

Summary
Neovascularization (NV), as a cardinal complication of several ocular diseases, has been intensively studied, and research has shown its close association with inflammation and immune cells. In the present study, the role of interleukin‐17A (IL‐17A) in angiogenesis in the process of ocular NV both in vivo and in vitro was investigated. Also, a paracrine role of IL‐17A was demonstrated in the crosstalk between endothelial cells and macrophages in angiogenesis. In the retinas of mice with retinopathy of prematurity, the IL‐17A expression increased significantly at postnatal day 15 (P15) and P18 during retinal NV. Mice given IL‐17A neutralizing antibody (NAb) developed significantly reduced choroidal NV and retinal NV. Studies on vascular endothelial growth factor (VEGF) over‐expressing mice suggested that IL‐17A modulated NV through the VEGF pathway. Furthermore, IL‐17A deficiency shifted macrophage polarization toward an M2 phenotype during retinal NV with significantly reduced M1 cytokine expression compared with wild‐type controls. In vitro assays revealed that IL‐17A treated macrophage supernatant gave rise to elevated human umbilical vascular endothelial cell proliferation, tube formation and VEGF receptor 1 and receptor 2 expression. Therefore, IL‐17A could potentially serve as a novel target for treating ocular NV diseases. The limitation of this study involved the potential mechanisms, such as which transcription accounted for macrophage polarization and how the subsequent cytokines were modulated when macrophages were polarized. Further studies need to be undertaken to definitively determine the extent to which IL‐17A neutralizing anti‐angiogenic activity depends on macrophage modulation compared with anti‐VEGF treatment.
Keywords: interleukin‐17A; M1, M2 macrophage polarization; ocular neovascularization; vascular endothelial cells; vascular endothelial growth factor
Abbreviations
- DMEM
Dulbecco's modified Eagle's medium
- ERK
extracellular signal‐regulated kinase
- FBS
fetal bovine serum
- GM‐CSF
granulocyte–macrophage colony‐stimulating factor
- HUVECs
human umbilical vascular endothelial cells
- IL‐17
interleukin‐17
- JNK
Jun N‐terminal kinase
- KC
keratinocyte‐derived chemokine
- MAPK
mitogen‐activated protein kinase
- MCP‐1
monocyte chemoattractant protein 1
- NAb
neutralizing antibody
- NV
neovascularization
- Pn
postnatal day n
- RANTES
regulated upon activation normal T‐cell expressed and secreted
- RNV/CNV
retinal/choroidal neovascularization
- ROP
retinopathy of prematurity
- TNF‐α
tumour necrosis factor‐α
- VEGF
vascular endothelial growth factor
- WT
wild‐type
Introduction
Formation of blood vessels is an essential process for the development of organism and tissue regeneration. However, angiogenesis occurring during postnatal development, usually abnormal angiogenesis, is probably associated with inflammation.1 Ocular neovascularization (NV) includes retinal and choroidal NV (RNV/CNV), which account for most moderate and severe vision losses in developed countries.2 Treatments for these diseases, such as age‐related macular degeneration and diabetic retinopathy, include numerous intravitreal injections of anti‐vascular endothelial growth factor (anti‐VEGF) antibody to inhibit NV, which costs patients repeated office visits. However, the treatments take effect in only some patients, indicating that anti‐VEGF alone is not effective enough to modulate the NV process.3 Recently, a growing number of studies have revealed that NV is regulated by a complex interplay between inflammatory cytokines and immune cells. During the process, immune cells such as macrophages and granulocytes are recruited to the hypoxic area to produce a variety of cytokines to make a pro‐angiogenic microenvironment.4
Interleukin‐17 (IL‐17) has been reported as a main effector cytokine of Th17 cells and has become increasingly interesting following the identification of Th17 immunology. Six IL‐17 family members have been identified currently, IL‐17A to IL‐17F; IL‐17A, which is often recognized as the signature cytokine of T helper type 17 cells, has been identified as the prototypic family member.5 Interleukin‐17, an inflammatory cytokine, has been studied in immunity, infection, transplantation and tumours, and has been shown to enhance tumorigenesis through the up‐regulation of angiogenesis‐related molecules. Previous investigations by Chung et al.6 revealed a paracrine network mediated by IL‐17‐promoted tumour resistance to anti‐angiogenic therapy. Pickens et al.7 found an increased concentration of IL‐17 in rheumatoid arthritis joints, and it induced human lung microvascular endothelial cell migration. Other research has shown that IL‐17 stimulated tumour growth by up‐regulating pro‐angiogenic factors such as VEGF and matrix metalloproteinase‐9, suggesting an indirect role of IL‐17 in angiogenesis.8
Recently, many reports have demonstrated the involvement of IL‐17 in the pathogenesis of age‐related macular degeneration. These studies identified elevated levels of IL‐17 in patients with age‐related macular degeneration.9, 10 Besides, Hasegawa et al.11 demonstrated that IL‐17 promoted CNV in a VEGF‐independent manner in a laser‐induced CNV mouse model. According to these findings, it is reasonable to support that IL‐17 plays an important role in ocular NV in addition to VEGF. However, further studies on how IL‐17 modulates ocular NV remained to be carried out. Therefore, in this study, the role of IL‐17A in regulating ocular NV and its potential interactions with macrophages and inflammatory cytokines in several ocular NV mouse models and cell assays were investigated.
Materials and methods
Mice and ethics statement
All procedures and animal care were performed in accordance with the Guide for the Care and Use of Laboratory Animals, by the National Institutes of Health, with the approval (SYXK‐2011‐0026) of the Scientific Investigation Board of Shanghai Jiao Tong University School of Medicine, Shanghai, China. Mice used in this study included specific pathogen‐free C57BL/6 mice (Charles River Laboratories, Wilmington, MA), IL‐17A−/− mice (purchased from Jackson Laboratory, Bar Harbor, ME), rhodopsin promoter/VEGF (rho/VEGF) transgenic mice,12 and double transgenic mice with doxycycline‐inducible expression of VEGF in photoreceptors (Tet/opsin/VEGF double transgenic mice),13 all on a C57BL/6 background. All efforts were made to minimize animal suffering.
Mouse model of oxygen‐induced RNV and immunostaining
Mice on a C57BL/6 background were exposed to 75 ± 3% oxygen from postnatal day 7 (P7) to P12 with their nursing mother and returned to room air at P12, as described in a previous study.14 At P12, the mice were divided into several groups, with one eye intravitreously injected with IL‐17A neutralizing antibody (NAb) at 0·1, 0·5 or 1 μg/μl or with recombinant IL‐17A (rIL‐17A) at 0·01, 0·1 or 0·5 μg/μl and the other eye intravitreously injected with PBS. Intraocular injection was performed using a dissecting microscope with a Harvard Pump Microinjection System (Harvard Apparatus, Holliston, MA) and pulled glass micropipettes as described in a previous study.15 At P17, the mice were killed and their eyes were removed and fixed in 4% phosphate‐buffered formalin at room temperature for at least 5 hr. The retinas were then dissected and incubated in fluorescein Griffonia Simplicifolia Lectin‐B4 (GSA‐Lectin, 1 : 50; Vector Laboratories, Inc., Burlingame, CA) labelled with FITC for 45 min at room temperature. After this, the retinas were washed three times in PBS and whole mounted with radial incision from the edge of the retina to the equator on glass slides in a mounting medium (Aquamount; Polysciences, Warrington, PA). Retinal flat‐mounts were examined and captured with a fluorescence microscope (Nikon Instruments Inc., New York, NY), and images were analysed using the image‐pro Plus software (Media Cybernetics, Silver Spring, MD).
Mouse model of CNV and flat‐mounts analysis
Choroidal NV was induced by laser photocoagulation with rupture of Bruch's membrane, as described in a previous study.16 Briefly, 5‐ to 8‐week‐old C57BL/6 mice were anaesthetized with ketamine hydrochloride (100‐mg/kg body weight), and their pupils were dilated with 1% tropicamide. Laser spots were delivered using a system of an OcuLight GL diode laser (100‐μm spot size, 0·1‐second duration, 100 mW; Iridex, Mountain View, CA, USA), with a cover slide placed on the cornea as a contact lens to view the retina, to perform burns at 3, 9 and 12 o'clock positions in the retina per eye. A bubble at the impact site was confirmed as the disruption of the Bruch's membrane. As a result, only laser spots produced with a bubble were included in the study. At days 1 and 7 after photocoagulation, mice were given 0·1, 0·5 or 1 μg/μl of IL‐17A NAb or 0·01, 0·1 or 0·5 μg/μl of rIL‐17A in one eye and PBS in the other eye. Two weeks after photocoagulation, mice were anaesthetized and perfused with 1 ml of PBS containing 50 mg/ml of fluorescein‐labelled dextran (2 × 106 average molecular weight; Sigma‐Aldrich, St Louis, MO) into the left ventricle. The mice were then killed and their eyes were removed and fixed in 4% formalin at room temperature for at least 5 hr. Choroidal membranes were carefully dissected, flat‐mounted, examined and captured with a fluorescence microscope, and images were analysed using the image‐pro plus software, with investigators masked with respect to the experimental groups.
Mouse model of VEGF over‐expressed transgenic mice
Rho/VEGF transgenic mice, in which the rhodopsin promoter drives the expression of human VEGF165 in photoreceptors so that new vessels sprout from the deep capillary bed of the retina starting at P10,12 were intravitreously injected with 0·5 μg/μl IL‐17A NAb or 0·1 μg/μl rIL‐17A in one eye and PBS in the other eye. At P21, the rho/VEGF transgenic mice were anaesthetized and perfused with fluorescein‐labelled dextran (Sigma‐Aldrich). Their eyes were removed and fixed in 4% formalin for at least 5 hr. The cornea and lens were removed and the entire retina was carefully dissected from the eyecup with a radial cut from the edge of the retina to the equator and flat‐mounted in a mounting medium. The retinal flat‐mounts were examined by fluorescence microscopy at a 200 × magnification so that easy delineation of the NV on the outer edge of the retina was accessible, with the remainder of the retinal vessels out of focus. The image‐pro plus software was used to measure the number and the total area of NV lesions per retina. Double‐hemizygous Tet/opsin/VEGF double‐transgenic mice (4–6 weeks old) with doxycyclin‐inducible expression of human VEGF165 were given 2 mg/ml of doxycycline in their drinking water to trigger high expression of VEGF (at least 30‐fold higher than that seen in the rho/VEGF mice) in photoreceptors within 5 days.13 After 5 days, retinas were pooled for RNA isolation.
Immunofluorescence staining of IL‐17A and GSA‐lectin
C57BL/6 mice with retinopathy of prematurity (ROP) or age‐matched controls were killed at P15 and P18. Their eyes were enucleated and rapidly frozen in optimum cutting temperature embedding compound (Miles Laboratories, Elkhart, IN) for frozen section. Sections (10 μm) were thawed, air‐dried and fixed in acetone at −20°C for 20 min. These sections were then incubated in 5% BSA followed by overnight incubation at 4°C in polyclonal rabbit anti‐mouse IL‐17A antibody (Santa Cruz Biotechnology, Inc., Dallas, TX). They were then incubated in Alexa 555 anti‐rabbit IgG (Cell Signaling Technology, Inc., Danvers, MA) and GSA‐Lectin. Antibodies were diluted with an antibody diluent (Dako, Glostrup, Denmark). The sections were finally washed with PBS between these incubations, and examined and captured with a fluorescence microscope.
Cell culture
Human umbilical vascular endothelial cells (HUVECs) and RAW264.7 cells (Cell Bank of Chinese Academy of Science, Shanghai, China) were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS). The cells were resuspended in DMEM containing 2% FBS and seeded in triplicate to six‐well plates until the cells adhered to the plate after 24 hr. The medium of RAW264.7 cells was replaced with 1 ml of fresh DMEM containing 0·2% FBS and different concentrations (0, 10, 50 and 100 ng/ml) of rIL‐17A (R&D Systems, Minneapolis, MN). After 24 hr, the supernatant of RAW264.7 cells was collected to replace the medium of HUVECs for the following incubation. The cells were harvested and subjected to RNA and protein isolation as described under Real‐time RT‐PCR and Immunoblots of retina tissues and cell lysate.
Cell proliferation
The HUVECs were resuspended in DMEM containing 0·2% FBS. Approximately 4 × 103 cells in 100 μl of medium were added in triplicate to each well and incubated at 37°C in 5% CO2 for 24 hr until the cells adhered to the plate. Then the medium was replaced with 100 μl per well of the supernatant of RAW264.7 cells cultured with different concentrations of rIL‐17A or HUVEC culture medium containing different concentrations of rIL‐17A. After 24, 48 or 72 hr of incubation, 10 μl of Cell Counting Kit‐8 (CCK‐8; Dojindo, Kumamoto, Japan) was added to each well in accordance with the manufacturer's instructions, and the cells were incubated for another 90 min at 37°C. The absorbance of 450 nm was measured with a microplate reader (Model 450; Bio‐Rad, Hercules, CA). The independent experiments were repeated three times.
Tube formation assay
Growth factor‐reduced basement membrane matrix (Matrigel; BD Bioscience, San Jose, CA) was added at 150 μl per well into 48‐well plates on ice and the plates were placed at 37°C for 30 min for polymerization. HUVECs (2 × 104) in 100 μl of DMEM containing 0·2% FBS with or without 50 ng/ml rIL‐17A or supernatant of RAW264.7 cells treated with 50 ng/ml rIL‐17A were plated onto the gel surface and incubated at 37°C for 6 hr. VEGF (10 ng/ml) was used as a positive control. Cell tube formation was examined by phase‐contrast microscopy and images of five random fields per well were taken. An endothelial tube number was quantified as described in a previous study.17
Real‐time RT‐PCR
Mice with ROP or age‐matched controls were killed, and their eyes were removed at specific times for observation. The retinas were dissected after an annular incision along the limbus, leaving the anterior segments removed. Total RNA of retinas and harvested cells was isolated using a Trizol reagent (Invitrogen, Carlsbad, CA), in accordance with the manufacturer's instructions.18 Pre‐treated with DNase I (Promega, Fitchburg, WI), 2 μg of each sample of RNA was reverse transcribed into complementary DNA with M‐MLV Transcriptase and oligo dT Primers (Promega) as instructed by the manufacturers. Quantitative RT‐PCR analysis was performed as described in a previous study.19 Briefly, each PCR was carried out in a 20‐μl volume using iQ SYBR Green mix (Roche, Basel, Switzerland) for 10 min at 95°C denaturation, followed by 95°C for 30 seconds and 60°C for 1 min for 40 cycles in ABI 7500, and normalized by housekeeping genes. Two retinas were considered as one sample. The ΔΔC T method was used for relative quantification. Primers used included VEGFR1 (forward: 5′‐TAGTGTTGTGGGCTCTGTATTC‐3′, reverse: 5′‐AGCTTCCTCAGCACACTATTT‐3′), VEGFR2 (forward: 5′‐AGCAGGATGGCAAAGACTAC‐3′, reverse: 5′‐TACTTCCTCCTCCTCCATACAG‐3′), cyclophilin A (forward: 5′‐CAGACGCCACTGTCGCTTT‐3′, reverse: 5′‐TGTCTTTGGAACTTTGTCTGCAA‐3′). Others are listed in Table 1.
Table 1.
Primer sequences and fold changes for real‐time RT‐PCR analysis
| Gene | Accession ID | Forward primer (5′–3′) | Reverse primer (5′–3′) | IL‐17A−/−/WT | |
|---|---|---|---|---|---|
| Fold | P‐value | ||||
| MCP‐1 | NM_011333.3 | CTCGGACTGTGATGCCTTAAT | TAAATGCAAGGTGTGGATCCA | 0·69 | 0·028 (P15) |
| ICAM‐1 | NM_010493.2 | TTCTCATGCCGCACAGAACT | TCCTGGCCTCGGAGACATTA | 0·47 | 0·031 (P15) |
| VEGFa | NM_001287058.1 | CACTTCCAGAAACACGACAAAC | TGGAACCGGCATCTTTATCTC | 0·38 | 3·8E‐04 (P15) |
| CD11c | NM_021334.2 | GTGCCCATCAGTTCCTTACA | GAGAAGAACTGTGGAGCTGAC | 0·303 | 0·014 (P15) |
| iNOS | NM_001313922.1 | CCCTTCAATGGTTGGTACATGG | ACATTGATCTCCGTGACAGCC | 0·66 | 0·27 (P15) |
| IL‐1β | NM_008361.4 | TGCCACCTTTTGACAGTGATG | AAGGTCCACGGGAAAGACAC | 0·32 | 0·0062 (P15) |
| CD206 | NM_008625.2 | GGAATCAAGGGCACAGAGTTA | ATTGTGGAGCAGATGGAA | 5·14 | 4·02E‐06 (P17) |
| IL‐10 | NM_010548.2 | TAACTGCACCCACTTCCCAG | AAGGCTTGGCAACCCAAGTA | 6·37 | 0·014 (P17) |
| CD163 | NM_001170395.1 | CAGACTGGTTGGAGGAGAAATC | TGACTTGTCTCTGGAAGCTG | 2·28 | 0·18 (P17) |
| TGF‐β | NM_025609.2 | ATTCCTGGCGTTACCTTGG | AGCCCTGTATTCCGTCTCCT | 2·20 | 0·011 (P17) |
| MHC II | NM_007575.2 | GTCTCTGTCCTGGTGGCTCT | AGGTTCTGGGAGGTGATGG | 0·31 | 0·026 (P17) |
| IL‐1ra | NM_001159562.1 | TAGTGTGTTCTTGGGCATCC | CGCTTGTCTTCTTCTTTGTTCT | 1·49 | 0·048 (P17) |
| Fizz1 | NM_020509.3 | CGTGGAGAATAAGGTCAAGGA | CAGTAGCAGTCATCCCAGCA | 4·54 | 0·045 (P17) |
| Ym1 | NM_009892.3 | TCTCTACTCCTCAGAACCGTCAG | CGCATTTCCTTCACCAGAAC | 1·19 | 0·59 (P17) |
| TLR4 | NM_021297.3 | TCAGAGCCGTTGGTGTATCTT | GCTTTCTTGGGCTTCCTCTT | 0·50 | 0·042 (P15) |
| TLR2 | NM_011905.3 | GTGTCTGGAGTCTGCTGTGC | GCTTTCTTGGGCTTCCTCTT | 1·12 | 0·73 (P15) |
| CD80 | NM_009855.2 | TGTCCAAGGCTCATTCTTCTC | TAACGGCAAGGCAGCAATA | 0·91 | 0·67 (P15) |
| CD86 | NM_019388.3 | AGCACGGACTTGAACAACCA | TGTAAATGGGCACGGCAGAT | 0·48 | 0·031 (P15) |
| IFN‐γ | NM_008337.4 | AGCAAGGCGAAAAAGGATGC | TCATTGAATGCTTGGCGCTG | 0·26 | 0·041 (P15) |
| IL‐12a | NM_001159424.2 | TCTTCTCACCGTGCACATCC | TGGCCAAACTGAGGTGGTTT | 0·16 | 0·083 (P15) |
| IL‐23 | NM_031252.2 | CAAAGGATCCGCCAAGGTCT | GGAGGTGTGAAGTTGCTCCA | 1·04 | 0·91 (P15) |
| IL‐6 | NM_001314054.1 | GACAAAGCCAGAGTCCTTCAGA | TGTGACTCCAGCTTATCTCTTGG | 0·19 | 0·0032 (P15) |
| 0·23 | 0·035 (P17) | ||||
| TNF‐α | NM_013693.3 | GAACTGGCAGAAGAGGCACT | AGGGTCTGGGCCATAGAACT | 0·33 | 0·0083 (P15) |
| 0·27 | 0·032(P17) | ||||
Messenger RNA expression of macrophage polarization‐associated genes in the wild‐type (WT) and interleukin‐17A‐deficient (IL‐17A−/−) mouse model of retinopathy of prematurity. To investigate the effect of IL‐17A blockade on macrophage polarization, M1‐ and M2‐associated cytokines were analysed at P15 and P17. Relative gene expressions were analysed using the ΔΔCT method and endogenously normalized to cyclophilin A. Statistical analysis were followed with the Student's t‐test (n = 12 mice/group).
Immunoblots of retina tissues and cell lysate
C57BL/6 mice with ROP or age‐matched controls were killed at P13, P15, P18 and P21. The retinas were immediately dissected and pooled for protein isolation. The retina tissues were sonicated for 5 seconds at 4°C, and RAW264.7 cells were lysed in ice‐cold protein lysis buffer containing 2% proteinase inhibitor and 1% phosphatase inhibitor (Roche), in accordance with the manufacturer's instructions. The protein concentration of the supernatant was measured using a BCA Protein Quantification Assay Kit (Thermofisher Scientific, Göteborg, Sweden). Protein (100 μg) was loaded in wells of a 10% SDS gel, and separated proteins were transferred to a nitrocellulose membrane after electrophoresis. The membrane was incubated in 0·05 m Tris‐buffered saline (TBS), pH 7·6, containing 5% skim milk, for 2 hr at room temperature to block non‐specific binding sites, and then probed with primary antibodies overnight at 4°C, followed by incubation with horseradish peroxidase‐conjugated goat anti‐rabbit polyclonal antibody (Amersham Pharmacia Biotech, Livingston, NJ) at room temperature for 1 hr then washing in TBST three times. Membranes were then incubated in an Enhanced Chemoluminescence‐Plus Western Blotting Detection Reagent (Amersham Pharmacia Biotech) for signal detection. Primary antibodies used included rabbit anti‐mouse IL‐17A antibody, Jun N‐terminal kinase (JNK), phospho‐JNK, Akt, phospho‐Akt (Thr308), extracellular signal‐regulated kinase 1/2 (ERK1/2), phospho‐ERK1/2, Notch1, p38, phospho‐p38 (Cell Signaling Technology), and β‐actin (Sigma‐Aldrich).
Antibody array detection of cytokine levels
Cytokine levels in culture supernatant of RAW264.7 treated with rIL‐17A were measured using a Quantibody Cytokine Array Kit (QAM‐CYT‐1‐1; RayBiotech, Inc., Norcross, GA). Cytokines including IL‐1α, IL‐1β, IL‐2, IL‐3, IL‐4, IL‐5, IL‐6, IL‐9, IL‐10, IL‐12, IL‐13, IL‐17, keratinocyte‐derived chemokine (KC), monocyte chemotactic protein 1 (MCP‐1), macrophage colony‐stimulating factor (M‐CSF,) regulated upon activation normal T‐cell expressed and secreted (RANTES), tumour necrosis factor‐α (TNF‐α), VEGF, granulocyte–macrophage colony‐stimulating factor (GM‐CSF) and interferon‐γ were quantitatively detected according to the manufacturer's instructions, and the data were analysed with an accessible software provided by the company.
Quantification of NO production in cell supernatant
RAW264.7 macrophages (5 × 104/ml) were cultured in DMEM with different concentrations of rIL‐17A for 24 hr. The supernatant was then collected for measuring the accumulation of nitrite according to the Griess reaction.20 Briefly, equal volumes of culture supernatant from each well sample of medium were mixed with Griess reagent in a 96‐well plate. After 15 min of incubation at room temperature, the absorbance at 550 nm was examined and the nitrite concentration in the supernatants was calculated using nitrite as a standard.
Isolation of mouse retina CD11b+ cells
Mouse retinas of P13, P15, P18 and P21 from wild‐type (WT) mice and IL‐17−/− mice with oxygen‐induced retinopathy (each time‐point included 6–10 mice, one mouse as one sample) were carefully dissected out and digested in pre‐warmed 16·5‐U/ml papain solution (Worthington Biochemical, Lakewood, NJ) for 30 min with gentle pipetting. The cell digestion suspensions were then transferred and passed through cell strainers (BD Falcon, Franklin Lakes, NJ, USA) to ensure single‐cell suspension. Trypsin inhibitor was added to stop digestion, and then cells were spun down at 900 rpm (89g). After gently removing the supernatant, the cell pellet was resuspended with 90‐μl MACS buffer (BD Biosciences) and mixed well with 10‐μl anti‐mouse CD11b magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany), incubated at 4°C for 20 min, washed once, and resuspended in 500‐μl MACS buffer. The cell pellet was then loaded on a pre‐moisturized MS column (BD Biosciences) and washed twice. Then the columns were taken off the magnetite and residue cells were flushed out of the column, according to the manufacturer's protocol. The selected cells were collected and analysed by flow cytometry. All retina cell samples from different mice were treated as mentioned separately.
Flow cytometry analysis
The CD11b+ cells from the retinas of mice with ROP were resuspended in MACS buffer (BD Biosciences) and incubated with phycoerythrin‐conjugated anti‐mouse CD11c (BD Biosciences), F4/80 (eBioscience, Vienna, Austria), Alexa Fluor 647‐conjugated CD206 (AbD), and the matching control isotype IgGs for 30 min at 4°C. Then the cells were washed and rinsed again and incubated with secondary antibodies for 30 min at 4°C. The cells were then washed and resuspended in FACS buffer (BD Biosciences) and analysed by flow cytometry (FACSCalibur cytometer; BD Biosciences, Heidelberg, Germany). M1 macrophages were identified as F4/80‐positive/CD11c‐positive and M2 macrophages as F4/80‐positive/CD206‐positive cells.21 Data analyses were performed using flowjo software (Tree Star, Ashland, OR).
Statistical analysis
Quantitative data were presented as mean values ± standard error of mean (SEM). Statistical significance was determined by the two‐tailed Student's t‐test or one‐way analysis of variance with the Student–Newman–Keuls method for multiple comparisons. Differences were considered to be statistically significant at P values of 0·05. Statistical analysis was performed using SAS 9.0 software (SAS Institute Inc., Cary, NC, USA).
Results
Detection of high levels of IL‐17A in ROP mice retinas as RNV progressed
Immunofluorescence staining, quantitative PCR and immunoblot assays were performed to investigate if IL‐17A played a role in RNV progress. Quantitative PCR and immunoblot assays demonstrated that IL‐17A elevated significantly at P15 and P18 in retinas of mice with ROP compared with age‐matched controls (Fig. 1b–d). Immunofluorescence staining also showed high IL‐17A expression in retinas of mice with ROP at P15 and P18. Merged images did not show co‐localization of IL‐17A and lectin, which stained for endothelial cells (Fig. 1a). These findings indicated that IL‐17A played a supportive role in RNV progress, but it might not be directly through endothelial cells.
Figure 1.
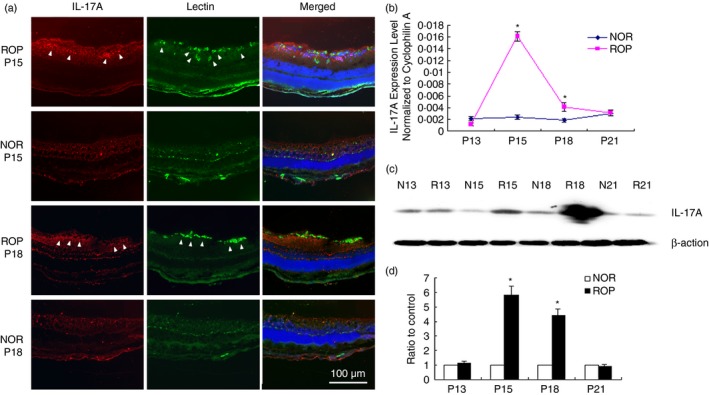
Interleukin‐17A (IL‐17A) expression examined by immunofluorescence staining, quantitative PCR and immunoblots in retinas of mice with retinopathy of prematurity (ROP) at postnatal day 13 (P13), P15, P18 and P21. Mice with ROP were killed at P13, P15, P18 and P21 and the eyes of ROP mice and age‐matched controls at P15 and P18 were enucleated, fixed and prepared for frozen sections. Immunofluorescence staining of IL‐17A (red) and lectin (green) was performed at P15 and P18 in the retinas of mice with ROP (arrowheads) as neovascularization (NV) progressed (a). Quantitative PCR and immunoblot assays were performed to evaluate the expression level of IL‐17A from P13 to P21 in mice with ROP and age‐matched controls (b and c). Quantification of the protein level of IL‐17A in mouse retinas (d). Data were mean ± SEM from three independent experiments. Statistical analysis was performed using the two‐tailed Student's t‐test (n = 6–8 mice/group, *P < 0·05).
Pro‐angiogenic effects of IL‐17A on NV both in RNV and CNV mouse models
The IL‐17A−/− and WT mice, both on C57BL/6 background, were subjected to both ROP and CNV models as described in the Materials and methods section. The WT mice were divided into several groups at random to evaluate the effects of rIL‐17A and IL‐17A NAb on RNV and CNV. To investigate the effects as well as the effective concentration, dose–effect experiments were performed with these two mouse models. Results revealed that IL‐17A neutralization significantly inhibited RNV and CNV at a concentration of 0·5 μg/μl, whereas rIL‐17A promoted NV with a significant difference at a concentration of 0·1 μg/μl compared with PBS‐injected eyes. Retinal and choroidal flat‐mounts were performed as displayed in Fig. 2. The area of CNV and RNV decreased significantly in IL‐17A−/− and 0·5‐μg/μl IL‐17A Nab‐treated mice, but increased in mice treated with 0·1 μg/μl rIL‐17A (Fig. 2a–e and g–k). High magnification of retinal flat‐mounts (arrows) showed less and smaller NV sprouts of IL‐17A−/− (Fig. 2h) and IL‐17A Nab‐treated mice (Fig. 2j). These findings demonstrated that IL‐17A probably acted as a pro‐angiogenic cytokine in the ROP and CNV mouse models.
Figure 2.
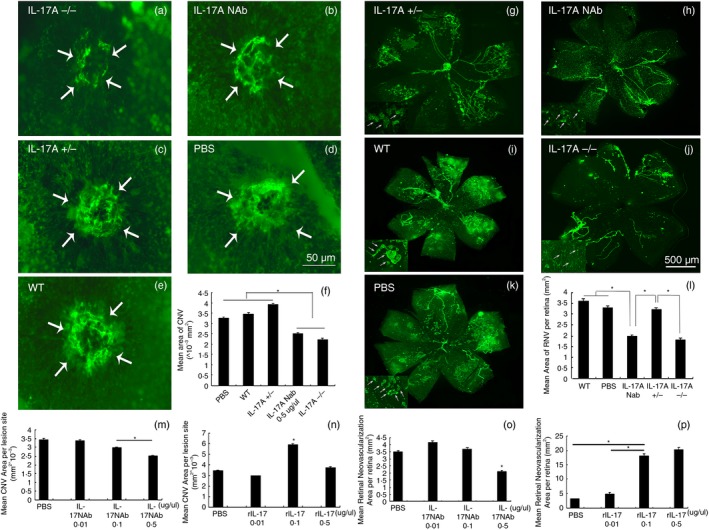
Immunofluorescent staining of retinas and choroidal flat‐mounts of models of mice with retinopathy of prematurity (ROP) and choroidal neovascularization (CNV). Interleukin‐17A‐deficient (IL‐17A−/−) and wild‐type (WT) C57BL/6 mice were subjected to hypoxic retinopathy and laser‐induced CNV as described in the Materials and methods section. Among them, the WT mice were divided into several groups at random, with one eye intravitreously injected with rIL‐17A or IL‐17A neutralizing antibody (NAb) and the other eye injected with PBS at postnatal day 12 (P12) for mice with ROP, and at days 1 and 7 after laser photocoagulation for mice with CNV. The retinas of mice with ROP were flat‐mounted and stained with FITC‐lectin at P17, while choroidal flat‐mounts from mice with CNV were performed at day 14 after photocoagulation as described (a–e: n = 10 mice/group; g–k: n = 10 mice/group). Dose–effect experiments of IL‐17 NAb and rIL‐17A were performed (m–p). The image‐pro plus software was used to quantify the area of NV (f and l). Statistics were analysed using the Student–Newman–Keuls method. Data were mean ± SEM; *P < 0·05.
Reduction of sub‐retinal NV by IL‐17A neutralization in VEGF over‐expressed mice, but not conversely
Rho/VEGF mice were intravitreously injected with IL‐17A NAb and rIL‐17A in one eye and PBS in the other at P12. Retinas were dissected, flat‐mounted, and stained with FITC‐lectin at P21. An examination of retina flat‐mounts under fluorescence microscopy revealed that IL‐17A NAb inhibited sub‐RNV significantly, whereas rIL‐17A did the opposite (Fig. 3a–h). Further investigation as to whether high levels of VEGF expression stimulated IL‐17A expression was carried out using VEGF‐over‐expressed and VEGF‐neutralized mice. Quantitative PCR assays of these mouse retinas demonstrated no significant difference of IL‐17A expression in VEGF‐over‐expressed and VEGF‐neutralized mice compared with controls (Fig. 3i–k). These findings revealed that VEGF‐induced ocular NV seemed independent of IL‐17A, but IL‐17A might modulate ocular NV through VEGF.
Figure 3.
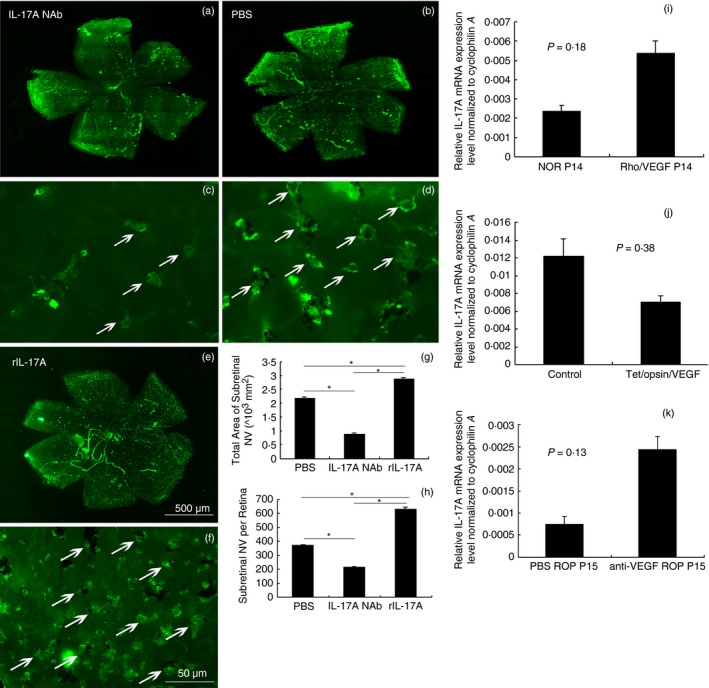
Immunofluorescence staining of retinal flat‐mounts of rho/vascular endothelial growth factor (VEGF) mice treated with interleukin‐17A neutralizing antibody (IL‐17A NAb), rIL‐17A, and IL‐17A mRNA expression in the retinas of VEGF‐over‐expressed and VEGF‐neutralized mice. IL‐17A NAb and rIL‐17A were intravitreously injected into the eyes of rho/VEGF mice at postnatal day 12 (P12), with control eyes injected with PBS; the retinas were dissected, stained with FITC‐lectin, and flat‐mounted at P21 (n = 12 mice/group). The total area of sub‐retinal neovascularization (RNV) was reckoned by the image‐pro plus software (a–h). Data were analysed with the Student–Newman–Keuls test. The retinas of rho/VEGF mice and Tet/opsin/VEGF double‐transgenic mice were dissected for RNA isolation as described in the Materials and methods section, including the retinas of mice with retinopathy of prematurity (ROP) treated with anti‐VEGF NAb (n = 12 mice/group). The real‐time RT‐PCR demonstrated the difference of IL‐17A mRNA expression in these VEGF‐over‐expressed and anti‐VEGF‐neutralized mice compared with controls (i–k). Data were analysed using the Student's t‐test and shown as mean ± SEM (*P < 0·05).
Promotion of M1‐polarization‐associated cytokine expression by IL‐17A in mice with ROP
The retinas of IL‐17A−/− and WT C57BL/6 mice with ROP were collected for RNA isolation, and quantitative PCR was performed to investigate the macrophage polarization‐associated gene expression. Cluster analysis of ROP mouse gene expression by Sato et al.22 had already demonstrated that genes associated with inflammation expressed high values during the whole process of oxygen‐induced retinopathy, and up‐regulated before the formation of RNV. Spiller KL et al.23 suggested that M1 macrophages appeared at the early stages of wound healing (1–3 days), and M2 macrophages appeared at a later stage (4–7 days). Therefore, in this study, inflammation‐associated genes, most probably M1 cytokines, were examined at P15 and genes relevant to later‐stage macrophage polarization at P17. The results suggested that IL‐17A−/− mice with ROP expressed relatively low levels of inflammatory cytokines with statistical significance compared with WT controls (MCP‐1, intercellular adhesion molecule 1, VEGF, TNF‐α, inducible nitric oxide synthase, interferon‐γ, IL‐1β, IL‐6, Toll‐like receptor 4 and IL‐12), indicating potential roles of IL‐17A in M1 polarization. Also, according to the functional profiles of M1 and M2 macrophages introduced by Haruaki Tomioka et al.24 and the findings we detected, significant high levels of Fizz1, transforming growth factor‐β, IL‐1RA and CD206, and low levels of TNF‐α, MHC II and IL‐6 at P17 suggested a predominant mixed M2a and M2c phenotype in IL‐17A deficiency (Table 1).
Shift of macrophage polarization toward an M2 phenotype during RNV due to IL‐17A deficiency
Flow cytometry analysis was applied to exploring the role of IL‐17A in macrophage polarization in the process of RNV. The retinas of WT and IL‐17A−/− mice with ROP were dissected and digested with papain for flow cytometry analysis at P13, P15, P17 and P21. After sorting by CD11b beads, F4/80‐positive CD11c‐positive cells were identified as M1 whereas F4/80‐positive CD206‐positive cells were identified as M2, as suggested in previous studies.21, 25 Results showed fewer M1 polarized macrophages in IL‐17A−/− mice than in WT controls, especially at P15 and P17, but more M2 polarized macrophages in IL‐17A−/− mice than in WT controls, especially at P17. The M1/M2 ratio revealed a shift of macrophage polarization toward M2 in IL‐17A−/− mice compared with WT controls during the RNV process (Fig. 4).
Figure 4.
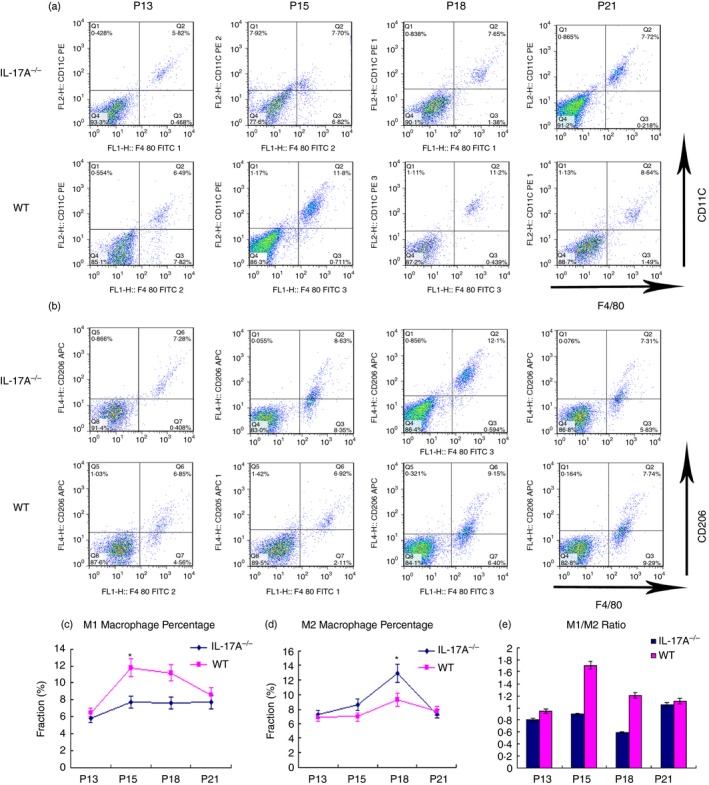
Macrophage polarization shift in the retinas of the interleukin‐17A‐deficient (IL‐17A−/−) mice with retinopathy of prematurity (ROP) mouse model and wild‐type (WT) ROP controls. WT and IL‐17A−/− mice were subjected to the ROP mouse model. At postnatal day 13 (P13), P15, P18 and P21, the retinas were digested with papain and all cells were sorted by incubation with CD11b beads before staining with F4/80, CD11c and CD206. The cells were identified as M1 or M2 macrophages with F4/80+ CD11c+ or F4/80+ CD206+, respectively. Representative panels of flow cytometry (a, b). (c, d) Cell distribution patterns of F4/80+ CD11c+ and F4/80+ CD206+ cells. (e) M1/M2 ratio of WT (pink) and IL‐17A−/− mice (dark blue). Data were analysed using the flowjo software. Statistical difference was shown using a non‐parametric Mann–Whitney test. Experiments were repeated three times. Bars represent mean ± SEM (*P < 0·05) (n = 3 mice/group).
Enhancement of M1 proliferation signalling but mitigation of M2 proliferation signalling in macrophages by IL‐17A
The mitogen‐activated protein kinase (MAPK) family consisted of three parallel signal‐transduction modules converging on the serine/threonine kinases JNK, p38, and ERK, and research has shown MAPK signalling to be involved in the promotion of M2 macrophage polarization.26 Studies by Chiu et al.27 revealed that the inhibition of Akt and nuclear factor‐κB diminished M2 induction in tumour‐associated macrophages in oral squamous cell carcinoma. Wang et al.28 found Scriptaid shifted macrophage polarization toward the protective M2 phenotype by enhancing phosphorylation of AKT signalling in severe traumatic brain injury. Also, Singla et al.29 indicated that Notch1 signalling played a pivotal role in M1 macrophage differentiation and enhanced inflammatory responses, and inhibition of Notch1 subsequent downstream signalling enhanced M2 polarized macrophage outcomes and promoted anti‐inflammatory mediation. In an attempt to explore the role of IL‐17A in macrophage polarization in vitro, the expression level and phosphorylation status of these kinases in RAW264.7 macrophages were examined. As shown in Fig. 5, phosphorylation levels of MAPK signalling decreased in the presence of rIL‐17A, whereas expression of M1 polarization‐associated Notch1 increased with a statistical significance. These findings suggested that IL‐17A tended to be an M1 polarization promoter that might heighten inflammatory responses.
Figure 5.
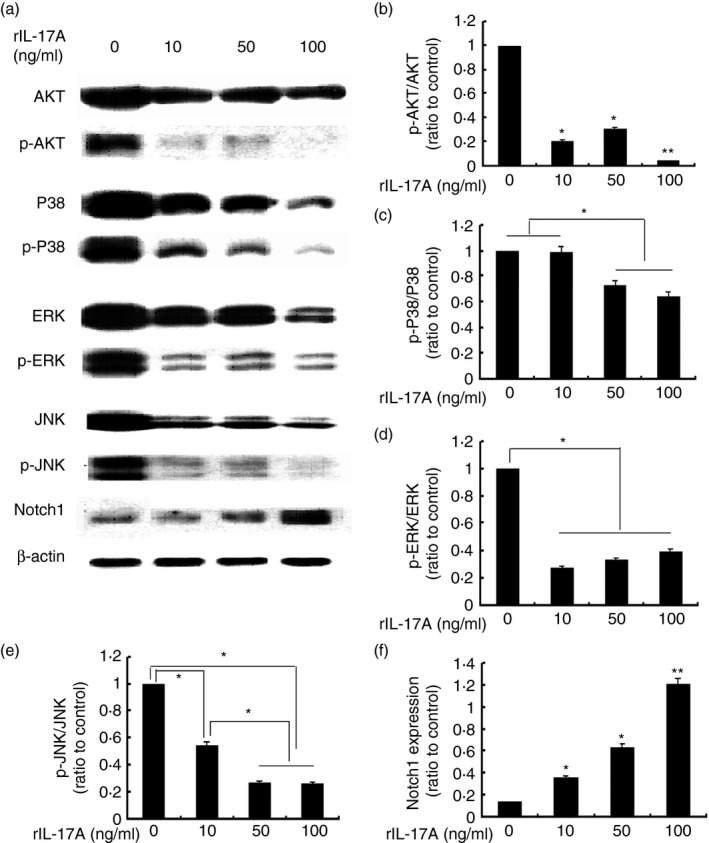
Immunoblot analysis of AKT/p‐AKT, extracellular signal‐regulated kinase (ERK)/p‐ERK, Jun N‐terminal kinase (JNK)/p‐JNK, and Notch1 protein expression in RAW264.7 cells. RAW264.7 cells were treated with different concentrations of recombinant interleukin‐17A (rIL‐17A; 0, 10, 50 and 100 ng/ml) for 24 hr. Equal amounts of whole‐cell lysate were subjected to SDS–PAGE and Western blotting to detect levels of phosphorylated (p) AKT and total AKT, p‐ERK1/2 and total ERK1/2, p‐JNK and JNK, and Notch1. Levels of β‐actin were considered as endogenous control. Representative Western blots (a). Quantification of AKT, ERK1/2, JNK phosphorylation, and Notch1 expression levels in RAW264.7 in response to 0, 10, 50 and 100 ng/ml rIL‐17A are shown, respectively. Data are expressed as mean ± SEM from three independent experiments (b–f, *P < 0·05, **P < 0·01). Statistical analysis was performed by the Student–Newman–Keuls method for multiple comparisons.
Expression of inflammatory M1associated cytokines rather than M2‐associated cytokines by RAW264.7 macrophages due to IL‐17A
RAW264.7 cells were treated with 0, 10, 50 and 100 ng/ml rIL‐17A for 24 hr. Cell lysate and culture supernatant were collected for quantitative PCR and protein detection. Quantitative analysis of cell mRNA revealed that mRNA expression of inducible nitric oxide synthase and TNF‐α was up‐regulated after stimulation by rIL‐17A, whereas IL‐10 and CD206 decreased. Protein concentrations of GM‐CSF, IL‐2, KC, IL‐5, IL‐9, RANTES and VEGF in the culture supernatants were detected by the Quantibody Cytokine Array Kit. Interleukin‐17A significantly induced the expression of these cytokines. Also, NO production was determined by measuring the accumulation of nitrite in the supernatant using Griess reagent, which showed an increment in a dose‐dependent manner. These results indicated that IL‐17A stimulated the macrophage polarization shift to M1 phenotype, which resulted in a pro‐angiogenic microenvironment (Fig. 6).
Figure 6.
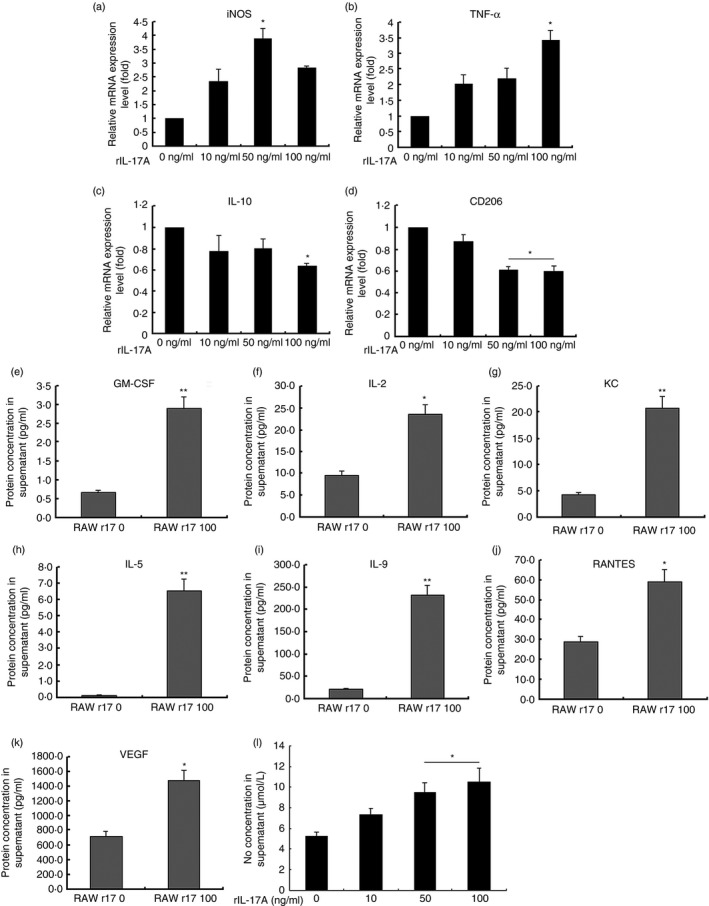
Messenger RNA and protein expression levels of recombinant interleukin‐17A (rIL‐17A) ‐treated RAW264.7 macrophages and supernatant. RAW264.7 macrophages were treated with different concentrations of rIL‐17A for 24 hr. Cell lysate and cytokine concentrations in the culture medium were detected. The mRNA expression of inducible nitric oxide synthase (iNOS), tumour necrosis factor‐α (TNF‐α), IL‐10 and CD206 were detected by real‐time RT‐PCR using the ΔΔCT method and endogenously normalized to cyclophilin A (a–d) (Student–Newman–Keuls method). Protein levels of granulocyte–macrophage colony‐stimulating factor (GM‐CSF) (e), IL‐2 (f), keratinocyte‐derived chemokine (KC) (g), IL‐5 (h), IL‐9 (i), regulated upon activation normal T‐cell expressed and secreted (RANTES) (j), and vascular endothelial growth factor (VEGF) (k) were detected by the Quantibody Cytokine Array Kit. The 0 ng/ml rIL‐17A‐treated groups served as controls (Student's t‐test). NO concentration in supernatant was determined by measuring the accumulation of nitrite using Griess reagent (l) (Student–Newman–Keuls method). The values represented mean ± SD of three independent experiments (n = 5/group, *P < 0·05, **P < 0·01).
Promotion of HUVEC proliferation and tube formation by IL‐17A‐treated macrophage supernatant
Human umbilical vascular endothelial cells were incubated in culture supernatant of rIL‐17A‐treated RAW264.7 macrophages or a culture medium containing different concentrations of rIL‐17A cytokine for 24 hr. After 24 hr, the cells were subjected to RNA isolation. Quantitative PCR suggested significantly high expression of VEGFR1 and VEGFR2 in HUVECs incubated in 50 ng/ml rIL‐17A‐treated RAW264.7 macrophage supernatant (Fig. 7a, b). But incubation in the presence of rIL‐17A cytokine did not cause a significantly increased expression level of VEGFR1 and VEGFR2 in HUVECs (Fig. 7d, e). CCK‐8 assays were performed at 24, 48 and 72 hr to test endothelial proliferation, and the results showed that supernatant of 50 and 100 ng/ml rIL‐17A‐treated RAW264.7 macrophages stimulated HUVEC proliferation significantly at these observation time‐points (Fig. 7c). However, rIL‐17A cytokine did not induce significantly high proliferation of HUVECs. Tube formation assays (Fig. 7f) suggested that 50 ng/ml rIL‐17A‐treated RAW264.7 supernatant and 50 ng/ml rIL‐17A cytokine stimulated similar levels of endothelial cell tube formation (Fig. 7g–k). Considering the aforementioned tested protein levels, the results showed that IL‐17A might stimulate macrophages to pro‐angiogenic status so as to promote endothelial proliferation.
Figure 7.
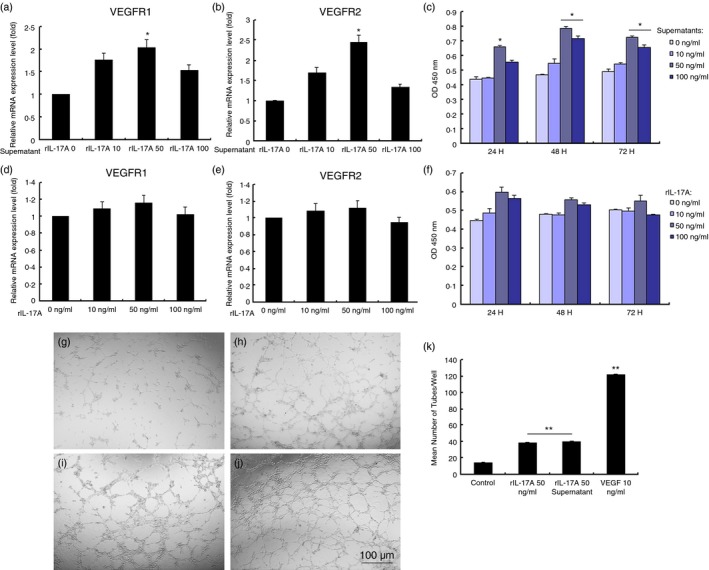
Vascular endothelial growth factor receptor 1 (VEGFR1) and VEGFR2 mRNA expression, cell proliferation, and tube formation of human umbilical vascular endothelial cells (HUVECs) incubated in the culture medium or culture supernatants of recombinant interleukin‐17A (rIL‐17A) ‐treated RAW264.7 macrophages. VEGFR1 and VEGFR2 mRNA expression in HUVECs was up‐regulated after incubation in supernatants of RAW264.7 cells treated with rIL‐17A (a and b). VEGFR1 and VEGFR2 mRNA expression levels in HUVECs cultured in media containing different concentrations of rIL‐17A cytokine (d and e). CCK‐8 assays showed different proliferative effects of rIL‐17A‐treated RAW264.7 cell supernatant on HUVECs for 24, 48 and 72 hr, respectively (c and f). Tube formation assays were taken in HUVECs in the presence of 50 ng/ml rIL‐17A cytokine (h) and rIL‐17A‐treated RAW264.7 cell supernatant (i). PBS (g) and 10‐ng/ml VEGF (j) served as controls. Data presented demonstrate mean number of tubes in each treatment group (k). Each condition was performed in triplicate and the experiments were repeated three times. Bars represented mean ± SEM (n = 3/group, *P < 0·05, **P < 0·01). Statistical analysis was followed with the Student–Newman–Keuls method.
Discussion
The function of IL‐17A covers a wide range of biological activities and is well known to participate in various inflammatory reactions.30, 31, 32 Roles of IL‐17A in angiogenesis have also been reported in tumour studies. Research has shown that IL‐17A expression increased in 58% of prostate cancer,33 and a higher percentage of Th17 cells in blood correlated to poorer outcome.34 Meng et al.35 demonstrated that over‐expression of IL‐17 in tumour‐associated macrophage correlated with the differentiation and angiogenesis of laryngeal squamous cell carcinoma. Studies by Suryawanshi et al. found that corneal herpes simplex virus infection results in IL‐17A production, which participates in corneal NV.36 Animals lacking responses to IL‐17A signalling, either because of IL‐17 receptor A knockout or WT animals receiving NAb to IL‐17A, had diminished corneal NV compared with controls.36 As IL‐17 participates in both inflammation and angiogenesis, which are essential pathological processes in NV eye diseases, this study investigated the potential role of IL‐17A in modulating ocular NV with the application of several animal models. This study demonstrated an increased expression of IL‐17A during the pathological process and indicated a promotive role of rIL‐17A in NV. These results confirmed the present notion that IL‐17A plays a stimulative role in ocular angiogenesis.
This study next explored the counteraction of IL‐17A with potential inflammatory and angiogenic cytokines involved in ocular NV. The results showed that pro‐inflammatory cytokines were significantly reduced in IL‐17A−/− mice with ROP compared with WT controls, but M2‐associated cytokines such as IL‐10 and M2 macrophage marker CD206 increased significantly in IL‐17A−/− mice with ROP. These findings directed the efforts to understanding the potential angiogenic effects of IL‐17A on the polarization shift of macrophages. Therefore, macrophage polarization by flow cytometry was investigated. The results indicated a macrophage polarization shift to M2 phenotype in IL‐17A−/− mice under hypoxic retinopathy. To further validate the effects of IL‐17A on macrophages, in vitro assays were performed in the presence of different concentrations of rIL‐17A. The results were consistent with the present in vivo study, indicating a polarization shift toward M1 phenotype in macrophages cultured in high concentrations of rIL‐17A. As is well accepted, M1 macrophages produce high levels of pro‐inflammatory cytokines such as TNF‐α, IL‐1β, IL‐6 and reactive oxygen species but low levels of anti‐inflammatory cytokines such as IL‐10. M2 macrophages produce high levels of IL‐10 and increased expression of mannose and galactose receptors.37 Investigations of sub‐retinal fluid analysis of patients with advanced ROP by Jie et al. showed significantly high expression of such pro‐inflammatory cytokines as interferon‐γ, IL‐4, IL‐6, IL‐8, IL‐12p70, TNF‐α, G‐CSF, and GM‐CSF compared with that of acute retinal detachment within 48 hr. In vitro assays for capillary formation and endothelial proliferation showed that sub‐retinal fluid from ROP induced robust support for these cellular processes. Furthermore, retrolental membranes bluntly dissected from patients with advanced ROP displayed overwhelming M1 macrophages compared with M2 macrophages, clearly demonstrating that the retinal microenvironment of ROP is pro‐angiogenic and pro‐inflammatory.4 Although there have been several polarization types of macrophage populations reported with different expression and production profiles according to the stimuli, cytokines like MHC II, IL‐6 and TNF‐α that are characteristically highly expressed in M2b macrophages are also produced by M1 macrophages. But actually, macrophages usually do not form a stable subtype in response to a combination of factors present in the tissue.38, 39 As a result, it is preferable that signatures of macrophage subtypes may not exclude one another but display an overlapping effect. Based on the data presented and the functional profiles of M1 and M2 macrophages introduced by Haruaki Tomioka et al.,24 the study believed that IL‐17A neutralization alleviated inflammation and facilitated tissue repair in ocular NV by modulating macrophage polarization toward the M2 phenotype and reduced expression of VEGF from M1 macrophages.
However, NV is the proliferation of endothelial cells from pre‐existing vessels. The study then investigated whether IL‐17A interfering macrophages would modulate endothelial cells. The study detected elevated concentrations of pro‐angiogenic VEGF and NO in high concentrations of rIL‐17A‐treated supernatants in addition to increased pro‐inflammatory cytokines (such as GM‐CSF, IL‐2, KC, IL‐5, IL‐9 and RANTES). Furthermore, investigations on HUVECs cultured in medium containing rIL‐17A cytokines and rIL‐17A‐treated macrophage supernatant suggested an indirect effect of IL‐17A on endothelial cells that might orchestrate with macrophages to modulate ocular NV.
As VEGF is a pivotal cytokine modulating angiogenesis, the study also explored the interaction of IL‐17A and VEGF applying two VEGF over‐expressed animal models, rho/VEGF transgenic mice and Tet/opsin/VEGF double‐transgenic mice. As shown in the results, these findings suggested that IL‐17A might be an upstream cytokine that modulated ocular NV through the VEGF pathway.
The remarkable plasticity of macrophages has made it an interesting target in immunomodulation in inflammation and tissue regeneration. Research supported the view that controlled recruitment of appropriate phenotypes of macrophages promoted functional recovery in tissue inflammation and remodelling.36 The present study demonstrated that IL‐17A neutralization alleviated ocular NV through a polarization shift toward the M2 phenotype and reduced VEGF expression from M1 macrophages. Other research suggested that M2 macrophages secreted endothelial cell growth and angiogenic factors such as VEGF and CXCL1 and were considered to promote wound repair and NV.40, 41 Studies by Richards et al.42 also demonstrated that IL‐19 accelerated revascularization in ischaemic hind limbs by polarization of macrophages to M2 together with the induction of VEGF expression in these cells.
The limitation of this study involved the potential mechanisms by which transcription accounted for macrophage polarization and how the subsequent cytokines were modulated when macrophages were polarized. Also, further studies are needed to definitively determine the extent to which IL‐17A neutralizing anti‐angiogenic activity depends on macrophage modulation compared with anti‐VEGF treatment.
To conclude, the present study implicated for the first time that IL‐17A neutralization alleviated ocular NV by promoting the macrophage polarization shift towards the M2 phenotype and reducing VEGF expression from M1 macrophages, whereas rIL‐17A administration increased ocular NV by stimulating M1 polarization and endothelial cell proliferation and tube formation triggered by increased expression of VEGF from M1. The present study revealed an indirect effect of IL‐17A on NV through macrophage polarization modulation, indicating that IL‐17A might play a paracrine role in endothelial cell–macrophage crosstalk in angiogenesis. This would shed light on the therapeutic potential for this cytokine in treating NV eye diseases in addition to anti‐VEGF therapy.
Disclosures
The authors declare no financial or commercial conflict of interest.
Acknowledgements
The authors thank the Shanghai Institute of Burns for field and facility assistance. The study is supported by the National Nature Science Foundation of China 81470639 and 81570853; Shanghai Nature Science Foundation Grant 14411968400; National Major Scientific Equipment program (2012YQ12008003); Shanghai Charity Cancer Research Centre Programme 2013, SY‐2012‐131, 2013‐3036, Guizhou; and 2015 Doctoral Innovation Fund Projects BXJ201414 from Shanghai Jiao Tong University School of Medicine, China.
References
- 1. Szade A, Grochot‐Przeczek A, Florczyk U, Jozkowicz A, Dulak J. Cellular and molecular mechanisms of inflammation‐induced angiogenesis. IUBMB Life 2015; 67:145–59. [DOI] [PubMed] [Google Scholar]
- 2. Campochiaro PA. Ocular neovascularization. J Mol Med (Berl) 2013; 91:311–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rofagha S, Bhisitkul RB, Boyer DS, Sadda SR, Zhang K. Seven‐year outcomes in ranibizumab‐treated patients in ANCHOR, MARINA, and HORIZON: a multicenter cohort study (SEVEN‐UP). Ophthalmology 2013; 120:2292–9. [DOI] [PubMed] [Google Scholar]
- 4. Ma J, Mehta M, Lam G, Cyr D, Ng TF, Hirose T et al Influence of subretinal fluid in advanced stage retinopathy of prematurity on proangiogenic response and cell proliferation. Mol Vis 2014; 20:881–93. [PMC free article] [PubMed] [Google Scholar]
- 5. Yang B, Kang H, Fung A, Zhao H, Wang T, Ma D. The role of interleukin 17 in tumour proliferation, angiogenesis, and metastasis. Mediators Inflamm 2014; 2014:623759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chung AS, Wu X, Zhuang G, Ngu H, Kasman I, Zhang J et al An interleukin‐17‐mediated paracrine network promotes tumor resistance to anti‐angiogenic therapy. Nat Med 2013; 19:1114–23. [DOI] [PubMed] [Google Scholar]
- 7. Pickens SR, Volin MV, Mandelin AM 2nd, Kolls JK, Pope RM, Shahrara S. IL‐17 contributes to angiogenesis in rheumatoid arthritis. J Immunol 2010; 184:3233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL‐17 can promote tumor growth through an IL‐6‐Stat3 signaling pathway. J Exp Med 2009; 206:1457–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu B, Wei L, Meyerle C, Tuo J, Sen HN, Li Z et al Complement component C5a promotes expression of IL‐22 and IL‐17 from human T cells and its implication in age‐related macular degeneration. J Transl Med 2011; 9:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tuo J, Cao X, Shen D, Wang Y, Zhang J, Oh JY et al Anti‐inflammatory recombinant TSG‐6 stabilizes the progression of focal retinal degeneration in a murine model. J Neuroinflammation 2012; 9:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hasegawa E, Sonoda KH, Shichita T, Morita R, Sekiya T, Kimura A et al IL‐23‐independent induction of IL‐17 from γδT cells and innate lymphoid cells promotes experimental intraocular neovascularization. J Immunol 2013; 190:1778–87. [DOI] [PubMed] [Google Scholar]
- 12. Okamoto N, Tobe T, Hackett SF, Ozaki H, Vinores MA, LaRochelle W et al Transgenic mice with increased expression of vascular endothelial growth factor in the retina: a new model of intraretinal and subretinal neovascularization. Am J Pathol 1997; 151:281–91. [PMC free article] [PubMed] [Google Scholar]
- 13. Ohno‐Matsui K, Hirose A, Yamamoto S, Saikia J, Okamoto N, Gehlbach P et al Inducible expression of vascular endothelial growth factor in adult mice causes severe proliferative retinopathy and retinal detachment. Am J Pathol 2002; 160:711–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shen J, Xie B, Dong A, Swaim M, Hackett SF, Campochiaro PA. In vivo immunostaining demonstrates macrophages associate with growing and regressing vessels. Invest Ophthalmol Vis Sci 2007; 48:4335–41. [DOI] [PubMed] [Google Scholar]
- 15. Mori K, Duh E, Gehlbach P, Ando A, Takahashi K, Pearlman J et al Pigment epithelium‐derived factor inhibits retinal and choroidal neovascularization. J Cell Physiol 2001; 188:253–63. [DOI] [PubMed] [Google Scholar]
- 16. Zhu Y, Lu Q, Shen J, Zhang L, Gao Y, Shen X et al Improvement and optimization of standards for a preclinical animal test model of laser induced choroidal neovascularization. PLoS ONE 2014; 9:e94743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haas CS, Amin MA, Ruth JH, Allen BL, Ahmed S, Pakozdi A et al In vivo inhibition of angiogenesis by interleukin‐13 gene therapy in a rat model of rheumatoid arthritis. Arthritis Rheum 2007; 56:2535–48. [DOI] [PubMed] [Google Scholar]
- 18. Shen J, Yang X, Xie B, Chen Y, Swaim M, Hackett SF et al MicroRNAs regulate ocular neovascularization. Mol Ther 2008; 16:1208–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fujimura S, Takahashi H, Yuda K, Ueta T, Iriyama A, Inoue T et al Angiostatic effect of CXCR3 expressed on choroidal neovascularization. Invest Ophthalmol Vis Sci 2012; 53:1999–2006. [DOI] [PubMed] [Google Scholar]
- 20. Szliszka E, Skaba D, Czuba ZP, Krol W. Inhibition of inflammatory mediators by neobavaisoflavone in activated RAW264.7 macrophages. Molecules 2011; 16:3701–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lumeng CN, DelProposto JB, Westcott DJ, Saltiel AR. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 2008; 57:3239–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sato T, Kusaka S, Hashida N, Saishin Y, Fujikado T, Tano Y. Comprehensive gene‐expression profile in murine oxygen‐induced retinopathy. Br J Ophthalmol 2009; 93:96–103. [DOI] [PubMed] [Google Scholar]
- 23. Spiller KL, Anfang RR, Spiller KJ, Ng J, Nakazawa KR, Daulton JW et al The role of macrophage phenotype in vascularization of tissue engineering scaffolds. Biomaterials 2014; 35:4477–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tomioka H, Tatano Y, Maw WW, Sano C, Kanehiro Y, Shimizu T. Characteristics of suppressor macrophages induced by mycobacterial and protozoal infections in relation to alternatively activated M2 macrophages. Clin Dev Immunol 2012; 2012:635451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bao B, Chen YG, Zhang L, Na Xu YL, Wang X, Liu J et al Momordica charantia (Bitter Melon) reduces obesity‐associated macrophage and mast cell infiltration as well as inflammatory cytokine expression in adipose tissues. PLoS ONE 2013; 8:e84075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang J, Cao J, Ma S, Dong R, Meng W, Ying M et al Tumor hypoxia enhances non‐small cell lung cancer metastasis by selectively promoting macrophage M2 polarization through the activation of ERK signaling. Oncotarget 2014; 5:9664–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chiu K‐C, Lee C‐H, Liu S‐Y, Chou Y‐T, Huang R‐Y, Huang S‐M et al Polarization of tumor‐associated macrophages and Gas6/Axl signaling in oral squamous cell carcinoma. Oral Oncol 2015; 51(7):683–9. [DOI] [PubMed] [Google Scholar]
- 28. Wang G, Shi Y, Jiang X, Leak RK, Hu X, Wu Y et al HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3β/PTEN/Akt axis. Proc Natl Acad Sci USA 2015; 112:2853–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Singla RD, Wang J, Singla DK. Regulation of Notch 1 signaling in THP‐1 cells enhances M2 macrophage differentiation. Am J Physiol Heart Circ Physiol 2014; 307:H1634–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Suryawanshi A, Veiga‐Parga T, Rajasagi NK, Reddy PB, Sehrawat S, Sharma S et al Role of IL‐17 and Th17 cells in herpes simplex virus‐induced corneal immunopathology. J Immunol 2011; 187:1919–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shen F, Gaffen SL. Structure‐function relationships in the IL‐17 receptor: implications for signal transduction and therapy. Cytokine 2008; 41:92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Molesworth‐Kenyon SJ, Yin R, Oakes JE, Lausch RN. IL‐17 receptor signaling influences virus‐induced corneal inflammation. J Leukoc Biol 2008; 83:401–8. [DOI] [PubMed] [Google Scholar]
- 33. Steiner GE, Newman ME, Paikl D, Stix U, Memaran‐Dagda N, Lee C et al Expression and function of pro‐inflammatory interleukin IL‐17 and IL‐17 receptor in normal, benign hyperplastic, and malignant prostate. Prostate 2003; 56:171–82. [DOI] [PubMed] [Google Scholar]
- 34. Derhovanessian E, Adams V, Hahnel K, Groeger A, Pandha H, Ward S et al Pretreatment frequency of circulating IL‐17+ CD4+ T‐cells, but not Tregs, correlates with clinical response to whole‐cell vaccination in prostate cancer patients. Int J Cancer 2009; 125:1372–9. [DOI] [PubMed] [Google Scholar]
- 35. Meng CD, Zhu DD, Jiang XD, Li L, Sha JC, Dong Z et al Overexpression of interleukin‐17 in tumor‐associated macrophages is correlated with the differentiation and angiogenesis of laryngeal squamous cell carcinoma. Chin Med J (Engl) 2012; 125:1603–7. [PubMed] [Google Scholar]
- 36. Suryawanshi A, Veiga‐Parga T, Reddy PB, Rajasagi NK, Rouse BT. IL‐17A differentially regulates corneal vascular endothelial growth factor (VEGF)‐A and soluble VEGF receptor 1 expression and promotes corneal angiogenesis after herpes simplex virus infection. J Immunol 2012; 188:3434–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mokarram N, Bellamkonda RV. A perspective on immunomodulation and tissue repair. Ann Biomed Eng 2014; 42:338–51. [DOI] [PubMed] [Google Scholar]
- 38. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep 2014; 6:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S et al Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 2014; 41:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lamagna C, Aurrand‐Lions M, Imhof BA. Dual role of macrophages in tumor growth and angiogenesis. J Leukoc Biol 2006; 80:705–13. [DOI] [PubMed] [Google Scholar]
- 41. Ligresti G, Aplin AC, Zorzi P, Morishita A, Nicosia RF. Macrophage‐derived tumor necrosis factor‐α is an early component of the molecular cascade leading to angiogenesis in response to aortic injury. Arterioscler Thromb Vasc Biol 2011; 31:1151–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Richards J, Gabunia K, Kelemen SE, Kako F, Choi ET, Autieri MV. Interleukin‐19 increases angiogenesis in ischemic hind limbs by direct effects on both endothelial cells and macrophage polarization. J Mol Cell Cardiol 2015; 79:21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]