] [Google Scholar]
- 99. Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, Avruch J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF‐4E BP1 through a common effector mechanism. J Biol Chem 1998;273:14484–14494. [DOI] [PubMed] [Google Scholar]
- 100. Drummond MJ, Rasmussen BB. Leucine‐enriched nutrients and the regulation of mammalian target of rapamycin signalling and human skeletal muscle protein synthesis. Curr Opin Clin Nutr Metab Care 2008;11:222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Ditch S, Paull TT. The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Trends Biochem Sci 2012;37:15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer 2006;6:729–734. [DOI] [PubMed] [Google Scholar]
- 103. Lira VA, Okutsu M, Zhang M, Greene NP, Laker RC, Breen DS, et al. Autophagy is required for exercise training‐induced skeletal muscle adaptation and improvement of physical performance. FASEB J 2013;27:4184–4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Sanchez AM, Bernardi H, Py G, Candau RB. Autophagy is essential to support skeletal muscle plasticity in response to endurance exercise. Am J Physiol Regul Integr Comp Physiol 2014;307:R956–R969. [DOI] [PubMed] [Google Scholar]
- 105. Couillard A, Maltais F, Saey D, Debigare R, Michaud A, Koechlin C, et al. Exercise‐induced quadriceps oxidative stress and peripheral muscle dysfunction in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2003;167:1664–1669. [DOI] [PubMed] [Google Scholar]
- 106. Wilborn CD, Taylor LW, Greenwood M, Kreider RB, Willoughby DS. Effects of different intensities of resistance exercise on regulators of myogenesis. J Strength Cond Res 2009;23:2179–2187. [DOI] [PubMed] [Google Scholar]
- 107. Hill M, Goldspink G. Expression and splicing of the insulin‐like growth factor gene in rodent muscle is associated with muscle satellite (stem) cell activation following local tissue damage. J Physiol 2003;549:409–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Snijders T, Verdijk LB, McKay BR, Smeets JS, van Kranenburg J, Groen BB, et al. Acute dietary protein intake restriction is associated with changes in myostatin expression after a single bout of resistance exercise in healthy young men. J Nutr 2014;144:137–145. [DOI] [PubMed] [Google Scholar]
- 109. Zhang L, Rajan V, Lin E, Hu Z, Han HQ, Zhou X, et al. Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2011;25:1653–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Lach‐Trifilieff E, Minetti GC, Sheppard K, Ibebunjo C, Feige JN, Hartmann S, et al. An antibody blocking activin type II receptors induces strong skeletal muscle hypertrophy and protects from atrophy. Mol Cell Biol 2014;34:606–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Bonetto A, Penna F, Minero VG, Reffo P, Costamagna D, Bonelli G, et al. Glutamine prevents myostatin hyperexpression and protein hypercatabolism induced in C2C12 myotubes by tumor necrosis factor‐alpha. Amino Acids 2011;40:585–594. [DOI] [PubMed] [Google Scholar]
- 112. Pan L, Wang M, Xie X, Du C, Guo Y. Effects of anabolic steroids on chronic obstructive pulmonary disease: a meta‐analysis of randomised controlled trials. PLoS One. 2014;9:e84855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Schols AM. Nutrition as a metabolic modulator in COPD. Chest 2013;144:1340–1345. [DOI] [PubMed] [Google Scholar]
- 114. Schoffelen PF, Westerterp KR, Saris WH, Ten Hoor F. A dual‐respiration chamber system with automated calibration. J Appl Physiol (1985) 1997;83:2064–2072. [DOI] [PubMed] [Google Scholar]
- 115. Roberts SB. Use of the doubly labeled water method for measurement of energy expenditure, total body water, water intake, and metabolizable energy intake in humans and small animals. Can J Physiol Pharmacol 1989;67:1190–1198. [DOI] [PubMed] [Google Scholar]
- 116. Schoeller DA, Ravussin E, Schutz Y, Acheson KJ, Baertschi P, Jequier E. Energy expenditure by doubly labeled water: validation in humans and proposed calculation. Am J Physiol 1986;250:R823–R830. [DOI] [PubMed] [Google Scholar]
- 117. Hugli O, Schutz Y, Fitting JW. The daily energy expenditure in stable chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1996;153:294–300. [DOI] [PubMed] [Google Scholar]
- 118. Slinde F, Ellegard L, Gronberg AM, Larsson S, Rossander‐Hulthen L. Total energy expenditure in underweight patients with severe chronic obstructive pulmonary disease living at home. Clin Nutr 2003;22:159–165. [DOI] [PubMed] [Google Scholar]
- 119. Baarends EM, Schols AM, Westerterp KR, Wouters EF. Total daily energy expenditure relative to resting energy expenditure in clinically stable patients with COPD. Thorax 1997;52:780–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Mineo TC, Pompeo E, Mineo D, Ambrogi V, Ciarapica D, Polito A. Resting energy expenditure and metabolic changes after lung volume reduction surgery for emphysema. Ann Thorac Surg 2006;82:1205–1211. [DOI] [PubMed] [Google Scholar]
- 121. Kao CC, Hsu JW, Bandi V, Hanania NA, Kheradmand F, Jahoor F. Resting energy expenditure and protein turnover are increased in patients with severe chronic obstructive pulmonary disease. Metabolism 2011;60:1449–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Schols AM, Fredrix EW, Soeters PB, Westerterp KR, Wouters EF. Resting energy expenditure in patients with chronic obstructive pulmonary disease. Am J Clin Nutr 1991;54:983–987. [DOI] [PubMed] [Google Scholar]
- 123. Goldstein S, Askanazi J, Weissman C, Thomashow B, Kinney JM. Energy expenditure in patients with chronic obstructive pulmonary disease. Chest 1987;91:222–224. [DOI] [PubMed] [Google Scholar]
- 124. Cohen RI, Marzouk K, Berkoski P, O'Donnell CP, Polotsky VY, Scharf SM. Body composition and resting energy expenditure in clinically stable, non‐weight‐losing patients with severe emphysema. Chest 2003;124:1365–1372. [DOI] [PubMed] [Google Scholar]
- 125. Green JH, Muers MF. The thermic effect of food in underweight patients with emphysematous chronic obstructive pulmonary disease. Eur Respir J 1991;4:813–819. [PubMed] [Google Scholar]
- 126. Vermeeren MA, Schols AM, Wouters EF. Effects of an acute exacerbation on nutritional and metabolic profile of patients with COPD. Eur Respir J 1997;10:2264–2269. [DOI] [PubMed] [Google Scholar]
- 127. Ryan CF, Road JD, Buckley PA, Ross C, Whittaker JS. Energy balance in stable malnourished patients with chronic obstructive pulmonary disease. Chest 1993;103:1038–1044. [DOI] [PubMed] [Google Scholar]
- 128. Kurpad AV, Muthayya S, Vaz M. Consequences of inadequate food energy and negative energy balance in humans. Public Health Nutr 2005;8:1053–1076. [DOI] [PubMed] [Google Scholar]
- 129. Fredrix EW. Staal‐van den Brekel AJ, Wouters EF. Energy balance in nonsmall cell lung carcinoma patients before and after surgical resection of their tumors. Cancer 1997;79:717–723. [PubMed] [Google Scholar]
- 130. Harvie MN, Campbell IT, Thatcher N, Baildam A. Changes in body composition in men and women with advanced nonsmall cell lung cancer (NSCLC) undergoing chemotherapy. J Hum Nutr Diet 2003;16:323–326. [DOI] [PubMed] [Google Scholar]
- 131. Staal‐van den Brekel AJ, Schols AM, Dentener MA, ten Velde GP, Buurman WA, Wouters EF. Metabolism in patients with small cell lung carcinoma compared with patients with non‐small cell lung carcinoma and healthy controls. Thorax 1997;52:338–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Cao DX, Wu GH, Zhang B, Quan YJ, Wei J, Jin H, et al. Resting energy expenditure and body composition in patients with newly detected cancer. Clin Nutr 2010;29:72–77. [DOI] [PubMed] [Google Scholar]
- 133. Van Remoortel H, Hornikx M, Demeyer H, Langer D, Burtin C, Decramer M, et al. Daily physical activity in subjects with newly diagnosed COPD. Thorax 2013;68:962–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Andersson M, Slinde F, Gronberg AM, Svantesson U, Janson C, Emtner M. Physical activity level and its clinical correlates in chronic obstructive pulmonary disease: a cross‐sectional study. Respir Res 2013;14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Troosters T, Sciurba F, Battaglia S, Langer D, Valluri SR, Martino L, et al. Physical inactivity in patients with COPD, a controlled multi‐center pilot‐study. Respir Med 2010;104:1005–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Pitta F, Troosters T, Spruit MA, Probst VS, Decramer M, Gosselink R. Characteristics of physical activities in daily life in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2005;171:972–977. [DOI] [PubMed] [Google Scholar]
- 137. Ehsan M, Khan R, Wakefield D, Qureshi A, Murray L, Zuwallack R, et al. A longitudinal study evaluating the effect of exacerbations on physical activity in patients with chronic obstructive pulmonary disease. Ann Am Thorac Soc 2013;10:559–564. [DOI] [PubMed] [Google Scholar]
- 138. Pitta F, Troosters T, Probst VS, Spruit MA, Decramer M, Gosselink R. Physical activity and hospitalization for exacerbation of COPD. Chest 2006;129:536–544. [DOI] [PubMed] [Google Scholar]
- 139. Waschki B, Spruit MA, Watz H, Albert PS, Shrikrishna D, Groenen M, et al. Physical activity monitoring in COPD: compliance and associations with clinical characteristics in a multicenter study. Respir Med 2012;106:522–530. [DOI] [PubMed] [Google Scholar]
- 140. Ferguson GT, Fernandez E, Zamora MR, Pomerantz M, Buchholz J, Make BJ. Improved exercise performance following lung volume reduction surgery for emphysema. Am J Respir Crit Care Med 1998;157:1195–1203. [DOI] [PubMed] [Google Scholar]
- 141. Hartman JE, Boezen HM, Heintzbergen S, de Greef MH, Klooster K, ten Hacken NH, et al. Daily physical activity after bronchoscopic lung volume reduction: a pilot study. Eur Respir J 2012;40:1566–1567. [DOI] [PubMed] [Google Scholar]
- 142. Lahaije AJ, van Helvoort HA, Dekhuijzen PN, Heijdra YF. Physiologic limitations during daily life activities in COPD patients. Respir Med 2010;104:1152–1159. [DOI] [PubMed] [Google Scholar]
- 143. Vaes AW, Wouters EF, Franssen FM, Uszko‐Lencer NH, Stakenborg KH, Westra M, et al. Task‐related oxygen uptake during domestic activities of daily life in patients with COPD and healthy elderly subjects. Chest 2011;140:970–979. [DOI] [PubMed] [Google Scholar]
- 144. Calvert LD, Steiner MC, Morgan MD, Singh SJ. Plasma ammonia response to incremental cycling and walking tests in COPD. Respir Med 2010;104:675–681. [DOI] [PubMed] [Google Scholar]
- 145. Criner GJ, Belt P, Sternberg AL, Mosenifar Z, Make BJ, Utz JP, et al. Effects of lung volume reduction surgery on gas exchange and breathing pattern during maximum exercise. Chest 2009;135:1268–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Budweiser S, Heinemann F, Meyer K, Wild PJ, Pfeifer M. Weight gain in cachectic COPD patients receiving noninvasive positive‐pressure ventilation. Respir Care 2006;51:126–132. [PubMed] [Google Scholar]
- 147. Granger CL, McDonald CF, Irving L, Clark RA, Gough K, Murnane A, et al. Low physical activity levels and functional decline in individuals with lung cancer. Lung Cancer 2014;83:292–299. [DOI] [PubMed] [Google Scholar]
- 148. Hummler S, Thomas M, Hoffmann B, Gartner P, Zoz M, Huber G, et al. Physical performance and psychosocial status in lung cancer patients: results from a pilot study. Oncol Res Treat 2014;37:36–41. [DOI] [PubMed] [Google Scholar]
- 149. Hugli O, Frascarolo P, Schutz Y, Jequier E, Leuenberger P, Fitting JW. Diet‐induced thermogenesis in chronic obstructive pulmonary disease. Am Rev Respir Dis 1993;148:1479–1483. [DOI] [PubMed] [Google Scholar]
- 150. Dore MF, Laaban JP, Orvoen‐Frija E, Kouchakji B, Joubert M, Rochemaure J. Role of the thermic effect of food in malnutrition of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1997;155:1535–1540. [DOI] [PubMed] [Google Scholar]
- 151. Tang NL, Chung ML, Elia M, Hui E, Lum CM, Luk JK, et al. Total daily energy expenditure in wasted chronic obstructive pulmonary disease patients. Eur J Clin Nutr 2002;56:282–287. [DOI] [PubMed] [Google Scholar]
- 152. Schols A, Mostert R, Cobben N, Soeters P, Wouters E. Transcutaneous oxygen saturation and carbon dioxide tension during meals in patients with chronic obstructive pulmonary disease. Chest 1991;100:1287–1292. [DOI] [PubMed] [Google Scholar]
- 153. Broekhuizen R, Wouters EF, Creutzberg EC, Schols AM. Raised CRP levels mark metabolic and functional impairment in advanced COPD. Thorax 2006;61:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Staal‐van den Brekel AJ, Schols AM, ten Velde GP, Buurman WA, Wouters EF. Analysis of the energy balance in lung cancer patients. Cancer Res 1994;54:6430–6433. [PubMed] [Google Scholar]
- 155. Langen RC, Haegens A, Vernooy JH, Wouters EF, de Winther MP, Carlsen H, et al. NF‐kappaB activation is required for the transition of pulmonary inflammation to muscle atrophy. Am J Respir Cell Mol Biol 2012;47:288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Tkacova R, Ukropec J, Skyba P, Ukropcova B, Pobeha P, Kurdiova T, et al. Increased adipose tissue expression of proinflammatory CD40, MKK4 and JNK in patients with very severe chronic obstructive pulmonary disease. Respiration 2011;81:386–393. [DOI] [PubMed] [Google Scholar]
- 157. Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 2011;12:489–495. [DOI] [PubMed] [Google Scholar]
- 158. Dahlman I, Mejhert N, Linder K, Agustsson T, Mutch DM, Kulyte A, et al. Adipose tissue pathways involved in weight loss of cancer cachexia. Br J Cancer 2010;102:1541–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Schols AM, Creutzberg EC, Buurman WA, Campfield LA, Saris WH, Wouters EF. Plasma leptin is related to proinflammatory status and dietary intake in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1999;160:1220–1226. [DOI] [PubMed] [Google Scholar]
- 160. Brusik M, Ukropec J, Joppa P, Ukropcova B, Skyba P, Balaz M, et al. Circulatory and adipose tissue leptin and adiponectin in relationship to resting energy expenditure in patients with chronic obstructive pulmonary disease. Physiol Res 2012;61:469–480. [DOI] [PubMed] [Google Scholar]
- 161. Kajimura S, Saito M. A new era in brown adipose tissue biology: molecular control of brown fat development and energy homeostasis. Annu Rev Physiol 2014;76:225–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Cinti S. Transdifferentiation properties of adipocytes in the adipose organ. Am J Physiol Endocrinol Metab 2009;297:E977–E986. [DOI] [PubMed] [Google Scholar]
- 163. Petruzzelli M, Schweiger M, Schreiber R, Campos‐Olivas R, Tsoli M, Allen J, et al. A switch from white to brown fat increases energy expenditure in cancer‐associated cachexia. Cell Metab 2014;20:433–447. [DOI] [PubMed] [Google Scholar]
- 164. Vijgen GH, Bouvy ND, Teule GJ, Brans B, Schrauwen P, van Marken Lichtenbelt WD. Brown adipose tissue in morbidly obese subjects. PLoS One 2011;6:e17247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Yoneshiro T, Aita S, Matsushita M, Okamatsu‐Ogura Y, Kameya T, Kawai Y, et al. Age‐related decrease in cold‐activated brown adipose tissue and accumulation of body fat in healthy humans. Obesity (Silver Spring) 2011;19:1755–1760. [DOI] [PubMed] [Google Scholar]
- 166. Lee P, Greenfield JR, Ho KK, Fulham MJ. A critical appraisal of the prevalence and metabolic significance of brown adipose tissue in adult humans. Am J Physiol Endocrinol Metab 2010;299:E601–E606. [DOI] [PubMed] [Google Scholar]
- 167. Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med 2009;360:1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Shellock FG, Riedinger MS, Fishbein MC. Brown adipose tissue in cancer patients: possible cause of cancer‐induced cachexia. J Cancer Res Clin Oncol 1986;111:82–85. [DOI] [PubMed] [Google Scholar]
- 169. Huang YC, Chen TB, Hsu CC, Li SH, Wang PW, Lee BF, et al. The relationship between brown adipose tissue activity and neoplastic status: an (18)F‐FDG PET/CT study in the tropics. Lipids Health Dis 2011;10:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Saure EW, Eagan TM, Jensen RL, Voll‐Aanerud M, Aukrust P, Bakke PS, et al. Explained variance for blood gases in a population with COPD. Clin Respir J 2012;6:72–80. [DOI] [PubMed] [Google Scholar]
- 171. Nakayama K, Frew IJ, Hagensen M, Skals M, Habelhah H, Bhoumik A, et al. Siah2 regulates stability of prolyl‐hydroxylases, controls HIF1alpha abundance, and modulates physiological responses to hypoxia. Cell 2004;117:941–952. [DOI] [PubMed] [Google Scholar]
- 172. van den Borst B, Schols AM, de Theije C, Boots AW, Kohler SE, Goossens GH, et al. Characterization of the inflammatory and metabolic profile of adipose tissue in a mouse model of chronic hypoxia. J Appl Physiol (1985). 2013;114:1619–1628. [DOI] [PubMed] [Google Scholar]
- 173. Sun K, Wernstedt Asterholm I, Kusminski CM, Bueno AC, Wang ZV, Pollard JW, et al. Dichotomous effects of VEGF‐A on adipose tissue dysfunction. Proc Natl Acad Sci U S A 2012;109:5874–5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Elias I, Franckhauser S, Ferre T, Vila L, Tafuro S, Munoz S, et al. Adipose tissue overexpression of vascular endothelial growth factor protects against diet‐induced obesity and insulin resistance. Diabetes 2012;61:1801–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Franssen FM, Sauerwein HP, Ackermans MT, Rutten EP, Wouters EF, Schols AM. Increased postabsorptive and exercise‐induced whole‐body glucose production in patients with chronic obstructive pulmonary disease. Metabolism 2011;60:957–964. [DOI] [PubMed] [Google Scholar]
- 176. Engelen MP, Schols AM, Does JD, Deutz NE, Wouters EF. Altered glutamate metabolism is associated with reduced muscle glutathione levels in patients with emphysema. Am J Respir Crit Care Med 2000;161:98–103. [DOI] [PubMed] [Google Scholar]
- 177. Engelen MP, Schols AM, Does JD, Gosker HR, Deutz NE, Wouters EF. Exercise‐induced lactate increase in relation to muscle substrates in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2000;162:1697–1704. [DOI] [PubMed] [Google Scholar]
- 178. Perez de Heredia F, Wood IS, Trayhurn P. Hypoxia stimulates lactate release and modulates monocarboxylate transporter (MCT1, MCT2, and MCT4) expression in human adipocytes. Pflugers Arch 2010;459:509–518. [DOI] [PubMed] [Google Scholar]
- 179. Carriere A, Jeanson Y, Berger‐Muller S, Andre M, Chenouard V, Arnaud E, et al. Browning of white adipose cells by intermediate metabolites: an adaptive mechanism to alleviate redox pressure. Diabetes 2014;63:3253–3265. [DOI] [PubMed] [Google Scholar]
- 180. Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, et al. Functional brown adipose tissue in healthy adults. N Engl J Med 2009;360:1518–1525. [DOI] [PubMed] [Google Scholar]
- 181. Soderlund V, Larsson SA, Jacobsson H. Reduction of FDG uptake in brown adipose tissue in clinical patients by a single dose of propranolol. Eur J Nucl Med Mol Imaging 2007;34:1018–1022. [DOI] [PubMed] [Google Scholar]
- 182. Hofford JM, Milakofsky L, Vogel WH, Sacher RS, Savage GJ, Pell S. The nutritional status in advanced emphysema associated with chronic bronchitis. A study of amino acid and catecholamine levels. Am Rev Respir Dis 1990;141:902–908. [DOI] [PubMed] [Google Scholar]
- 183. Schiffelers SL, Blaak EE, Baarends EM, Van Baak MA, Saris WH, Wouters EF, et al. beta‐Adrenoceptor‐mediated thermogenesis and lipolysis in patients with chronic obstructive pulmonary disease. Am J Physiol Endocrinol Metab 2001;280:E357–E364. [DOI] [PubMed] [Google Scholar]
- 184. Vosselman MJ, van der Lans AA, Brans B, Wierts R, van Baak MA, Schrauwen P, et al. Systemic beta‐adrenergic stimulation of thermogenesis is not accompanied by brown adipose tissue activity in humans. Diabetes 2012;61:3106–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Parysow O, Mollerach AM, Jager V, Racioppi S, San Roman J, Gerbaudo VH. Low‐dose oral propranolol could reduce brown adipose tissue F‐18 FDG uptake in patients undergoing PET scans. Clin Nucl Med 2007;32:351–357. [DOI] [PubMed] [Google Scholar]
- 186. Lee H, Kim S, Lim Y, Gwon H, Kim Y, Ahn JJ, et al. Nutritional status and disease severity in patients with chronic obstructive pulmonary disease (COPD). Arch Gerontol Geriatr 2013;56:518–523. [DOI] [PubMed] [Google Scholar]
- 187. Goris AH, Vermeeren MA, Wouters EF, Schols AM, Westerterp KR. Energy balance in depleted ambulatory patients with chronic obstructive pulmonary disease: the effect of physical activity and oral nutritional supplementation. Br J Nutr 2003;89:725–731. [DOI] [PubMed] [Google Scholar]
- 188. Schols AM, Soeters PB, Mostert R, Saris WH, Wouters EF. Energy balance in chronic obstructive pulmonary disease. Am Rev Respir Dis 1991;143:1248–1252. [DOI] [PubMed] [Google Scholar]
- 189. Creutzberg EC, Wouters EF, Vanderhoven‐Augustin IM, Dentener MA, Schols AM. Disturbances in leptin metabolism are related to energy imbalance during acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2000;162:1239–1245. [DOI] [PubMed] [Google Scholar]
- 190. Bruera E. ABC of palliative care. Anorexia, cachexia, and nutrition. BMJ 1997;315:1219–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Pilhatsch M, Scheuing H, Kroemer N, Kobiella A, Bidlingmaier M, Farger G, et al. Nicotine administration in healthy non‐smokers reduces appetite but does not alter plasma ghrelin. Hum Psychopharmacol 2014;29:384–387. [DOI] [PubMed] [Google Scholar]
- 192. Eckerblad J, Todt K, Jakobsson P, Unosson M, Skargren E, Kentsson M, et al. Symptom burden in stable COPD patients with moderate or severe airflow limitation. Heart Lung 2014;43:351–357. [DOI] [PubMed] [Google Scholar]
- 193. Hilmarsen CW, Wilke S, Engan H, Spruit MA, Rodenburg J, Janssen DJ, et al. Impact of symptoms of anxiety and depression on COPD Assessment Test scores. Eur Respir J 2014;43:898–900. [DOI] [PubMed] [Google Scholar]
- 194. Gronberg AM, Slinde F, Engstrom CP, Hulthen L, Larsson S. Dietary problems in patients with severe chronic obstructive pulmonary disease. J Hum Nutr Diet 2005;18:445–452. [DOI] [PubMed] [Google Scholar]
- 195. Sarna L, Lindsey AM, Dean H, Brecht ML, McCorkle R. Weight change and lung cancer: relationships with symptom distress, functional status, and smoking. Res Nurs Health 1994;17:371–379. [DOI] [PubMed] [Google Scholar]
- 196. Lyer S, Roughley A, Rider A, Taylor‐Stokes G. The symptom burden of non‐small cell lung cancer in the USA: a real‐world cross‐sectional study. Support Care Cancer 2014;22:181–187. [DOI] [PubMed] [Google Scholar]
- 197. Bentsen SB, Rustoen T, Miaskowski C. Prevalence and characteristics of pain in patients with chronic obstructive pulmonary disease compared to the Norwegian general population. J Pain 2011;12:539–545. [DOI] [PubMed] [Google Scholar]
- 198. Roberts MH, Mapel DW, Hartry A, Von Worley A, Thomson H. Chronic pain and pain medication use in chronic obstructive pulmonary disease. A cross‐sectional study. Ann Am Thorac Soc 2013;10:290–298. [DOI] [PubMed] [Google Scholar]
- 199. Ketwaroo GA, Cheng V, Lembo A. Opioid‐induced bowel dysfunction. Curr Gastroenterol Rep 2013;15:344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Portenoy RK, Ahmed E. Principles of opioid use in cancer pain. J Clin Oncol 2014;32:1662–1670. [DOI] [PubMed] [Google Scholar]
- 201. Jamshed N, Lee ZE, Olden KW. Diagnostic approach to chronic constipation in adults. Am Fam Physician 2011;84:299–306. [PubMed] [Google Scholar]
- 202. Tielemans MM, Jaspers Focks J, van Rossum LG, Eikendal T, Jansen JB, Laheij RJ, et al. Gastrointestinal symptoms are still prevalent and negatively impact health‐related quality of life: a large cross‐sectional population based study in The Netherlands. PLoS One 2013;8:e69876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Veldhuizen MG, Albrecht J, Zelano C, Boesveldt S, Breslin P, Lundstrom JN. Identification of human gustatory cortex by activation likelihood estimation. Hum Brain Mapp 2011;32:2256–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Oliveira‐Maia AJ, Roberts CD, Simon SA, Nicolelis MA. Gustatory and reward brain circuits in the control of food intake. Adv Tech Stand Neurosurg 2011;36:31–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Norden J, Gronberg AM, Bosaeus I, Berteus Forslund H, Hulthen L, Rothenberg E, et al. Nutrition impact symptoms and body composition in patients with COPD. Eur J Clin Nutr 2015;69:256–261. [DOI] [PubMed] [Google Scholar]
- 206. Dewan NA, Bell CW, Moore J, Anderson B, Kirchain W, O'Donohue WJ Jr. Smell and taste function in subjects with chronic obstructive pulmonary disease. Effect of long‐term oxygen via nasal cannulas. Chest 1990;97:595–599. [DOI] [PubMed] [Google Scholar]
- 207. Wardwell L, Chapman‐Novakofski K, Brewer MS. Effects of age, gender and chronic obstructive pulmonary disease on taste acuity. Int J Food Sci Nutr 2009;60:84–97. [DOI] [PubMed] [Google Scholar]
- 208. Janssen P, Vanden Berghe P, Verschueren S, Lehmann A, Depoortere I, Tack J. Review article: the role of gastric motility in the control of food intake. Aliment Pharmacol Ther 2011;33:880–894. [DOI] [PubMed] [Google Scholar]
- 209. Karapanagiotou EM, Tsochatzis EA, Dilana KD, Tourkantonis I, Gratsias I, Syrigos KN. The significance of leptin, adiponectin, and resistin serum levels in non‐small cell lung cancer (NSCLC). Lung Cancer 2008;61:391–397. [DOI] [PubMed] [Google Scholar]
- 210. Terzidis A, Sergentanis TN, Antonopoulos G, Syrigos C, Efremidis A, Polyzos A, et al. Elevated serum leptin levels: a risk factor for non‐small‐cell lung cancer? Oncology 2009;76:19–25. [DOI] [PubMed] [Google Scholar]
- 211. Itoh T, Nagaya N, Yoshikawa M, Fukuoka A, Takenaka H, Shimizu Y, et al. Elevated plasma ghrelin level in underweight patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2004;170:879–882. [DOI] [PubMed] [Google Scholar]
- 212. Karapanagiotou EM, Polyzos A, Dilana KD, Gratsias I, Boura P, Gkiozos I, et al. Increased serum levels of ghrelin at diagnosis mediate body weight loss in non‐small cell lung cancer (NSCLC) patients. Lung Cancer 2009;66:393–398. [DOI] [PubMed] [Google Scholar]
- 213. Sescousse G, Caldu X, Segura B, Dreher JC. Processing of primary and secondary rewards: a quantitative meta‐analysis and review of human functional neuroimaging studies. Neurosci Biobehav Rev 2013;37:681–696. [DOI] [PubMed] [Google Scholar]
- 214. Murdaugh DL, Cox JE, Cook EW 3rd, Weller RE. fMRI reactivity to high‐calorie food pictures predicts short‐ and long‐term outcome in a weight‐loss program. Neuroimage 2012;59:2709–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Patra SK, Arora S. Integrative role of neuropeptides and cytokines in cancer anorexia‐cachexia syndrome. Clin Chim Acta 2012;413:1025–1034. [DOI] [PubMed] [Google Scholar]
- 216. Luo N, Marcelin G, Liu SM, Schwartz G, Chua S Jr. Neuropeptide Y and agouti‐related peptide mediate complementary functions of hyperphagia and reduced energy expenditure in leptin receptor deficiency. Endocrinology 2011;152:883–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Briggs DI, Andrews ZB. Metabolic status regulates ghrelin function on energy homeostasis. Neuroendocrinology 2011;93:48–57. [DOI] [PubMed] [Google Scholar]
- 218. Sanchez‐Lara K, Arrieta O, Pasaye E, Laviano A, Mercadillo RE, Sosa‐Sanchez R, et al. Brain activity correlated with food preferences: a functional study comparing advanced non‐small cell lung cancer patients with and without anorexia. Nutrition 2013;29:1013–1019. [DOI] [PubMed] [Google Scholar]
- 219. Sahebjami H, Vassallo CL. Influence of starvation on enzyme‐induced emphysema. J Appl Physiol Respir Environ Exerc Physiol 1980;48:284–288. [DOI] [PubMed] [Google Scholar]
- 220. Sahebjami H, MacGee J. Effects of starvation on lung mechanics and biochemistry in young and old rats. J Appl Physiol (1985) 1985;58:778–784. [DOI] [PubMed] [Google Scholar]
- 221. Sahebjami H, Domino M. Effects of starvation and refeeding on elastase‐induced emphysema. J Appl Physiol (1985). 1989;66:2611–2616. [DOI] [PubMed] [Google Scholar]
- 222. Bishai JM, Mitzner W. Effect of severe calorie restriction on the lung in two strains of mice. Am J Physiol Lung Cell Mol Physiol 2008;295:L356–L362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Coxson HO, Chan IH, Mayo JR, Hlynsky J, Nakano Y, Birmingham CL. Early emphysema in patients with anorexia nervosa. Am J Respir Crit Care Med 2004;170:748–752. [DOI] [PubMed] [Google Scholar]
- 224. Thurlbeck WM. Diaphragm and body weight in emphysema. Thorax 1978;33:483–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Golgeli A, Ozesmi C, Suer C. Effect of acute starvation on rat diaphragm function. Jpn J Physiol 1994;44:743–747. [DOI] [PubMed] [Google Scholar]
- 226. Efthimiou J, Fleming J, Gomes C, Spiro SG. The effect of supplementary oral nutrition in poorly nourished patients with chronic obstructive pulmonary disease. Am Rev Respir Dis 1988;137:1075–1082. [DOI] [PubMed] [Google Scholar]
- 227. Weekes CE, Emery PW, Elia M. Dietary counselling and food fortification in stable COPD: a randomised trial. Thorax 2009;64:326–331. [DOI] [PubMed] [Google Scholar]
- 228. Engelen MP, Rutten EP, De Castro CL, Wouters EF, Schols AM, Deutz NE. Casein protein results in higher prandial and exercise induced whole body protein anabolism than whey protein in chronic obstructive pulmonary disease. Metabolism 2012;61:1289–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Engelen MP, De Castro CL, Rutten EP, Wouters EF, Schols AM, Deutz NE. Enhanced anabolic response to milk protein sip feeding in elderly subjects with COPD is associated with a reduced splanchnic extraction of multiple amino acids. Clin Nutr 2012;31:616–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. van de Bool C, Mattijssen‐Verdonschot C, van Melick PP, Spruit MA, Franssen FM, Wouters EF, et al. Quality of dietary intake in relation to body composition in patients with chronic obstructive pulmonary disease eligible for pulmonary rehabilitation. Eur J Clin Nutr 2014;68:159–165. [DOI] [PubMed] [Google Scholar]
- 231. Rutten EP, Lenaerts K, Buurman WA, Wouters EF. Disturbed intestinal integrity in patients with COPD: effects of activities of daily living. Chest 2014;145:245–252. [DOI] [PubMed] [Google Scholar]
- 232. Engelen MP, Rutten EP, De Castro CL, Wouters EF, Schols AM, Deutz NE. Altered interorgan response to feeding in patients with chronic obstructive pulmonary disease. Am J Clin Nutr 2005;82:366–372. [DOI] [PubMed] [Google Scholar]
- 233. Pouw EM, Schols AM, Deutz NE, Wouters EF. Plasma and muscle amino acid levels in relation to resting energy expenditure and inflammation in stable chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998;158:797–801. [DOI] [PubMed] [Google Scholar]
- 234. Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth‐hormone‐releasing acylated peptide from stomach. Nature 1999;402:656–660. [DOI] [PubMed] [Google Scholar]
- 235. Sun Y, Wang P, Zheng H, Smith RG. Ghrelin stimulation of growth hormone release and appetite is mediated through the growth hormone secretagogue receptor. Proc Natl Acad Sci U S A 2004;101:4679–4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Miki K, Maekura R, Nagaya N, Kitada S, Miki M, Yoshimura K, et al. Effects of ghrelin treatment on exercise capacity in underweight COPD patients: a substudy of a multicenter, randomized, double‐blind, placebo‐controlled trial of ghrelin treatment. BMC Pulm Med 2013;13:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Garcia JM, Boccia RV, Graham CD, Yan Y, Duus EM, Allen S, et al. Anamorelin for patients with cancer cachexia: an integrated analysis of two phase 2, randomised, placebo‐controlled, double‐blind trials. Lancet Oncol 2015;16: 108–116. [DOI] [PubMed] [Google Scholar]
- 238. Currow DC, Abernethy AP. Anamorelin hydrochloride in the treatment of cancer anorexia‐cachexia syndrome. Future Oncol 2014;10:789–802. [DOI] [PubMed] [Google Scholar]
- 239. Ziauddeen H, Farooqi IS, Fletcher PC. Obesity and the brain: how convincing is the addiction model? Nat Rev Neurosci 2012;13:279–286. [DOI] [PubMed] [Google Scholar]
- 240. Stice E, Yokum S, Burger KS, Epstein LH, Small DM. Youth at risk for obesity show greater activation of striatal and somatosensory regions to food. J Neurosci 2011;31:4360–4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Roefs A, Stapert D, Isabella LA, Wolters G, Wojciechowski F, Jansen A. Early associations with food in anorexia nervosa patients and obese people assessed in the affective priming paradigm. Eat Behav 2005;6:151–163. [DOI] [PubMed] [Google Scholar]
- 242. Zipfel S, Wild B, Gross G, Friederich HC, Teufel M, Schellberg D, et al. Focal psychodynamic therapy, cognitive behaviour therapy, and optimised treatment as usual in outpatients with anorexia nervosa (ANTOP study): randomised controlled trial. Lancet 2014;383:127–137. [DOI] [PubMed] [Google Scholar]
- 243. Deckersbach T, Das SK, Urban LE, Salinardi T, Batra P, Rodman AM, et al. Pilot randomized trial demonstrating reversal of obesity‐related abnormalities in reward system responsivity to food cues with a behavioral intervention. Nutr Diabetes 2014;4:e129. [DOI] [PMC free article] [PubMed] [Google Scholar]