
Fig. 3.
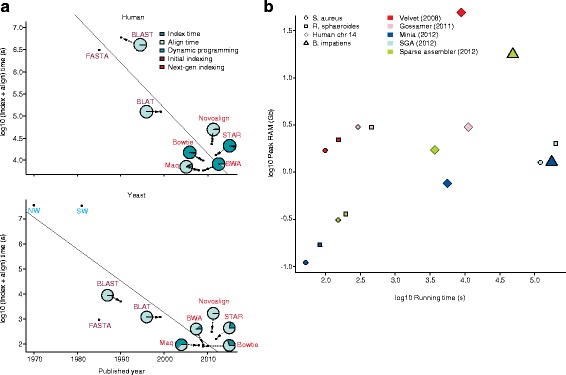
a Multiple advances in alignment algorithms have contributed to an exponential decrease in running time over the past 40 years. We synthesized one million single-ended reads of 75 bp for both human and yeast. The comparison only considers the data structure, algorithms, and speeds. There are many other factors, such as accuracy and sensitivity, which are not discussed here, but which are covered elsewhere [25]. Initial alignment algorithms based on dynamic programming were applicable to the alignment of individual protein sequences, but they were too slow for efficient alignment at a genome scale. Advances in indexing helped to reduce running time. Additional improvements in index and scoring structures enabled next generation aligners to further improve alignment time. A negative correlation is also observed between the initial construction of an index and the marginal mapping time per read. b Peak memory usage plotted against the running time for different genome assemblers on a log-log plot. Assembler performance was tested using multiple genomes, including Staphylococcus aureus, Rhodobacter sphaeroides, human chromosome 14, and Bombus impatiens. Data were obtained from Kleftogiannis et al. [33]