

The significance of the trifluoromethyl-substituted propargylic alcohol moiety, for example in the anti-HIV drug Efavirenz, has stimulated the development of several methods that furnish this motif in racemic form.[1] In contrast to the general advance with asymmetric alkynylations of aldehydes,[2] ketones,[3] and imines,[4] trifluoromethyl ketones have remained challenging substrates,[5] and initially required the use of stoichiometric amounts of lithium or zinc aminoalkoxides.[6] Shibasaki first demonstrated the feasibility of asymmetric catalysis, providing CF3-substituted propargylic alcohols in up to 52% ee.[7] Significant progress in this field emerged in 2011 when Carreira reported an intriguing autocatalytic protocol that is tailored to the production of an Efavirenz precursor.[8] At the same time, Ma introduced an alkynylation method that gives 55–98% yield and 65–94% ee with nonenolizable trifluoromethyl ketones when 2.5 equivalents of the alkyne, 3 equivalents of Me2Zn, and 2 equivalents of Ti(Oi-Pr)4 are used in addition to catalytic amounts of a cinchona alkaloid and BaF2.[9]
In recent years, terminal ynamides have become readily available surrogates of the highly reactive and less practical parent ynamines, and have been applied in a variety of cycloadditions[10] and several other reactions.[11] By contrast, the usefulness of terminal ynamides in nucleophilic addition reactions has rarely been explored,[12] and the reaction with trifluoromethyl ketones has not been reported to date. We hypothesized that a catalytic asymmetric method that allows addition of terminal ynamides to trifluoroacetophenone and derivatives thereof has potential to overcome the remaining drawbacks of the reaction with alkynes, in particular the use of excess of pyrophoric dimethylzinc. At the same time, the enantioselective synthesis of ynamide derived, CF3-substituted propargylic alcohols would provide unprecedented access to a variety of highly functionalized chiral building blocks if one could exploit the unique reactivity of the polarized N-substituted triple bond (Scheme 1).[13] The recent introduction of a very practical two-step synthesis of terminal ynamides from tosylamides and trichloroethylene by Anderson et al. provided an excellent starting point for our study.[14]
Scheme 1.

Catalytic asymmetric ynamide addition to trifluoromethyl ketones provides practical access to chiral enamides, amides and N,O-ketene acetals carrying a tertiary CF3-substituted alcohol group.
At the onset of this investigation we employed N-ethynyl-N-butylbenzenesulfonamide, 1, and other ynamides in several literature protocols previously developed for catalytic enantioselective alkynylations of carbonyl electrophiles. While these screening efforts were mostly unsuccessful, we were excited to find that asymmetric ynamide addition to trifluoroactophenone occurs in the presence of catalytic amounts of zinc triflate, N-methyl ephedrine and excess of either Et3N or i-Pr2NEt. Further investigation then revealed that the yield and ee varied substantially based on the source of the tertiary amine employed.[15] Careful purification of the amines used as well as investigation of possible effects of impurities and amine degradation products that may be present in small amounts but could possibly affect the ynamide addition did not resolve this problem. We therefore decided to develop a practical catalytic method that avoids both organozinc reagents and amine additives. Comprehensive screening of zinc and copper complexes, a large variety of chiral ligands in several solvents and the analysis of the effect of (MeO)3PO, (EtO)3PO, Ph3PO, Ph3PS, t-Bu3P, HMPA or other additives on the asymmetric induction and turnover gave mixed results. Initially, moderate ee’s were obtained with 10 mol% of zinc triflate and L1–L4. But we were delighted to find that the reaction between the readily available ynamide 1 and 2 occurs with these catalysts even in the absence of triethylamine or Hünig’s base, providing 3 with up to 95% yield at 25 °C (Table 1, entries 1–4).
Table 1.
Optimization of the zinc triflate catalyzed asymmetric ynamide addition to trifluoroacetophenone.
 | |||||
|---|---|---|---|---|---|
| entry | ligand | R | conditions | yield (%) | ee (%) |
| 1 | L1 | n-Bu | DCE, 25 °C, 20 h | 75 | 48 |
| 2 | L2 | n-Bu | DCE, 25 °C, 20 h | 95 | 44 |
| 3 | L3 | n-Bu | DCE, 25 °C, 20 h | 89 | 33 |
| 4 | L4 | n-Bu | DCE, 25 °C, 16 h | 27 | 3 |
| 5 | L5 | n-Bu | DCE, 25 °C, 20 h | 15 | 22 |
| 6 | L6 | n-Bu | DCE, 25 °C, 16 h | 25 | 14 |
| 7 | L7 | n-Bu | DCE, 25 °C, 16 h | 72 | 45 |
| 8 | L8 | n-Bu | DCE, 25 °C, 16 h | 87 | 89 |
| 9 | L9 | n-Bu | DCE, 25 °C, 20 h | 15 | 0 |
| 10 | L10 | n-Bu | DCE, 25 °C, 16 h | 93 | 80 |
| 11 | L11 | n-Bu | DCE, 25 °C, 16 h | 22 | 9 |
| 12 | L8 | allyl | DCE, 25 °C, 16 h | 88 | 86 |
| 13 | L8 | n-Bu | CH2Cl2, 25 °C, 20 h | 95 | 87 |
| 14 | L8 | n-Bu | CHCl3, 25 °C, 20 h | 95 | 86 |
| 15 | L8 | n-Bu | toluene, 25 °C, 16 h | 70 | 78 |
| 16 | L8 | n-Bu | THF, 25 °C, 16 h | 55 | 66 |
| 17 | L8 | n-Bu | ACN, 25 °C, 20 h | 90 | 31 |
| 18 | L8 | n-Bu | EtOH, 25 °C, 20 h | 5 | n.d. |
| 19 | L8 | n-Bu | DCE, 0 °C, 45 h | 78 | 93 |
| 20 | L8 | n-Bu | DCE, −10 °C, 51 h | 73 | 91 |
| 21a | L8 | n-Bu | DCE, −20 °C, 24 h | 96 | 96 |
DCE=1,2-dichloroethane.
(EtO)3PO (20 mol%).
Although encouraging results were obtained with quinine,[16] we turned our attention to Trost’s Bis-ProPhenols which can be more easily modified if ligand fine-tuning becomes necessary.[17] The employment of L5–7 carrying either N-methyl ephedrine or diphenylprolinol units attached to a phenol core did not show improvement (entries 5–7). However, the introduction of C2-symmetric L8 gave 3 in 87% yield and 89% ee within 16 hours and essentially the same results were obtained with the N-allyl analogue of 1 indicating that this method tolerates different ynamides (compare entries 8 and 12). The presence of the free phenol group appears to be essential to the catalytic activity of L8 and very low yields were obtained with L9. The use of the naphthyl analogue L10 further improved the yield but at the expense of the enantioselectivity while to our surprise L11 gave poor results (entries 10 and 11). We therefore continued to evaluate different solvents and the effect of temperature on this reaction using 10 mol% of Zn(OTf)2 and L8 (entries 13–20). Chlorinated solvents proved superior and gave consistently high yields and ee’s. Using dichloroethane as solvent, we observed that ee’s improve above 90% when the temperature was decreased to at least 0 °C but the reaction time increased to approximately 2 days. This was addressed with the addition of catalytic amounts of triethyl phosphate to affect catalytic turnover and we obtained 3 in 96% yield and 96% ee at −20 °C in 24 hours (entry 21).
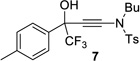
The ynamide addition is ligand-accelerated and does not occur in the absence of a zinc catalyst (SI).[18] When we employed phenylacetylene in our optimized base-free protocol we observed sluggish conversion producing the propargylic alcohol in low yields. 1H NMR analysis of a stoichiometric mixture of zinc triflate, the Bis-ProPhenol ligand and 1 showed formation of a Zn-L8 complex in deuterated chloroform but no sign of coordination and activation of the ynamide (SI). The reaction presumably involves an intermediate side-on or end-on zinc-ynamide species which does not form in the absence of the trifluoromethyl ketone. In accordance with a study by Cozzi et al.,[19] the substrate may therefore play a pivotal role in promoting the ynamide activation and its own consumption. Altogether, these observations reveal the strikingly different reactivity of terminal ynamides compared to simple alkynes and the distinct behavior of trifluoromethyl ketones. Having developed a base-free catalytic method for the asymmetric addition of readily available ynamides to 2 and excluding the use of pyrophoric dialkylzinc reagents as well as other stoichiometric additives, we began with the evaluation of the substrate scope. The introduction of simple trifluoroacetophenone analogues to our procedure gave the β-hydroxy ynamides 5, 7 and 9 in 96–97% yield and in 94–96% ee (entries 2–4 in Table 2). Similar results were obtained with a series of functionalized analogues (entries 5–12).
Table 2.
Asymmetric catalytic addition of ynamides to trifluoromethyl ketones.
 | ||||
|---|---|---|---|---|
| entry | CF3 ketone | β-hydroxy ynamide | yielda (%) | ee (%) |
| 1 |  |
 |
96% | 96% |
| 2 |  |
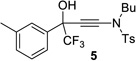 |
97% | 95% |
| 3 | 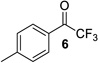 |
 |
97% | 94% |
| 4 | 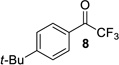 |
 |
95% | 96% |
| 5 | 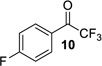 |
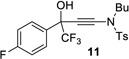 |
97% | 94% |
| 6 | 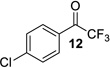 |
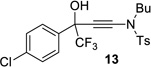 |
95% | 93% |
| 7 | 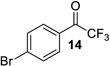 |
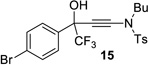 |
97% | 90% |
| 8 | 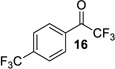 |
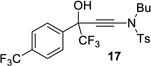 |
99% | 92% |
| 9 |  |
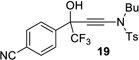 |
91% | 90% |
| 10 |  |
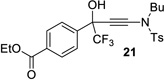 |
99% | 89% |
| 11 |  |
 |
99% | 90% |
| 12b |  |
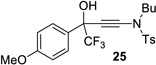 |
91% | 85% |
Isolated yields.
0 °C, 50 h.
The absolute configuration of 3 was determined by crystallographic analysis of the partial reduction and hydroacyloxylation derivatives 28 and 30 (see SI).
The intrinsic synthetic value of the β-hydroxy ynamides shown above stems from the high density of functional groups that are combined into a relatively small chiral building block. The trifluoromethylated tertiary chiral alcohol moiety is an increasingly attractive motif with potential use in medicinal applications[20] and the adjacent ynamide unit provides unique synthetic versatility that is orthogonal to the chemistry of C-substituted propargylic alcohols. With this in mind we set out to investigate selective transformations that would generate unprecedented access to new or generally challenging structures. In contrast to alkynes, the ynamide unit can be considered a masked amide bond and we therefore decided to develop a catalytic method that exploits regioselective hydration. After screening of several acids and solvents we found that smooth conversion of 3 toward 26 occurs in the presence of dilute sulfuric acid at room temperature. Using this protocol, we obtained the corresponding β-hydroxy sulfonamide 26 in 91% yield and without compromising the ee of the starting material (equation 1 in Scheme 2). Next, we turned our attention to N-tosyl β-hydroxy enamines which are viable substrates for the synthesis of a variety of compounds, including aminocyclopropyl carbinols and 1,3-amino alcohols.[21] Urabe originally developed a diastereoselective method that utilizes a chiral sulfonamide auxiliary to afford N-tosyl (E)-β-hydroxy enamines via Ti mediated ynamide addition to aldehydes.[22] Walsh et al. introduced an asymmetric route toward a series of (E)-isomers that is based on sequential hydroboration of internal N-tosyl ynamides, boron-to-zinc transmetalation and catalytic nucleophilic addition to aldehydes in one pot.[21a] We now complement these methods, which afford (E)-enamines with a secondary alcohol moiety, by providing stereoselective access to both (Z)- and (E)-N-tosyl β-hydroxy enamines exhibiting a tertiary chiral carbinol group. Selective reduction of 3 with either Red-Al or by Pd catalyzed hydrogenation gave 27 and 28 in one step in 82–86% yield and 93% ee (equations 3 and 4).
Scheme 2.

Selective transformations of β-hydroxy ynamides.
Finally, we explored the possibility of mild diastereoselective addition of carboxylic acids to ynamide 3. To the best of our knowledge, few achiral α-acyloxyenamides have been prepared to date. Lam’s group was first to prepare α-acyloxyenamides by palladium catalyzed hydroacyloxylation of ynamides at 70 °C and demonstrated the synthetic utility of these N,O-ketene acetals in rearrangement reactions.[23] Recently, a metal free procedure that gives moderate to high yields albeit at even higher temperatures (100 °C) was reported.[24] We found that the hydroacyloxylation of 3 with acetic and benzoic acid can be accomplished in dichloromethane at room temperature and without loss in the enantiomeric purity. In both cases, the (E)-isomers were produced with high diastereoselectivity (E/Z>99:1) and we obtained 29 and 30 in 89% and 93% yield, respectively (equations 4 and 5).25
Slow evaporation of concentrated chloroform and dichloromethane solutions of 28 and 30, respectively, gave single crystals suitable for X-ray determination of the absolute and relative configurations (Figure 1).[26] These enamides have relatively short C=C bonds ranging from 1.319 to 1.325 Å and significantly longer C-N bonds (1.421–1.437 Å) in comparison to typical enamines, which explains the increased thermal stability and ease of isolation.
Figure 1.

X-ray structures of (S,Z)-28 (left) and (S,E)-30 (right). Selected bond lengths for 28 [Å]: C=C: 1.325, N-C(sp2): 1.437; 30: C=C: 1.319, N-C(sp2): 1.421, O-C(sp2): 1.411.
In summary, we have introduced the first catalytic enantioselective addition of terminal ynamides to trifluoromethyl ketones. The reaction occurs in the presence of catalytic amounts of Zn(OTf)2, a bis(prolinol)phenol ligand and triethyl phosphate, and it provides practical access to synthetically versatile propargylic CF3-substituted tertiary alcohols that are obtained in high yields and ee. The utility of the tertiary β-hydroxy-β-trifluoromethyl ynamides was demonstrated with highly regioselective hydration, stereoselective reductions and hydroacyloxylations, producing trifluoromethylated chiral alcohols with adjacent (Z)- and (E)-enamide, amide and N,O-ketene acetal functionalities.
Experimental Section
Zinc triflate (7.4 mg, 0.02 mmol), (R,R)-(−)-L8 (14.8 mg, 0.022 mmol), 1 (75.0 mg, 0.30 mmol), 2 (35 mg, 0.20 mmol) and triethyl phosphate (7.3 mg, 0.04 mmol) were dissolved in 1,2-dichloroethane (0.2 mL) under nitrogen atmosphere. The mixture was stirred at −20 °C for 24 h. Purification of the crude mixture by flash chromatography using 1% Et3N in CH2Cl2 as mobile phase on silica gel gave 82.0 mg (0.19 mmol, 96%, 96% ee) of 3 as colorless oil.1H NMR (400 MHz) δ = 7.76 (d, J = 8.1 Hz, 2H), 7.69 (m, 2H), 7.43 – 7.36 (m, 3H), 7.32 (d, J = 8.1 Hz, 2H), 3.44 – 3.32 (m, 2H), 3.05 (s, 1H), 3.05 (s, 3H), 1.67 – 1.58 (m, 2H), 1.40 – 1.29 (m, 2H), 0.90 (t, J = 7.3 Hz, 3H).13C NMR (100 MHz) δ = 145.0, 135.4, 134.3, 129.9, 129.4, 128.2, 127.7, 127.2, 123.4 (q, J = 285.6 Hz), 81.7, 73.4 (q, J = 32.5 Hz), 67.4, 50.9, 29.8, 21.7, 19.4, 13.5. The ee was determined by HPLC on Phenomenex® Cellulose-3. Anal. Calcd. for C21H22F3NO3S: C, 59.28; H, 5.21; N, 3.29. Found: C, 59.02; H, 5.12; N, 3.29.
Supplementary Material
Acknowledgments
Acknowledgment is made to the National Institutes of Health (GM106260) for finacial support.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) Chintareddy VR, Wadhwa K, Verkade JG. J. Org. Chem. 2011;76:4482. doi: 10.1021/jo200314g. [DOI] [PubMed] [Google Scholar]; b) Correia CA, Gilmore K, McQuade DT, Seeberger PH. Angew. Chem., Int. Ed. 2015;54:4945. doi: 10.1002/anie.201411728. [DOI] [PubMed] [Google Scholar]; c) Wang L, Liu N, Dai B, Ma X, Shi L. RSC Adv. 2015;5:10089. [Google Scholar]
- 2.a) Anand NK, Carreira EM. J. Am. Chem. Soc. 2001;123:9687. doi: 10.1021/ja016378u. [DOI] [PubMed] [Google Scholar]; b) Li X, Lu G, Kwok WH, Chan ASC. J. Am. Chem. Soc. 2002;124:12636. doi: 10.1021/ja025541y. [DOI] [PubMed] [Google Scholar]; c) Gao G, Xie R-G, Pu L. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5417. doi: 10.1073/pnas.0307136101. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Takita R, Yakura K, Ohshima T, Shibasaki M. J. Am. Chem. Soc. 2005;127:13760. doi: 10.1021/ja053946n. [DOI] [PubMed] [Google Scholar]; e) Gao G, Wang Q, Yu X-Q, Xie R-G, Pu L. Angew. Chem. Int. Ed. 2006;45:122. doi: 10.1002/anie.200500469. [DOI] [PubMed] [Google Scholar]; f) Wolf C, Liu S. J. Am. Chem. Soc. 2006;128:10996. doi: 10.1021/ja062711o. [DOI] [PubMed] [Google Scholar]
- 3.a) Cozzi PG. Angew. Chem. Int. Ed. 2003;42:2895. doi: 10.1002/anie.200351230. [DOI] [PubMed] [Google Scholar]; b) Lu G, Li X, Jia X, Chan WL, Chan ASC. Angew. Chem., Int. Ed. 2003;42:5057. doi: 10.1002/anie.200352013. [DOI] [PubMed] [Google Scholar]; c) Ohshima T, Kawabata T, Takeuchi Y, Kakinuma T, Iwasaki T, Yonezawa T, Murakami H, Nishiyama H, Mashima K. Angew. Chem., Int. Ed. 2011;50:6296. doi: 10.1002/anie.201100252. [DOI] [PubMed] [Google Scholar]
- 4.a) Wei C, Li CJ. J. Am. Chem. Soc. 2002;124:5638. doi: 10.1021/ja026007t. [DOI] [PubMed] [Google Scholar]; b) Gommermann N, Koradin C, Polborn K, Knochel P. Angew. Chem. Int. Ed. Engl. 2003;42:5763. doi: 10.1002/anie.200352578. [DOI] [PubMed] [Google Scholar]; c) Knöpfel TF, Aschwanden P, Ichikawa T, Watanabe T, Carreira EM. Angew. Chem., Int. Ed. 2004;43:5971. doi: 10.1002/anie.200461286. [DOI] [PubMed] [Google Scholar]; d) Ji J-X, Wu J, Chan ASC. Proc. Natl. Acad. Sci. U.S.A. 2005;102:11196. doi: 10.1073/pnas.0502105102. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) de Armas P, Tejedor D, Garcia-Tellado F. Angew. Chem. Int. Ed. 2010;49:1013. doi: 10.1002/anie.200906018. [DOI] [PubMed] [Google Scholar]
- 5.For excellent reviews: Trost BM, Weiss AH. Adv. Synth. Catal. 2009;351:963. doi: 10.1002/adsc.200800776. Nie J, Guo H-C, Cahard D, Ma J-A. Chem. Rev. 2011;111:455. doi: 10.1021/cr100166a. Li S, Ma J-A. Chem. Soc. Rev. 2015;44:7439. doi: 10.1039/c5cs00342c.
- 6.a) Thompson AS, Corley EG, Huntington MF, Grabowski EJJ. Tetrahedron Lett. 1995;36:8937. [Google Scholar]; b) Tan L, Chen C-Y, Tillyer RD, Grabowski EJJ, Reider PJ. Angew. Chem., Int. Ed. 1999;38:711. doi: 10.1002/(SICI)1521-3773(19990301)38:5<711::AID-ANIE711>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 7.Motoki R, Kanai M, Shibasaki M. Org. Lett. 2007;9:2997. doi: 10.1021/ol071003k. [DOI] [PubMed] [Google Scholar]
- 8.Chinkov N, Warm A, Carreira EM. Angew. Chem., Int. Ed. 2011;50:2957. doi: 10.1002/anie.201006689. [DOI] [PubMed] [Google Scholar]
- 9.Zhang G-W, Meng W, Ma H, Nie J, Zhang W-Q, Ma J-A. Angew. Chem., Int. Ed. 2011;50:3538. doi: 10.1002/anie.201007341. [DOI] [PubMed] [Google Scholar]
- 10.a) Li H, Hsung RP. Org. Lett. 2009;11:4462. doi: 10.1021/ol901860b. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang X, Hsung RP, Li H, Zhang Y, Johnson WL, Figueroa R. Org. Lett. 2008;10:3477. doi: 10.1021/ol801257j. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Martinez-Esperon MF, Rodriguez D, Castedo L, Saá C. Org. Lett. 2005;7:2213. doi: 10.1021/ol050609a. [DOI] [PubMed] [Google Scholar]; d) Witulski B, Stengel T. Angew. Chem. Int. Ed. 1999;38:2426. [PubMed] [Google Scholar]; e) Witulski B, Alayrac C. Angew. Chem. Int. Ed. 2002;41:3281. doi: 10.1002/1521-3773(20020902)41:17<3281::AID-ANIE3281>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]; f) Dateer RB, Shaibu BS, Liu R-S. Angew. Chem. Int. Ed. 2012;51:113. doi: 10.1002/anie.201105921. [DOI] [PubMed] [Google Scholar]
- 11.a) Tanaka R, Yuza A, Watai Y, Suzuki D, Takayama Y, Sato F, Urabe M. J. Am. Chem. Soc. 2005;127:7774. doi: 10.1021/ja050261e. [DOI] [PubMed] [Google Scholar]; b) Tanaka D, Sato Y, Mori M. J. Am. Chem. Soc. 2007;129:7730. doi: 10.1021/ja071954t. [DOI] [PubMed] [Google Scholar]; c) Tracey MR, Zhang Y, Frederick MO, Mulder JA, Hsung RP. Org. Lett. 2004;6:2209. doi: 10.1021/ol0493251. [DOI] [PubMed] [Google Scholar]; d) Couty S, Liegault B, Meyer C, Cossy J. Org. Lett. 2004;6:2511. doi: 10.1021/ol049302m. [DOI] [PubMed] [Google Scholar]; e) Al-Rashid ZF, Hsung RP. Org. Lett. 2008;10:661. doi: 10.1021/ol703083k. [DOI] [PubMed] [Google Scholar]; f) Frederick mO, Mulder JA, Tracey mR, Hsung RP, Huang J, Kurtz KCM, Shen L, Douglas CJ. J. Am. Chem. Soc. 2003;125:2368. doi: 10.1021/ja021304j. [DOI] [PubMed] [Google Scholar]
- 12.a) Egi M, Yamaguchi Y, Fujiwara N, Akai S. Org. Lett. 2008;10:1867. doi: 10.1021/ol800596c. [DOI] [PubMed] [Google Scholar]; b) Wang X-N, Winston-McPherson GN, Walton M, Zhang Y, Hsung RP, DeKorver KA. J. Org. Chem. 2013;78:6233. doi: 10.1021/jo400960e. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang X-N, Hsung RP, Rui Q, Fox SK, Lv M-C. Org. Lett. 2013;15:2514. doi: 10.1021/ol400989x. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Cook AM, Wolf C. Chem. Commun. 2014;50:3151. doi: 10.1039/c4cc00394b. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Zhang P, Cook AM, Liu Y, Wolf C. J. Org. Chem. 2014;79:4167. doi: 10.1021/jo500365h. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Cook AM, Wolf C. Tetrahedron Lett. 2015;56:2377. doi: 10.1016/j.tetlet.2015.03.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Excellent reviews on ynamide chemistry: Evano G, Coste A, Jouvin K. Angew. Chem. Int. Ed. 2010;49:2840. doi: 10.1002/anie.200905817. DeKorver KA, Li H, Lohse AG, Hayashi R, Lu Z, Zhang Y, Hsung RP. Chem. Rev. 2010;110:5064. doi: 10.1021/cr100003s. Wang X-N, Yeom H-S, Fang L-C, He S, Ma Z-X, Kedrowski BL, Hsung RP. Acc. Chem. Res. 2014;47:560. doi: 10.1021/ar400193g.
- 14.Prime examples of terminal ynamide synthesis: Witulski B, Goessmann M. Chem. Commun. 1999:1879. Bruckner D. Tetrahedron. 2006;62:3809. Rodriguez D, Castedo L, Saa C. Synlett. 2007:1963. Hamada T, Ye X, Stahl SS. J. Am. Chem. Soc. 2008;130:833. doi: 10.1021/ja077406x. Mansfield SJ, Campbell CD, Jones MW, Anderson EA. Chem. Commun. 2015;51:3316. doi: 10.1039/c4cc07876d.
- 15.For examples of asymmetric additions of alkynes to aldehydes in the presence of Zn(OTf)2, amino alcohol and amine additives: Frantz DE, Fassler R, Carreira EM. J. Am. Chem. Soc. 2000;122:1806. Jiang B, Chen Z, Xiong W. Chem. Commun. 2002:1524. doi: 10.1039/b203984b. Graham ER, Tykwinski RR. J. Org. Chem. 2011;76:6574. doi: 10.1021/jo2008719. Several groups reported inconsistent results using these methods which have been attributed to variations in the Zn(OTf)2 quality: Marshall JA, Bourbeau MP. Org. Lett. 2003;5:3197. doi: 10.1021/ol034918h. Kirkham JED, Courtney TDL, Lee V, Baldwin JE. Tetrahedron. 2005;61:7219. and references therein. Fenster MDB, Dake GR. Chem. Eur. J. 2005;11:639. doi: 10.1002/chem.200400749. g) For general review of this topic, see ref 5.
- 16.For cinchona alkaloid catalyzed alkynylation of ketones, see ref 9 and Liu L, Wang R, Kang Y-F, Chen C, Xu Z-Q, Zhou Y-F, Ni M, Cai H-Q, Gong M-Z. J. Org. Chem. 2005;70:1084. doi: 10.1021/jo0483522.
- 17.For Bis-ProPhenol catalyzed asymmetric alkynylations of aldehydes in the presence of dimethylzinc, see: Trost BM, Weiss AH, von Wangelin AJ. J. Am. Chem. Soc. 2006;128:8. doi: 10.1021/ja054871q. Trost BM, Chan VS, Yamamoto D. J. Am. Chem. Soc. 2010;132:5186. doi: 10.1021/ja910656b. Trost BM, Quintard BA. Angew. Chem., Int. Ed. 2012;51:6704. doi: 10.1002/anie.201203035. Trost BM, Bartlett MJ, Weiss AH, von Wangelin AJ, Chan VS. Chem. Eur. J. 2012;18:16498. doi: 10.1002/chem.201202085.
- 18.We used slight excess of the ynamide but essentially the same results are obtained with 1.0 equivalent of 1 and 1.5 equivalents of 2. The reaction is sluggish in the absence of L8 and yields 3 in only 47% after 26 h. The replacement of Zn(OTf)2 with Et2Zn gave racemic 3.
- 19.Cozzi PG, Rudolph J, Bolm C, Norrby P-O, Tomasini C. J. Org. Chem. 2005;70:5733. doi: 10.1021/jo050115r. [DOI] [PubMed] [Google Scholar]
- 20.Wang J, Sanchez-Rosello M, Acena JL, del Pozo C, Sorochinsky AE, Fustero S, Soloshonok VA, Liu H. Chem. Rev. 2014;114:2432. doi: 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]
- 21.a) Valenta P, Carroll PJ, Walsh PJ. J. Am. Chem. Soc. 2010;132:14179. doi: 10.1021/ja105435y. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gopalaiah K, Kagan HB. Chem. Rev. 2011;111:4599. doi: 10.1021/cr100031f. [DOI] [PubMed] [Google Scholar]
- 22.Hirano S, Fukudome Y, Tanaka R, Sato F, Urabe H. Tetrahedron. 2006;62:3896. [Google Scholar]
- 23.Smith DL, Goundry WRF, Lam HW. Chem. Commun. 2012;48:1505. doi: 10.1039/c1cc13595c. [DOI] [PubMed] [Google Scholar]
- 24.Xu S, Liu J, Hu D, Bi X. Green Chem. 2015;17:184. [Google Scholar]
- 25.The hydroacyloxylations were carried out using batches of 3 with slightly lower ee obtained during reaction optimization studies.
- 26.CCDC numbers 1438550 and 1438551 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; fax: (44) 1223-336-033; or deposit@ccdc.cam.ac.uk)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.