

ABSTRACT
In a follow-up to the modest efficacy observed in the RV144 trial, researchers in the HIV vaccine field seek to substantiate and extend the results by evaluating other poxvirus vectors and combinations with DNA and protein vaccines. Earlier clinical trials (EuroVacc trials 01 to 03) evaluated the immunogenicity of HIV-1 clade C GagPolNef and gp120 antigens delivered via the poxviral vector NYVAC. These showed that a vaccination regimen including DNA-C priming prior to a NYVAC-C boost considerably enhanced vaccine-elicited immune responses compared to those with NYVAC-C alone. Moreover, responses were improved by using three as opposed to two DNA-C primes. In the present study, we assessed in nonhuman primates whether such vaccination regimens can be streamlined further by using fewer and accelerated immunizations and employing a novel generation of improved DNA-C and NYVAC-C vaccine candidates designed for higher expression levels and more balanced immune responses. Three different DNA-C prime/NYVAC-C+ protein boost vaccination regimens were tested in rhesus macaques. All regimens elicited vigorous and well-balanced CD8+ and CD4+ T cell responses that were broad and polyfunctional. Very high IgG binding titers, substantial antibody-dependent cellular cytotoxicity (ADCC), and modest antibody-dependent cell-mediated virus inhibition (ADCVI), but very low neutralization activity, were measured after the final immunizations. Overall, immune responses elicited in all three groups were very similar and of greater magnitude, breadth, and quality than those of earlier EuroVacc vaccines. In conclusion, these findings indicate that vaccination schemes can be simplified by using improved antigens and regimens. This may offer a more practical and affordable means to elicit potentially protective immune responses upon vaccination, especially in resource-constrained settings.
IMPORTANCE Within the EuroVacc clinical trials, we previously assessed the immunogenicity of HIV clade C antigens delivered in a DNA prime/NYVAC boost regimen. The trials showed that the DNA prime crucially improved the responses, and three DNA primes with a NYVAC boost appeared to be optimal. Nevertheless, T cell responses were primarily directed toward Env, and humoral responses were modest. The aim of this study was to assess improved antigens for the capacity to elicit more potent and balanced responses in rhesus macaques, even with various simpler immunization regimens. Our results showed that the novel antigens in fact elicited larger numbers of T cells with a polyfunctional profile and a good Env-GagPolNef balance, as well as high-titer and Fc-functional antibody responses. Finally, comparison of the different schedules indicates that a simpler regimen of only two DNA primes and one NYVAC boost in combination with protein may be very efficient, thus showing that the novel antigens allow for easier immunization protocols.
INTRODUCTION
In order to develop an efficacious prophylactic vaccine against infection with human immunodeficiency virus type 1 (HIV-1), various approaches are being pursued to optimize the immune functions that might contribute to protection from infection or disease. Several factors are likely to be important for the potential success of a vaccine. Besides the choice of antigen as the core component of any vaccine, the mode of delivery, the immunization regimen, route, and dose, and the exploitation of immune-modulating factors, either added in trans as adjuvants or representing intrinsic properties of, e.g., vector systems, may also affect vaccine efficacy. Current approaches are mainly focused on the induction of antibody responses, as they are considered to prevent infection, while CD8+ cytotoxic T lymphocyte (CTL) responses are generally thought to modify disease progression by reducing viral loads (1). However, recent studies of rhesus macaques immunized with a novel cytomegalovirus (CMV) vector indicate the potentially protective role of CD8+ T cells, especially those with an effector memory phenotype (2–4). Moreover, given that helper CD4+ T cell responses are important for high-quality B cell responses, a vaccine candidate should likely elicit responses of all kinds—innate, B cell, helper T cell, and CTL—in a balanced manner.
The 31% protection observed in the RV144 Thai trial (5), which used the poxvirus ALVAC expressing Gag, Pro, and gp120-TM for the prime step and AIDSVAX B/E gp120 for the boost step, came as a surprise, as the AIDSVAX vaccine itself lacked efficacy (6, 7). This finding highlights the potential value of replication-deficient live recombinant viral vectors and heterologous prime-boost regimens to elicit protective immune responses. In particular, priming with DNA-vectored vaccines prior to the application of the viral vector, mostly by employing adenoviruses or poxviruses, has repeatedly been shown to considerably increase cellular and humoral immune responses compared to those obtained with the viral vector alone (8–10). In the context of the EuroVacc clinical trials EV01 and EV02 (11, 12), we tested HIV clade C antigens (GagPolNef and gp120) delivered via the poxvirus New York vaccinia virus (NYVAC), with or without priming with a DNA encoding the same set of antigens. These vectors previously showed promising immunogenicity profiles in preclinical assays and protective efficacy in primates against simian-human immunodeficiency virus SHIV89.6 challenge (13, 14). The clinical trials demonstrated that the vaccine candidates were safe and well tolerated and that DNA priming dramatically improved the T cell responses elicited by NYVAC. Both the proportion of responders and the number of HIV-specific T cells, as measured by enzyme-linked immunosorbent spot (ELISpot) analysis of gamma interferon (IFN-γ)-secreting cells, increased 2- to 4-fold (12, 15). Other studies have shown even better augmentation (16). The T cell responses were mainly of the CD4+ phenotype and directed against Env, although a balanced response against all antigens and a balanced ratio of CD4+ and CD8+ responses might be desirable, especially against Gag, as CD8 T cell responses against this antigen are associated with long-term disease control in some patients (17, 18). Regarding the immunization schedule, a regimen consisting of three DNA primes (at months 0, 1, and 2) plus a single NYVAC boost at month 6 (using the same generation of DNA and NYVAC as those in EV01 and EV02) was superior to one with two DNA primes (at months 0 and 1) followed by two NYVAC boosts (at months 5 and 6) (EV03 study) (19). Although macaques seem to be more responsive than humans in immunogenicity analyses, the rhesus macaque model at least allows a ranking of the immunogenicities of different vectors (20). Therefore, despite additional limitations, such as differences in the configuration of major histocompatibility complex class I (MHC-I) loci (21), monkey trials are instrumental in evaluating the numerous vaccine regimens and help in the design of human clinical trials. Importantly, the cellular immune responses observed in humans in the EV02 study were remarkably similar to those obtained in macaques immunized with the same DNA and NYVAC vaccine candidates and according to the same regimen (22), which supports the value of the rhesus macaque model.
Researchers in the HIV vaccine field plan to substantiate and extend the results observed in the RV144 trial by evaluating other poxvirus vectors, such as NYVAC, in combination with DNA and protein vaccines. A key question that has to be addressed is how to best schedule the immunizations. This is an especially important issue because complex immunization regimens comprising many immunizations (for instance, 7 in the AIDSVAX trials and 6 in the RV144 trial) are unlikely to be brought to market. Yet simple modifications in the number, timing, or sequence of heterologous vectors can have a major impact on immunogenicity (23, 24). Such modifications offer a lot of potential, as they can be tested quite easily without again performing safety analyses if the individual components have already been assessed. Therefore, the present study was designed to determine whether an accelerated DNA schedule or fewer DNA injections elicit equally effective priming responses if optimized vaccines are employed. Toward this goal, both the DNA and NYVAC vaccines were redesigned and thus represent a new generation of vaccine candidates with optimized antigens and vectors.
Specifically, the major optimization comprises modified antigens, mainly in regard to the weak Gag-specific responses observed in nonhuman primate (NHP) and clinical studies (EV01 to EV03) (11, 12, 15, 19). For this purpose, the original GagPolNef antigen, consisting of a 160-kDa fusion protein with modifications introduced for increased safety (i.e., abrogated myristoylation, lack of IN, inactivation of PR, splitting of RT, and scrambling of Nef [25]), was refined further to allow for efficient production and release of virus-like particles and to better balance the relative expression of Gag and PolNef antigens. For plasmid delivery of these next-generation antigens, Gag and PolNef were split into two DNA vectors in order to reduce the individual plasmid size, thus likely facilitating uptake in vivo. Moreover, a gp140 form was used instead of gp120 to more closely resemble the native trimeric envelope structure (26) and was included in a third DNA vector. Immunogenicity analyses in mice clearly showed superiority of this three-plasmid configuration over the parental EuroVacc vaccines regarding both the magnitude of antigen-specific T cells and the balance toward the different antigens (27). Improved responses after splitting of a different Gag-Pol-Nef antigen into separate parts were also observed in humans in clinical phase I trials (16, 28). For the immunological assessment of our next-generation antigens in rhesus macaques, the plasmid backbone was also changed to VRC-8400 (29) from the pORT constructs (30) to further increase expression of the antigens.
For the next generation of recombinant NYVAC vectors, the GagPolNef and Env antigens were inserted into two separate NYVAC vectors (bivalent) rather than a single vector as before (monovalent). For this purpose, the natural ribosomal (−1) frameshift between Gag and Pol was restored to skew Gag:PolNef expression to approximately 10:1 (31), and the N-terminal myristoylation signal was reintroduced to enable release of GagPolNef virus-like particles from infected cells. As for the DNA vaccine, gp120 was replaced by gp140 in the second NYVAC vector. In mice, these vectors triggered polyfunctional CD4+ and CD8+ T cell as well as humoral responses to the HIV antigens (32).
In this investigation, we demonstrate that the new generation of DNA and NYVAC vectors with the novel antigens show increased levels of immunogenicity compared to those of earlier-generation (NYVAC-Cold and DNA-Cold) vaccines, as assessed by both qualitative and quantitative endpoints. In addition, we found that by using these novel DNA vaccine candidates, a third DNA prime has no additional benefit for T cell responses and the NYVAC/protein boost can be given earlier, thus making the schedule leaner and more applicable for use in humans.
MATERIALS AND METHODS
Ethics statement.
This study was performed with male Indian rhesus macaques (Macaca mulatta mulatta) that were housed, fed, given environmental enrichment, and handled at the animal facility of Advanced BioScience Laboratories (ABL), Inc. (Rockville, MD). The study protocol and primate colony care strictly adhered to the guidance found in the 8th edition of the Guide for the Care and Use of Laboratory Animals (33), as well as the Public Health Services policy on the humane care and use of laboratory animals from the Office of Animal Welfare (part of the U.S. Department of Health and Human Services). These activities were also in full compliance with the regulations found in the Animal Welfare Act (9 CFR 3.81) and enforced by the U.S. Department of Agriculture. The study was approved by the ABL, Inc., Institutional Animal Care and Use Committee (animal use protocol 485). All procedures were carried out under anesthesia (ketamine administered at 10 mg/kg of body weight) by trained personnel under the supervision of veterinary staff, and all efforts were made to ameliorate animal welfare and to minimize animal suffering, in accordance with the recommendations found in the Weatherall report on the use of nonhuman primates (https://royalsociety.org/topics-policy/publications/2006/weatherall-report/). Enrichment and care beyond the specifications in the aforementioned regulations were done according to ABL's Primate Environmental Enrichment Program. During the study, monkeys were observed for general behavior, clinical symptoms, and local reactions at the injection sites twice daily the week after immunizations. When animals were sedated for immunizations or sample collections, body weight and temperature were measured. At selected time points, a physical examination was performed by a veterinarian, and clinical chemistry and hematology parameters were measured. A total of 24 animals were divided into three groups of eight animals each.
Antigens.
The Gag sequence was derived from the HIV-1 clade C isolate 96ZM651 (accession no. AF286224). The PolNef sequence was derived from the HIV clade C/B′ isolate 97CN54 (accession no. AX149647.1) and consists of p6* (amino acids [aa] 1 to 56) followed by protease (aa 57 to 155; inactivated by the D81N mutation), the N-terminal part of the reverse transcriptase (RT) (aa 156 to 320 and 357 to 360), scrambled Nef (aa 101 to 206 followed by aa 1 to 100), the C-terminal part of RT (aa 361 to 715), and then the middle part of RT (aa 321 to 356). The GagFSPolNef cassette inserted into NYVAC (see below) contains identical sequences on the amino acid level, though the two reading frames are connected by employing the natural ribosomal frameshift (FS; thus keeping the wild-type codon usage for the region from the slippery site to the stop codon of gag), leading to expression of Gag and GagPolNef, presumably at a ratio of about 10:1 (31). The gp140 sequence was derived from isolate 96ZM651, and it consists of aa 1 to 673, including the autologous signal sequence (incorporation of a strong Kozak initiation site leads to the R2G mutation within the signal sequence), and contains a mutated gp120-gp41 cleavage site (R516S).
All antigen open reading frames (ORFs) were (codon) optimized for human expression by using the GeneOptimizer algorithm (34), supplemented with a strong Kozak initiation site, and synthesized by GeneArt AG (Regensburg, Germany), with suitable restriction sites for insertion into the respective plasmids.
The plasmid VRC-8400 (G. Nabel, Vaccine Research Center, NIAID) was used as the vector backbone of the DNA vaccine. It contains a CMV immediate early enhancer/promoter followed by the R region from human T cell leukemia virus type 1 (HTLV-1) (with splice donor) and the CMV immediate early 3′ intron (with splice acceptor), with the antigen ORF inserted via SalI and NotI restriction sites, followed by the BGH polyadenylation sequence. Plasmids were produced in Escherichia coli strain DH5α and isolated and purified using a Qiagen EndoFree Plasmid Giga kit according to the manufacturer's instructions. Endotoxin levels were below 40 endotoxin units (EU)/mg.
GagPolNef and gp140 were inserted into NYVAC (35) as described by Perdiguero et al. (32). Viruses were produced in primary CEF cells and purified by sucrose cushion centrifugation twice. Titers were determined by plaque immunostaining of BSC-40 cells. Recombinant clade C gp120 proteins (isolates TV1 and 1086) were expressed from stably transfected CHO cell lines, purified, and characterized as previously described (36).
Vaccines and immunizations.
The DNA vaccine consists of a 1:1:1 mixture, by weight, of VRC-8400-Gag, VRC-8400-PolNef, and VRC-8400-gp140 at 2 mg/ml in phosphate-buffered saline (PBS). Two milliliters was injected intramuscularly into the upper left and upper right legs (1 ml per site). The NYVAC vaccine consists of a 1:1 mixture of NYVAC-GagFSPolNef and NYVAC-gp140 at 2 × 108 PFU/ml in Tris-buffered saline (TBS). One milliliter was injected intramuscularly into the deltoid of the right arm. The bivalent subtype C gp120 protein boost consisted of 50-μg doses each of the TV1 and 1086 gp120s adjuvanted with MF59 (1:1 by volume, mixed shortly before application) at a final protein concentration of 0.1 mg/ml. One milliliter was injected intramuscularly into the deltoid of the left arm.
Blood samples were taken at the time points indicated in Fig. 1. For T cell analyses, 22 ml EDTA-blood was collected, and for antibody analyses, 5 ml of plasma and clotted blood was collected.
FIG 1.
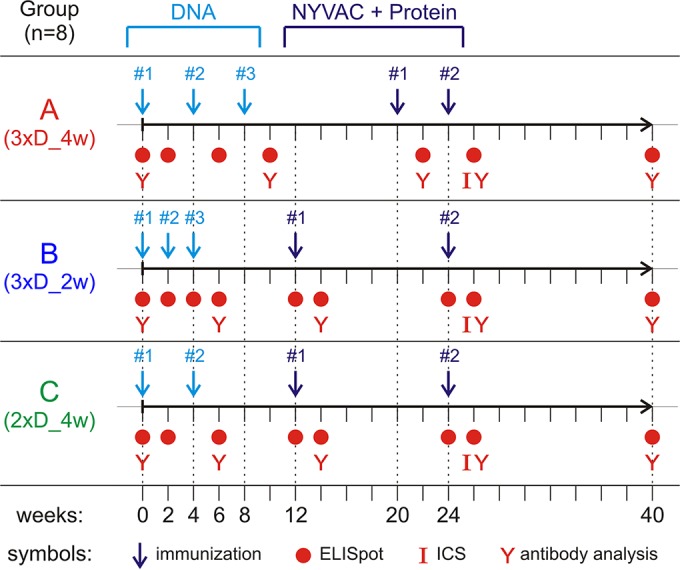
Immunization schedules for comparison of the three different priming regimens. Three groups of 8 macaques each were immunized two or three times with DNA (light blue) and twice with NYVAC/protein (dark blue). At the indicated time points, blood was collected for ELISpot analysis, intracellular cytokine staining (ICS), or antibody analysis (red symbols).
Immunological analyses. (i) Peptides.
Nine different peptide pools, consisting of 15-mers overlapping by 11 aa and matching the antigens and covering the regions Gag1, Gag/Pol, Gag2/Pol, Pol1, Pol2, Env1, Env2, Env3, and Nef, were used for T cell stimulations.
(ii) IFN-γ ELISpot assay.
Freshly isolated peripheral blood mononuclear cells (PBMCs) were stimulated in triplicate with peptide pools at 1 μg/ml, or with phytohemagglutinin (PHA) (2.5 μg/ml) as a positive control, while addition of medium only served as a negative control. Millipore 96-well filtration plates were coated with 5 μg/ml mouse anti-human IFN-γ antibody (BD Pharmingen) overnight at 4°C. After blocking with complete RPMI medium for 2 h at 37°C, 2 × 105 PBMCs and the peptides/PHA were added. Plates were incubated at 37°C for 18 to 24 h before washing with cold H2O twice and PBS plus Tween (PBST) five times. Biotinylated anti-IFN-γ antibody (Mabtech) was added at 1 μg/ml for 1 h at 37°C, and after washing, a 1:2,000 dilution of avidin-horseradish peroxidase (HRP) (Vector Laboratories) was added, again for 1 h at 37°C. After the final wash, stable DAB (Invitrogen) was added for 2 min. The reaction was stopped by washing with water. After drying, the number of spots in each well was counted with an automated ELISpot reader (CTL ImmunoSpot v5 reader). Animals with more than 50 spot-forming units (SFUs)/106 cells and 4 times the week 0 background values for any one of the peptide pools were considered responders (compare to reference 11).
(iii) ICS.
Intracellular cytokine staining (ICS) was carried out by the Nonhuman Primate Immunogenicity Core (Roederer lab), Vaccine Research Center, NIAID, as described previously (37). In brief, cryopreserved blood cells were thawed and rested overnight in medium. For stimulation, peptides or dimethyl sulfoxide (DMSO; negative control) was added at 2 μg/ml in the presence of 10 μg/ml brefeldin A to 1 × 106 to 3 × 106 cells in 96-well plates. After 6 h of incubation at 37°C, cells were stored at 4°C until staining. For staining, cells were fixed, permeabilized, and stained with fluorescence-labeled antibodies directed against CD3, CD4, CD8, IFN-γ, interleukin-2 (IL-2), and tumor necrosis factor (TNF) for flow cytometric analysis.
(iv) HIV-1-specific binding antibody assay.
HIV-1-specific IgG and IgA antibodies to gp120/gp140 proteins, scaffolded V1V2, p24, and p66(RT) were measured by an HIV-1 binding antibody multiplex assay (Tomaras lab) as previously described (38–40). All assays were run under good clinical laboratory practice-compliant conditions, including tracking of positive controls by use of Levy-Jennings charts. Positive controls included HIV immune globulin (HIVIG) and CH58 monoclonal antibody (MAb) IgG titration. Negative controls included in every assay were a blank, MuLVgp70_His6 (empty gp70 scaffold)-coupled beads, and HIV-1-negative sera. To control for antigen performance, we used the preset criterion that the positive-control titer (HIVIG) for each assay (and that for CH58 MAb, for assays with V1V2 antigens) had to be within 3 standard deviations of the mean for each antigen (tracked with a Levy-Jennings plot with preset acceptance of the titer [calculated with a four-parameter logistic equation]; SigmaPlot [Systat Software]). Antibody measurements were acquired on a Bio-Plex instrument (Bio-Rad, Hercules, CA) using 21CFR Part 11-compliant software, and the primary readout was the mean fluorescence intensity (MFI). Samples not matching the assay's predefined positivity criteria were considered nonresponders with a value of zero. For IgG, MFI data were transformed to values defining the area under the curve (AUC), which resemble the integral of the curve for MFI plotted against the 6-fold dilution series (41). The following antigens were examined: (i) consensus gp140 proteins of group M (Con S gp140 CF [42, 43]), clade A (A1.con.env03 140 CF), clade B (B.con.env03 140 CF), and clade C (C.con.env03 140 CF); (ii) primary Env variants 1086 gp120 (clade C), TV1 gp120 (clade C), JRFL gp140 (clade B), and MSA4076 gp140 (clade A1; also known as OOMSA); (iii) gp70_B.CaseA2 V1V2; and (iv) Gag p24 and RT p66.
(v) Neutralization.
Neutralization of sera was assessed using the TZM-bl cell assay and the more sensitive A3R5 cell assay, as described previously (44, 45), by the Comprehensive Antibody Vaccine Immune Monitoring Consortium (CA-VIMC). For the TZM-bl assay (Seaman lab), pseudoviruses carrying the following Envs were used: BaL.26 (tier 1B, clade B), Bx08.16 (tier 1B, clade B), MN (tier 1A, clade B), SS1196.1 (tier 1B, clade B), SHIV-SF162P4 (tier 1A, clade B), SHIV-SF162P3 (tier 2, clade B), MW965.26 (tier 1A, clade C), TV1.21 (tier 1, clade C), and murine leukemia virus (MuLV) (as a negative control). For SHIV-SF162.P3 and SHIV-SF162.P4, primary virus from PBMCs was used in some cases. The pseudoviruses were incubated with the macaque sera, and TZM-bl cells, which express luciferase in a Tat-dependent manner upon infection, were then added. The assay was carried out according to standardized protocols. For the A3R5 cell assay (Montefiori lab), also done according to standardized protocols, viral infectious molecular clones (IMCs) which encoded luciferase on the genome were used. Thus, the enzyme was introduced and expressed in the cells only upon infection. Six different IMCs were used, carrying the ectodomains of env genes of the following tier 2 clade C isolates: CAP45.2.00, Ce1086, Du151.2, Ce1176, Ce2010, and Du422.1. For both assays, the neutralization titer is given as the serum dilution leading to a 50% decrease (IC50) in relative light units between the respective virus and control after subtraction of the background values.
(vi) ADCC assay.
The antibody-dependent cellular cytotoxicity (ADCC) assay was carried out by the CA-VIMC ADCC core laboratory as described previously (46). Briefly, CEM.NKRCCR5 cells were used as target cells after coating with 10 μg/ml of recombinant gp120 (from isolate 1086 or TV1) for 90 min. During the last 15 min of incubation, the cells were labeled with fluorescent markers to assess viability and with the target cell fluorescent marker TFL4. PBMCs, including natural killer (NK) cells, were used as effector cells, at an effector-to-target (E:T) ratio of 30:1. The cells were mixed with a granzyme B (GrB) substrate, and finally, the plasma samples were added. After 15 min of incubation, the assay plates were centrifuged and again incubated for 1 h. After washing, cells were analyzed by flow cytometry for the fluorescence signal arising from cleavage of the GrB substrate, and the percent GrB-positive viable cells (minus the background) was calculated for each dilution. The titer of the ADCC-mediating antibodies was then calculated by interpolation of the dilution exhibiting a GrB activity matching the cutoff value for positivity of 8%.
(vii) ADCVI assay.
The antibody-dependent cell-mediated virus inhibition (ADCVI) assay (Forthal lab) was carried out as described previously (47). Briefly, CEM.NKRCCR5 cells were used as target cells. After infection with HIV-1 DU156 or HIV-1 DU422 for 1 h, cells were washed, incubated for 3 days, and then mixed with the respective plasma sample and PBMCs, as effector cells, at an E:T ratio of 10:1. After 3 days, cells were washed to remove unbound antibody, and after an additional 4 days, supernatants were harvested and assayed for p24 by enzyme-linked immunosorbent assay (ELISA). The percent virus inhibition was calculated as follows: % inhibition = {1 − [c(p24)_v/c(p24)_c)]} × 100, where c(p24)_v is the concentration of p24 in tests with plasmas from vaccinated animals and c(p24)_c is the concentration in the control plasmas (average for week 0 samples).
Statistics.
Statistical analysis was done using R, version 3.0.2 (The R Foundation for Statistical Computing). Comparisons within groups were done with the Wilcoxon signed-rank test, and differences between groups were assessed using the Wilcoxon rank sum test. P values of <0.05 were considered significant. Where applicable, Bonferroni correction was employed for multiple comparisons.
RESULTS
Building on the experience from the EuroVacc clinical trials EV01 to EV03 (11, 12, 19), as well as parallel confirmatory immunogenicity studies performed with rhesus macaques (22), we set out to refine several parameters of our vaccine concept to further augment the strength and quality of the anti-HIV-1 immune response, with a special emphasis on enhancing T cell responses. Optimized antigens were assessed in different immunization schedules. Based on the EV03 study, where three DNA primes and one NYVAC boost gave the best results, we sought to determine (i) if a second NYVAC boost is required to further increase responses, (ii) if an accelerated DNA prime (interval shortened to 2 weeks) gives responses similar to those with the previously applied 4-week interval, (iii) if the time to the NYVAC boost can be shortened, (iv) if the third DNA prime still improves immune responses with the improved antigens, and (v) if coadministering adjuvanted protein during the boost especially increases the humoral responses. With respect to the aim of establishing simplified regimens regarding the overall time frame, we chose to administer NYVAC and the protein component at the same time, into different arms, instead of using consecutive applications. The impacts of the above variations on the magnitude, breadth, and polyfunctionality of T cell responses and on the binding antibody titer, neutralization, ADCC, and ADCVI activity were assessed.
Immunizations.
Rhesus macaques were divided into three groups of eight animals each and received DNA primes according to the following schedules: group A, labeled 3xD_4w, received three immunizations with the DNA vaccine (3xD) at 4-week (4w) intervals (i.e., weeks 0, 4, and 8); group B, labeled 3xD_2w, also received three immunizations, but with an accelerated schedule with 2-week intervals (i.e., weeks 0, 2, and 4); and group C, labeled 2xD_4w, received only two immunizations, given 4 weeks apart (i.e., weeks 0 and 4). At each immunization, 2 doses of 2 mg DNA containing a mixture of three plasmids, encoding Gag, PolNef, and Env-gp140, were injected intramuscularly (i.m.) into the upper legs. All groups received two i.m. booster immunizations with a 1:1 mixture of NYVAC-GagPolNef and NYVAC-gp140 (2 × 108 PFU in total) in the upper right arm and, at the same time, a bivalent clade C gp120 boost with a total dose of 100 μg protein (50 μg each of 1086 and TV1 gp120s) adjuvanted with MF59 in the upper left arm. Group A was boosted at weeks 20 and 24 (4-week interval between boosts), and groups B and C were boosted at weeks 12 and 24 (12-week interval between boosts). The immunization schedules are summarized in Fig. 1.
The animal health monitoring program showed that the vaccinations were safe and well tolerated, with only mild or moderate adverse events. For the general cage-side observations, besides two episodes of mild emesis (group A; after 1st and 2nd DNA immunizations), reduced food consumption (between 11 and 50%) was observed on 11 occasions across groups and immunizations, always on the first day after the immunization. This was likely not a consequence of the immunization itself but of the anesthesia performed and is a commonly observed phenomenon. Only one local reaction was observed, for an animal from group C after the last immunization, with a pink skin discoloration of <5 cm in diameter at the injection site. Finally, the clinical pathology results showed incidental changes for some parameters (increased hemoglobin, urea nitrogen, and cholesterol levels and increased leukocyte counts), which were again distributed across the groups and immunizations such that an association with the vaccinations is unlikely.
Unfortunately, one animal from group C died from an acute gastric dilatation during the course of the study, 13 weeks after the final immunization. According to the veterinarian overseeing these studies, this incident was considered unrelated to the vaccinations, with the symptoms and onset being more consistent with an event occasionally occurring in this species.
Magnitude of T cell responses.
The overall strength of HIV-specific T cell responses elicited over the course of the study was analyzed by IFN-γ ELISpot analysis. Freshly isolated PBMCs were stimulated with 9 different peptide pools matching all antigens for 1 day, and the numbers of spot-forming units (SFUs) were determined as a measure of the magnitude of HIV-reactive T cells. The results over time are depicted in Fig. 2.
FIG 2.

All vaccination regimens induced similarly large numbers of IFN-γ-secreting T cells. Freshly isolated PBMCs collected at the indicated time points (weeks after the start of the study) were stimulated with nine different peptide pools and subjected to IFN-γ ELISpot analysis. The total numbers of spot-forming units (SFUs) per million cells for group A (3xD_4w; red), group B (3xD_2w; blue), and group C (2xD_4w; green) are shown, along with medians and interquartile ranges. (A) Responses after DNA prime immunizations. (B) Responses after NYVAC/protein boosting (note the different scales). Data sets obtained 2 weeks after the last DNA immunization and the last NYVAC/protein boost are highlighted by a shaded background.
Repeated DNA priming led to increasing numbers of HIV-specific T cells, reaching medians of 688 (group A; 3xD_4w), 785 (group B; 3xD_2w), and 703 (group C; 2xD_4w) SFUs/106 cells 2 weeks after the last DNA vaccination. Three animals, 1 in group A and 2 in group B, reached peak values of >2,000 SFUs/106 cells 2 weeks after the 3rd DNA vaccination. The administration of a third DNA prime in group A (4-week intervals) did not lead to a further increase in SFUs 2 weeks after the last DNA vaccination, and the final level was comparable to that obtained for group C, with only two DNA primes. For the accelerated regimen (group B), the numbers of SFUs were similar 2 weeks after the first (median, 210/106 cells) and second (median, 215/106 cells) primes and increased after the third prime, to levels obtained for the other regimens at the same point in time (6 weeks after the first or 2 weeks after the last DNA immunization). Thus, the different priming regimens performed equally well. All but one animal from group B and two animals from group C could be classified as responders, with SFU numbers exceeding the threshold criteria. Six weeks after the last DNA prime for groups B and C, the numbers of SFUs had decreased again, to medians of 535 and 250 SFUs/106 cells, respectively. Thus, neither the acceleration of the intervals between the three DNA immunizations from 4 weeks (group A; 8-week prime) to 2 weeks (group B; 4-week prime) nor the reduction from 3 to 2 DNA immunizations (group C; 4-week prime) had a negative impact on the total number of HIV-specific, IFN-γ-producing T cells.
Boosting with the NYVAC-protein combination led to a vigorous increase within 2 weeks, to median numbers of SFUs of 4,390, 3,088, and 3,453 per million PBMCs for groups A, B, and C, respectively. Similar to the responses upon priming, there were no statistically significant differences between the groups after the booster immunizations. Eight animals developed more than 5,000 SFUs/106 cells after the 1st NYVAC boost (three in group A, three in group B, and two in group C), with a top score of 9,880 SFUs/106 cells for one animal in group A. All animals in all groups could clearly be classified as responders. Over the 12 weeks following the first NYVAC/protein boost, the numbers of SFUs again declined (median of 1,218/106 cells for group B and 588/106 cells for group C at week 24). Reboosting at week 24 elicited responses that were similar in magnitude to those shortly after the first boost and were comparable between the groups (medians of 4,155, 4,508, and 3,098 SFUs/106 cells at week 26). Again, all animals were classifiable as responders. The T cell responses waned until the final assessment at week 40, to medians of 1,108, 1,145, and 920 SFUs/106 cells, again without exhibiting any statistically significant differences. Thus, the NYVAC/protein immunization clearly boosted the DNA-primed immune responses and was equally effective when applied 8 weeks after the final DNA prime as opposed to the conventional 12-week time point.
Quality of T cell responses.
Next, we assessed the quality of the T cell responses obtained 2 weeks after completing the regimen (week 26), i.e., after the final NYVAC/protein boost, when all animals exhibited strong responses in the ELISpot analysis. To assess the ratio of HIV-specific CD4+ and CD8+ T cells as well as their cytokine release profiles, cells were stained with fluorescently labeled antibodies against the surface markers CD3, CD4, and CD8 and the intracellular cytokines IFN-γ, IL-2, and TNF.
All three regimens elicited similar magnitudes of HIV-specific CD4+ and CD8+ cells (Fig. 3A), with no statistical differences. The overall frequency of HIV-specific cells was rather high, with medians of 1.5% CD4+ and 1.0% CD8+ T cells, on average. Thus, the ratio of CD4 to CD8 cells was balanced, with a factor of only 1.5 between them. Patterns of cytokine release were not significantly different between the groups (Fig. 3B). The level of polyfunctionality was high, with 61% of CD4+ and 44% of CD8+ cells, on average for all three groups, secreting all three of the cytokines assessed. Twenty-six percent and 13% of CD4+ cells and 39% and 17% of CD8+ cells secreted two and only one of the cytokines, respectively. Group C showed a trend toward a slightly smaller fraction of CD8+ cells (but not CD4+ cells) secreting all three tested cytokines (34%) than those for groups A (49%) and B (48%).
FIG 3.

All vaccine groups demonstrated similar T cell responses, characterized by a balanced ratio of CD4 to CD8 cells and large proportions of polyfunctional cells. PBMCs from vaccinated macaques were isolated at week 26, stimulated with the 9 peptide pools, stained for CD3, CD4, and CD8 and for the intracellular cytokines IFN-γ, IL-2, and TNF by use of fluorescently labeled antibodies, and analyzed by flow cytometry. (A) Percentages of CD4+ and CD8+ HIV-specific T cells (sums for all peptide pools and cytokines; medians with quartiles are shown). (B) CD4+ (upper graph) and CD8+ (lower graph) HIV-specific T cells separated for determination of cytokine release profiles (medians with quartiles are shown). Pie charts below show the relative fractions of trifunctional (green), bifunctional (blue), and monofunctional (red) T cells for the three groups. (C) Pie charts showing relative fractions of CD4+ (left) and CD8+ (right) T cells stimulated by the indicated peptide pools (sums for all cytokine profiles).
Regarding the distribution of HIV-specific T cells among the different antigens, Env-specific T cell responses were slightly more pronounced for all three groups than T cell responses directed against Gag/Pol and the small Nef protein. Considering the responses of all 24 animals of groups A, B, and C, 57% of all T cell responses were Env specific, whereas 41% were directed against GagPol and 2% against Nef (Fig. 3C). There was a tendency for slightly higher Nef and Gag/Pol responses for CD8+ T cells than for CD4+ T cells. Furthermore, group B seemed to show a slight trend toward more pronounced GagPol-specific CD8+ responses (52%) than those of groups C (42%) and A (37%). However, none of the observed trends reached statistical significance. In conclusion, all the tested immunization regimens elicited a balanced ratio of CD4 to CD8 cells and a high degree of polyfunctionality. The proportions of GagPolNef- versus Env-specific responses were well balanced compared with the Env polarization of the previous antigens (22).
Binding antibody responses.
Vaccine-induced IgG and IgA binding antibodies were measured using a multiclade envelope panel (gp140 consensus proteins of group M, clades A, B, and C; selected primary gp120/gp140 proteins) as well as MuLV gp70-scaffolded V1V2 (gp70V1V2), p24 Gag, and the p66 subunit of RT.
All three groups already showed measurable IgG responses to consensus clade C gp140 (Fig. 4A) after completion of the DNA primes, i.e., AUC value of 5,000 at week 10 for group A and AUC values of 700 and 400 at week 6 for groups B and C, respectively. The difference between group A and groups B and C is statistically significant (P = 0.0002 and 0.0006; Wilcoxon rank sum test). The magnitude of the antibody response further increased after the NYVAC/protein boosts (on average, 49-fold after the 1st boost and slightly [1.3-fold] after the 2nd boost) and declined again until the final measurement at week 40 (11-fold, on average). The time courses for the other three consensus gp140 proteins and the representative clade A and B gp140 proteins, as well as the gp120 proteins, exhibited very similar kinetics (Fig. 4C). We also detected significant antibody responses to gp70V1V2 for the time point after the second NYVAC/protein boost (Fig. 4B). At week 26 (2 weeks after completion of the immunization regimen), the magnitude of the mean IgG response was highest for the consensus clade C gp140 protein, followed by the group M gp140, clade C TV1 and 1086 gp120, consensus A, clade B JRFL gp140, consensus B gp70V1V2, and clade A MSA4076 gp140 proteins (Fig. 5A). Yet the median AUC values for IgGs toward the diverse HIV-1 envelope glycoproteins were within 1 order of magnitude (factor of 2.7 between the consensus C and MSA4076 proteins). The ranking was almost the same at week 40 (Fig. 5B). Mean p24 responses exceeded even the best Env responses (by a factor of 1.2).
FIG 4.

All vaccination regimens induced strong IgG antibody responses against several Env antigens, as well as V1V2-specific responses and responses against p24 and p66(RT). IgG binding magnitudes are indicated as AUC values, and medians and interquartile ranges are shown for the specified time points postvaccination. Shaded areas highlight time points 2 weeks after completion of the DNA and NYVAC/protein immunizations. (A) Time course for antibody responses against consensus clade C gp140. (B) Responses against gp70V1V2. (C) Responses against the other tested proteins, as indicated.
FIG 5.

Comparison of antibody responses against the different antigens at weeks 26 (A) and 40 (B). IgG binding magnitude data are from Fig. 4.
As observed for the T cell responses, the accelerated and DNA-sparing regimens were similar to the full regimen in comparisons of overall antibody titers after the two boosts.
Furthermore, the magnitude of serum IgA responses was measured simultaneously. As shown in Fig. 6A for clade C consensus gp140 and 1086 gp120, overall responses were rather low (barely above the background levels) and were observed only at the late time points after the NYVAC/protein boosts. Responses significantly above the week 0 preimmunization background were not observed. In general terms, the responses were negligible, and consequently, no differences between the groups were evident. Data for the other antigens are shown in Fig. 6B.
FIG 6.

The vaccination regimens elicited only negligible serum IgA responses. IgA binding magnitudes are indicated by MFI, and medians and interquartile ranges are shown for each time point postvaccination. (A) Responses against consensus clade C gp140 and 1086 gp120. (B) Responses against the indicated proteins.
Neutralizing antibody responses.
Furthermore, the capacity of the obtained sera to neutralize various Env isoforms was assessed with the TZM-bl cell assay and the more sensitive A3R5.7 cell assay. For the former, pseudotyped viruses carrying one of eight different Env isolates or MuLV (as a negative control) were incubated with sera obtained from immunized monkeys before addition of TZM-bl cells, which express luciferase upon infection. The serum titer which led to 50% neutralization was calculated, and the results are shown in Fig. 7. Only for the highly neutralization-sensitive tier 1A clade C isolate MW965.26 (48) was significant neutralization obtained for the late time points, i.e., after the first and, with a trend toward higher titers, second NYVAC/protein boosts (Fig. 7A). Some of the sera also showed a trend toward neutralizing the highly sensitive tier 1 clade B MN and SF162P4 Envs at week 26 (Fig. 7B). We also assessed the capacity of the sera to neutralize six different tier 2 clade C Envs in the context of infectious molecular clones by using the sensitive A3R5 neutralization assay. However, no neutralizing activity was detected (Fig. 8). In total, the serum neutralizing antibody response was very poor, and there were no differences between the various immunization regimens.
FIG 7.

Sera from immunized monkeys show poor neutralizing activity against a panel of Env isolates in a TZM-bl cell assay. Pseudoviruses carrying the indicated Env isolates were incubated with sera from the immunized monkeys before addition of TZM-bl cells. The serum dilution leading to a 50% decrease of luciferase activity (IC50) is shown for values above the assay's threshold of 20. (A) Time course of IC50 values over the study duration for the highly neutralization-sensitive tier 1A clade C isolate MW965.26 (top left) and for the other tested Env proteins, as indicated. (B) Week 26 IC50 values for all tested envelopes. MuLV was used as a negative control.
FIG 8.

Sera from immunized monkeys show poor neutralizing activity in an A3R5 cell assay. The graphs show time courses of serum neutralization activity in an A3R5 cell assay against infectious molecular clones carrying envelope proteins from the indicated isolates.
ADCC and ADCVI responses.
Finally, we analyzed plasma samples from the vaccinated macaques for the capacity to mediate nonneutralizing antibody effector functions dependent on Fc receptor engagement. For this purpose, we performed an ADCC assay that measures the antibody-mediated killing of antigen-coated cells by NK cells. CEM.NKRCCR5 target cells were coated with the gp120 protein of the 1086 or TV1 isolate, which was homologous to the proteins used for the booster immunizations. ADCC activity was not observed until boosting with NYVAC/protein (Fig. 9A). Two weeks after the first boost, only a trend toward some ADCC activity was apparent. However, the titers at week 26, after the second NYVAC/protein boost, were statistically significantly above the week 0 background titers for group C with 1086 gp120 and all groups with TV1 gp120. Interestingly, group C had significantly higher titers than those of group A at week 26, reaching median values of at least 105 for TV1, as opposed to 9 × 103 for group A. No significant differences in the magnitudes of group B and C ADCC responses were observed. Moreover, we performed an ADCVI assay on the week 26 plasma samples. In this assay, noncytolytic Fc receptor-mediated functions exerted by the effector cells are encompassed along with the cytolytic ADCC activity to gain a more comprehensive picture of the nonneutralizing antibody effector functions. The ADCVI antibody activities measured against two tier 2 viruses (DU156 and DU422) were very similar for all groups, exhibiting 34% inhibition, on average, for both viruses tested (Fig. 9B).
FIG 9.

Plasmas from immunized monkeys show moderate ADCC and ADCVI activities. (A) ADCC activity. CEM.NKRCCR5 target cells coated with gp120 of isolate 1086 (left) or TV1 (right) were mixed with PBMCs at an E:T ratio of 30:1. A granzyme B substrate and dilution series of the plasma samples were added, and the cells were incubated and assessed by flow cytometry for viable cells showing granzyme B activity. The antibody titer was calculated by interpolating the reciprocal plasma dilution causing a granzyme B activity matching the cutoff value of the assay. Horizontal arrows mark titers exceeding the upper limit of the assay (102,400). Diamonds indicate titers significantly above the week 0 values, and asterisks indicate significant between-group differences (P < 0.05, with appropriate Bonferroni correction). Time points 2 weeks after the last applications of DNA and NYVAC/protein are highlighted by shading. (B) ADCVI activity. CEM.NKRCCR5 target cells were infected with HIV-1 DU156 or DU422 for 3 days. Plasma samples obtained at week 26 and PBMCs at an E:T ratio of 10:1 were added. After 8 days, virus amounts were measured by p24 ELISA, and the % inhibition was calculated as the proportion of p24 obtained in the presence of plasma samples versus week 0 control plasma.
DISCUSSION
Within the EuroVacc program, our group has developed candidate HIV vaccines which have been tested in various DNA prime and poxvirus (NYVAC) boost regimens in macaques and humans. The immunizations were well tolerated and elicited good T cell responses, albeit mainly directed against Env, whereas antibody responses were rather low (11, 12, 19, 22). In the present study, we evaluated the immune responses elicited by a second generation of improved antigens in rhesus macaques. Different DNA-priming regimens in combination with NYVAC/protein boosts were compared in order to determine the optimal number of immunizations and to assess if the intervals between immunizations could be shortened. The magnitude and quality of T cell responses, as well as antibody titers, neutralization activity, and Fc-mediated nonneutralizing effector functions, were analyzed.
The concomitant general monkey health monitoring demonstrated that the vaccine formulations were safe and well tolerated. There was no apparent toxicity, and local reactions at the inoculation sites were generally mild in nature and resolved quickly. More intense preclinical safety analyses are ongoing or planned and will be conducted before initiation of clinical studies.
With respect to the magnitudes of HIV-specific T cell responses after application of the DNA prime immunizations, there were no statistically significant differences between the groups. The accelerated regimen, with intervals shortened to 2 weeks (group B), and the regimen sparing one dose (group C) led to comparable numbers of IFN-γ-producing cells, as did the reference regimen matching the priming scheme of the EV03 clinical trial (group A). Interestingly, in group B, T cell responses at week 4—2 weeks after the second DNA prime—resembled those at week 2, while a statistically significant increase was not evident until week 6—2 weeks after the third DNA prime. However, the values at the latter time point resemble those for groups A and B after only two immunizations. Thus, an accelerated regimen does not result in an accelerated increase in the magnitude of T cell immune responses. It is possible that saturating amounts of the antigens are expressed from the plasmid vectors over the 4-week interval and thus that an additional injection has no benefit during this time frame. In mice, antigen expression peaks 1 to 2 weeks after administration of DNA and then declines over several weeks (49), though this may vary depending on the choice of plasmid vector and on its promoter/enhancer elements (50). Moreover, in group A, there were no differences in responses after the second and third immunizations. This further confirms that the chosen combination of expression vector and antigen provides saturating amounts of antigen necessary for priming T cell responses. In fact, the absolute magnitude of T cell responses was rather high, with nearly 0.1% of all PBMCs being HIV-specific T cells at this early stage in the course of vaccination. Compared with the EV02 scheme (12), which elicited responses of similar magnitudes in both macaques and humans (22), the peak responses after DNA priming were >6-fold higher in the present study, even surpassing the levels obtained with the best first-generation DNA-NYVAC combination regimens at any time point.
Boosting with NYVAC/protein augmented T cell responses 5-fold, resulting in a nearly 10-fold improvement compared with that of the EV02 scheme in macaques (22). Shortening the time to boosting from 12 weeks to 8 weeks (groups B and C versus group A) was not detrimental, thus allowing for a shorter vaccination course. Interestingly, while the second boost clearly led to increased numbers of HIV-specific T cells within 2 weeks, the level was comparable to that observed 2 weeks after the first NYVAC/protein boost, without statistically significant differences within and between groups. Therefore, the T cell responses after the last vaccination might be only anamnestic in nature. However, it would be important to determine if T cell responses at the two time points differed qualitatively, which was not assessed in this study. If there are no differences, further narrowing of the vaccination course by omitting the second boost should be considered when focusing on T cell responses. Regarding the durability of the responses, 16 weeks after the final boost, i.e., at week 40, T cell magnitudes had declined again to levels similar to those beforehand. Another boost would likely again cause an anamnestic response of similar strength in a similar time frame. Employing poxvirus vectors with replication capacity in humans, such as the NYVAC-KC variant (51), or with optimized immunostimulatory capacity, e.g., by removal of Toll-like receptor (TLR)-inhibitory factors (52) or inhibitors of the interferon system (53), might further augment the immune responses.
A deeper analysis of the T cell responses at week 26 revealed a generally high quality. The responses were well balanced between CD4 and CD8 cells, and the majority of cells were polyfunctional, secreting all three cytokines assessed, and thus exhibited a phenotype associated with protective antiviral responses (54). Importantly, breaking down the responses to the individual target proteins also showed balance, with improved GagPol-specific responses compared to those in the previous studies, where anti-Env responses were primarily observed. Most likely, this is a consequence of the improved antigen configuration, i.e., splitting of Gag, PolNef, and gp140 in the context of the DNA vaccine or using GagFSPolNef in the context of NYVAC. This outcome was predicted by previous mouse immunogenicity studies of our redesigned antigens and is likely related to the increased expression levels, especially of Gag (27, 32). Similar observations were made in cynomolgus monkeys and humans after splitting of a differently designed Gag-Pol-Nef fusion protein into three parts which were subsequently delivered as a mixture of three plasmids rather than one (16, 28, 29).
Overall, for the T cell responses, the additional benefit of a third DNA prime as observed in the EV03 study seems to be compensated for by employing improved antigens, which per se elicit higher-magnitude and better-quality T cell responses. Moreover, a second NYVAC/protein boost seems to be dispensable.
The analyses of the humoral responses revealed that the highest antibody binding titers were against a clade C consensus gp140 protein, consistent with the use of clade C Env antigens in our study (96ZM651 gp140 in the case of DNA and NYVAC immunizations and TV1 and 1086 gp120s in the case of protein immunizations). Accordingly, the homologous TV1 and 1086 gp120 proteins were also bound at very high titers after the boosts, while the clade A and B sequences were inferior targets. Evaluation of IgG titers 2 weeks after the completion of DNA priming showed that group A (3xD_4w) exhibited significantly higher titers than those of groups B and C. This was likely a consequence of the longer time from the first immunization until measurement (10 weeks as opposed to 6 weeks) and might have been similar if all groups had been analyzed at week 10. Accordingly, at the late time points after the boosts when samples were collected in parallel, no differences were apparent between the groups. As for the T cell responses, antibody titers also waned until week 40. It would be interesting to see with what kinetics and to what level titers would rise after an additional boost; however, this was not part of the study design. Moreover, it would be interesting to assess how different adjuvants affect not only the magnitude of peak responses but also the subsequent kinetics of decline. Additionally, we found that IgG antibodies to V1V2 were induced. These responses are of special interest because V1V2-directed IgG correlated with a decreased risk of HIV-1 infection in the RV144 trial (55), especially when mediated by the IgG3 antibody subtype (40, 56). However, due to differences in rhesus IgG subclasses compared to those of humans, the equivalent to human IgG3 cannot, to date, be examined in the rhesus macaque model. Serum IgA levels, associated with mitigation of protective responses in RV144 (55), were very low in our study.
We also assessed the virus neutralization activity in macaque sera. With the exception of activity toward the neutralization-sensitive tier 1A MW965.26 Env, no neutralization activity was observed in either the TZM-bl cell assay or the more sensitive A3R5 cell assay, which employed tier 2 viruses. Thus, our vaccination regimens elicited conventional antibody responses rather than (broadly) neutralizing ones (57). This is not surprising considering the configuration of Env antigens employed here: while gp140 was used in the DNA and NYVAC vectors, the protein boosts used gp120 variants because they were the only good manufacturing practice (GMP) material available at the time of the study. Although gp120 is highly immunogenic, several studies have shown that gp140 elicits qualitatively better responses, especially with regard to the neutralizing capacity of the antibodies induced (58, 59). Currently, major efforts are being put into the rational design of next-generation envelope antigens (60), such as trimer-stabilized SOSIP variants (61, 62). Furthermore, strategies are being pursued to direct elicitation of broadly neutralizing antibodies by using sets of guide immunogens from activation of germ line B cell receptor precursors toward evolution of affinity-matured antibodies (57, 63). Thus, future HIV vaccine trials will hopefully benefit from such approaches toward development of better envelope antigens.
Finally, we assessed the capacity of macaque sera to mediate ADCC and ADCVI activities, both of which rely on Fc-FcγR interactions (64). These antibody functions have also been implicated in preventing and modulating lentiviral infections (23). ADCC titers significantly above the background level were observed, in a range previously described as being associated with virus inhibition (65), although we tested this only against the homologous gp120 antigens. The inhibition due to ADCVI was slightly lower than that in other studies (65, 66), but still in a range where it may contribute to a reduction of the plasma viral load.
Overall, for the humoral responses, the two NYVAC/protein boosts are necessary to obtain high-titer and functional responses. We believe that this is driven mainly by the very potent adjuvanted gp120 protein component. Overall, there were only small differences between the groups. Although group A seemed to be slightly superior before the boost, the group C ADCC response was significantly larger than that of group A (but there were no significant differences for group B and either group A or C) after the boost. Moreover, in several analyses, there were trends toward better responses for group C (2xD_4w). Thus, humoral responses do not profit from a third DNA prime immunization.
Looking at durability overall, though very high peak responses were observed after the final booster immunization, responses waned over the following 16 weeks. A decline in the absence of further stimulation is of course expected, yet it would be interesting to know whether the memory responses recalled in the case of an infection would be rapid and efficacious enough to prevent infection, or at least to mitigate the set-point viral load. Given the limited knowledge on correlates of protection, it is hard to rate the quality of the memory responses observed here. It was reported for the RV144 trial that efficacy peaked after 1 year, at 60.5% (67), and then declined, reaching 31.2% after 3.5 years (5). Analysis of the neutralizing antibody responses of trial participants also revealed a strong decline of these responses within 6 months (68). Therefore, it would be generally interesting to gather more data on the kinetics of immune responses to our vaccine candidates, especially for the rate of decline and the final long-term levels.
The efficacy of our vaccine candidates was not assessed because only limited conclusions would have been possible for the HIV-derived antigens by performing a challenge with an SHIV strain. Indeed, a repeated intrarectal low-dose SHIV challenge would have captured only the effects of Env-specific humoral and cellular responses and would have neglected a potential contribution of HIV-specific Gag-, Pol-, and Nef-specific T cell responses. Therefore, an analogous study using matched simian immunodeficiency virus (SIV) antigens might—within the borders of the applied challenge model—prove useful for assessing the protective efficacy of the vaccine concept. Thus, challenge with an infectious SIV strain might reveal whether the CTL or humoral responses, alone or in combination, do contribute to protection against infection, or at least against disease. In conclusion, our findings suggest the following. (i) A second NYVAC/protein boost might be dispensable for T cell responses, although further analyses of the T cell quality will be required for confirmation. (ii) In addition, both an accelerated priming regimen and a dose-sparing regimen perform as well as a conventional one for both T cell and humoral responses, suggesting that a third DNA prime may not be necessary with the improved antigens. (iii) Finally, the first NYVAC/protein boost can safely be applied 1 month earlier, without compromising the booster effect.
Therefore, we speculate that a simplified regimen consisting of two DNA/protein primes plus a single NYVAC boost in an overall scheme of only 12 weeks might elicit comparable or even superior immune responses. Because the humoral responses most likely profit from immunization with adjuvanted protein, we propose to apply the protein component alongside the DNA primes. It has been described that DNA-protein codelivery elicits humoral responses superior to those obtained with either component alone or with a DNA prime-protein boost scheme (69, 70). However, repeated administration of protein might skew the subclass ratio of HIV-specific IgGs in an undesired manner (40), so the number of protein immunizations should be chosen carefully. In addition, the optimal length of the interval between protein applications should be determined, as this may have a major impact on the quality of the memory responses. Moreover, antibody responses might benefit from employment of rationally designed next-generation envelope antigens, but these have yet to show their benefits and efficacy.
In summary, this work provides insights into how to streamline vaccination regimens which might be applicable to humans. Fewer immunizations and shorter intervals between immunizations will surely be of ultimate benefit for the purposes of raising protective immunity more quickly, improving compliance, and reducing costs, which are especially important factors in resource-constrained settings. However, the impacts of these vaccine candidates and proposed modified regimens on vaccine efficacy remain to be evaluated in humans.
ACKNOWLEDGMENTS
We thank the Vaccine Immune Monitoring Centers for the immune monitoring assays and the Vaccine Immunology Statistical Center for the statistical analysis. We thank Kelli Greene and Hongmei Gao for CA-VIMC program management, Marcella Sarzotti-Kelsoe for quality assurance oversight, and Hua-Xin Liao and Bart Haynes, Duke University, for providing envelope proteins for antibody assays.
This investigation was funded by the Bill & Melinda Gates Foundation Poxvirus T-Cell Vaccine Discovery Consortium (PTVDC) (grant 38599). The Vaccine Immune Monitoring Centers (grants OPP1032144 and OPP1032325) and the Vaccine Immunology Statistical Center (grant OPP1032317), as part of the Collaboration for AIDS Vaccine Discovery (CAVD), were funded by the Bill & Melinda Gates Foundation. Novartis Vaccines received support for this work under contract number HHSN266200500007C from DAIDS, NIAID, NIH.
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Schiffner T, Sattentau QJ, Dorrell L. 2013. Development of prophylactic vaccines against HIV-1. Retrovirology 10:72. doi: 10.1186/1742-4690-10-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, Coyne-Johnson L, Whizin N, Oswald K, Shoemaker R, Swanson T, Legasse AW, Chiuchiolo MJ, Parks CL, Axthelm MK, Nelson JA, Jarvis MA, Piatak M, Lifson JD, Picker LJ. 2011. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 473:523–527. doi: 10.1038/nature10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hansen SG, Sacha JB, Hughes CM, Ford JC, Burwitz BJ, Scholz I, Gilbride RM, Lewis MS, Gilliam AN, Ventura AB, Malouli D, Xu G, Richards R, Whizin N, Reed JS, Hammond KB, Fischer M, Turner JM, Legasse AW, Axthelm MK, Edlefsen PT, Nelson JA, Lifson JD, Früh K, Picker LJ. 2013. Cytomegalovirus vectors violate CD8+ T cell epitope recognition paradigms. Science 340:1237874. doi: 10.1126/science.1237874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Picker LJ, Hansen SG, Lifson JD. 2012. New paradigms for HIV/AIDS vaccine development. Annu Rev Med 63:95–111. doi: 10.1146/annurev-med-042010-085643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, Premsri N, Namwat C, de Souza M, Adams E, Benenson M, Gurunathan S, Tartaglia J, McNeil JG, Francis DP, Stablein D, Birx DL, Chunsuttiwat S, Khamboonruang C, Thongcharoen P, Robb ML, Michael NL, Kunasol P, Kim JH. 2009. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med 361:2209–2220. doi: 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- 6.Flynn NM, Forthal DN, Harro CD, Judson FN, Mayer KH, Para MF. 2005. Placebo-controlled phase 3 trial of a recombinant glycoprotein 120 vaccine to prevent HIV-1 infection. J Infect Dis 191:654–665. doi: 10.1086/428404. [DOI] [PubMed] [Google Scholar]
- 7.Pitisuttithum P, Gilbert P, Gurwith M, Heyward W, Martin M, van Griensven F, Hu D, Tappero JW, Choopanya K. 2006. Randomized, double-blind, placebo-controlled efficacy trial of a bivalent recombinant glycoprotein 120 HIV-1 vaccine among injection drug users in Bangkok, Thailand. J Infect Dis 194:1661–1671. doi: 10.1086/508748. [DOI] [PubMed] [Google Scholar]
- 8.Mascola JR, Sambor A, Beaudry K, Santra S, Welcher B, Louder MK, Vancott TC, Huang Y, Chakrabarti BK, Kong W-P, Yang Z-Y, Xu L, Montefiori DC, Nabel GJ, Letvin NL. 2005. Neutralizing antibodies elicited by immunization of monkeys with DNA plasmids and recombinant adenoviral vectors expressing human immunodeficiency virus type 1 proteins. J Virol 79:771–779. doi: 10.1128/JVI.79.2.771-779.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Casimiro DR, Chen L, Fu T-M, Evans RK, Caulfield MJ, Davies M-E, Tang A, Chen M, Huang L, Harris V, Freed DC, Wilson KA, Dubey S, Zhu D-M, Nawrocki D, Mach H, Troutman R, Isopi L, Williams D, Hurni W, Xu Z, Smith JG, Wang S, Liu X, Guan L, Long R, Trigona W, Heidecker GJ, Perry HC, Persaud N, Toner TJ, Su Q, Liang X, Youil R, Chastain M, Bett AJ, Volkin DB, Emini EA, Shiver JW. 2003. Comparative immunogenicity in rhesus monkeys of DNA plasmid, recombinant vaccinia virus, and replication-defective adenovirus vectors expressing a human immunodeficiency virus type 1 gag gene. J Virol 77:6305–6313. doi: 10.1128/JVI.77.11.6305-6313.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Girard MP, Osmanov S, Assossou OM, Kieny M-P. 2011. Human immunodeficiency virus (HIV) immunopathogenesis and vaccine development: a review. Vaccine 29:6191–6218. doi: 10.1016/j.vaccine.2011.06.085. [DOI] [PubMed] [Google Scholar]
- 11.Bart P-A, Goodall R, Barber T, Harari A, Guimaraes-Walker A, Khonkarly M, Sheppard NC, Bangala Y, Frachette M-J, Wagner R, Liljeström P, Kraehenbuhl J-P, Girard M, Goudsmit J, Esteban M, Heeney J, Sattentau Q, McCormack S, Babiker A, Pantaleo G, Weber J. 2008. EV01: a phase I trial in healthy HIV negative volunteers to evaluate a clade C HIV vaccine, NYVAC-C undertaken by the EuroVacc Consortium. Vaccine 26:3153–3161. doi: 10.1016/j.vaccine.2008.03.083. [DOI] [PubMed] [Google Scholar]
- 12.McCormack S, Stöhr W, Barber T, Bart P-A, Harari A, Moog C, Ciuffreda D, Cellerai C, Cowen M, Gamboni R, Burnet S, Legg K, Brodnicki E, Wolf H, Wagner R, Heeney J, Frachette M-J, Tartaglia J, Babiker A, Pantaleo G, Weber J. 2008. EV02: a phase I trial to compare the safety and immunogenicity of HIV DNA-C prime-NYVAC-C boost to NYVAC-C alone. Vaccine 26:3162–3174. doi: 10.1016/j.vaccine.2008.02.072. [DOI] [PubMed] [Google Scholar]
- 13.Gómez CE, Nájera JL, Jiménez V, Bieler K, Wild J, Kostic L, Heidari S, Chen M, Frachette M-J, Pantaleo G, Wolf H, Liljeström P, Wagner R, Esteban M. 2007. Generation and immunogenicity of novel HIV/AIDS vaccine candidates targeting HIV-1 Env/Gag-Pol-Nef antigens of clade C. Vaccine 25:1969–1992. doi: 10.1016/j.vaccine.2006.11.051. [DOI] [PubMed] [Google Scholar]
- 14.Mooij P, Balla-Jhagjhoorsingh SS, Koopman G, Beenhakker N, van Haaften P, Baak I, Nieuwenhuis IG, Kondova I, Wagner R, Wolf H, Gómez CE, Nájera JL, Jiménez V, Esteban M, Heeney JL. 2008. Differential CD4+ versus CD8+ T-cell responses elicited by different poxvirus-based human immunodeficiency virus type 1 vaccine candidates provide comparable efficacies in primates. J Virol 82:2975–2988. doi: 10.1128/JVI.02216-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harari A, Bart P-A, Stöhr W, Tapia G, Garcia M, Medjitna-Rais E, Burnet S, Cellerai C, Erlwein O, Barber T, Moog C, Liljestrom P, Wagner R, Wolf H, Kraehenbuhl J-P, Esteban M, Heeney J, Frachette M-J, Tartaglia J, McCormack S, Babiker A, Weber J, Pantaleo G. 2008. An HIV-1 clade C DNA prime, NYVAC boost vaccine regimen induces reliable, polyfunctional, and long-lasting T cell responses. J Exp Med 205:63–77. doi: 10.1084/jem.20071331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koup RA, Roederer M, Lamoreaux L, Fischer J, Novik L, Nason MC, Larkin BD, Enama ME, Ledgerwood JE, Bailer RT, Mascola JR, Nabel GJ, Graham BS, VRC 009 Study Team, VRC 010 Study Team 2010. Priming immunization with DNA augments immunogenicity of recombinant adenoviral vectors for both HIV-1 specific antibody and T-cell responses. PLoS One 5:e9015. doi: 10.1371/journal.pone.0009015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Connell KA, Bailey JR, Blankson JN. 2009. Elucidating the elite: mechanisms of control in HIV-1 infection. Trends Pharmacol Sci 30:631–637. doi: 10.1016/j.tips.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 18.Migueles SA, Sabbaghian MS, Shupert WL, Bettinotti MP, Marincola FM, Martino L, Hallahan CW, Selig SM, Schwartz D, Sullivan J, Connors M. 2000. HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc Natl Acad Sci U S A 97:2709–2714. doi: 10.1073/pnas.050567397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levy Y, Ellefsen K, Stöhr W, Bart P-A, Leliévre J-D, Launay O, Wolf H, Weber J, Chene G, Pantaleo G. 2010. Optimal priming of poxvirus vector (NYVAC)-based HIV vaccine regimens requires 3 DNA injections. Results of the EV03/ANRS Vac20 Phase I/II Trial. AIDS Res Hum Retroviruses 26:A10. [Google Scholar]
- 20.Bett AJ, Dubey SA, Mehrotra DV, Guan L, Long R, Anderson K, Collins K, Gaunt C, Fernandez R, Cole S, Meschino S, Tang A, Sun X, Gurunathan S, Tartaglia J, Robertson MN, Shiver JW, Casimiro DR. 2010. Comparison of T cell immune responses induced by vectored HIV vaccines in non-human primates and humans. Vaccine 28:7881–7889. doi: 10.1016/j.vaccine.2010.09.079. [DOI] [PubMed] [Google Scholar]
- 21.Hatziioannou T, Evans DT. 2012. Animal models for HIV/AIDS research. Nat Rev Microbiol 10:852–867. doi: 10.1038/nrmicro2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mooij P, Balla-Jhagjhoorsingh SS, Beenhakker N, van Haaften P, Baak I, Nieuwenhuis IG, Heidari S, Wolf H, Frachette M-J, Bieler K, Sheppard N, Harari A, Bart P-A, Liljeström P, Wagner R, Pantaleo G, Heeney JL. 2009. Comparison of human and rhesus macaque T-cell responses elicited by boosting with NYVAC encoding human immunodeficiency virus type 1 clade C immunogens. J Virol 83:5881–5889. doi: 10.1128/JVI.02345-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao P, Zhao J, Patterson LJ, Brocca-Cofano E, Venzon D, Kozlowski PA, Hidajat R, Demberg T, Robert-Guroff M. 2010. Multiple vaccine-elicited nonneutralizing antienvelope antibody activities contribute to protective efficacy by reducing both acute and chronic viremia following simian/human immunodeficiency virus SHIV89.6P challenge in rhesus macaques. J Virol 84:7161–7173. doi: 10.1128/JVI.00410-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marano N, Plikaytis BD, Martin SW, Rose C, Semenova VA, Martin SK, Freeman AE, Li H, Mulligan MJ, Parker SD, Babcock J, Keitel W, El Sahly H, Poland GA, Jacobson RM, Keyserling HL, Soroka SD, Fox SP, Stamper JL, McNeil MM, Perkins BA, Messonnier N, Quinn CP, Anthrax Vaccine Research Program Working Group. 2008. Effects of a reduced dose schedule and intramuscular administration of anthrax vaccine adsorbed on immunogenicity and safety at 7 months: a randomized trial. JAMA 300:1532–1543. doi: 10.1001/jama.300.13.1532. [DOI] [PubMed] [Google Scholar]
- 25.Wild J, Bieler K, Köstler J, Frachette M-J, Jeffs S, Vieira S, Esteban M, Liljeström P, Pantaleo G, Wolf H, Wagner R. 2009. Preclinical evaluation of the immunogenicity of C-type HIV-1-based DNA and NYVAC vaccines in the Balb/C mouse model. Viral Immunol 22:309–319. doi: 10.1089/vim.2009.0038. [DOI] [PubMed] [Google Scholar]
- 26.Yang X, Florin L, Farzan M, Kolchinsky P, Kwong PD, Sodroski J, Wyatt R. 2000. Modifications that stabilize human immunodeficiency virus envelope glycoprotein trimers in solution. J Virol 74:4746–4754. doi: 10.1128/JVI.74.10.4746-4754.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Böckl K, Wild J, Bredl S, Kindsmüller K, Köstler J, Wagner R. 2012. Altering an artificial Gagpolnef polyprotein and mode of ENV co-administration affects the immunogenicity of a clade C HIV DNA vaccine. PLoS One 7:e34723. doi: 10.1371/journal.pone.0034723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Catanzaro AT, Roederer M, Koup RA, Bailer RT, Enama ME, Nason MC, Martin JE, Rucker S, Andrews CA, Gomez PL, Mascola JR, Nabel GJ, Graham BS, VRC 007 Study Team 2007. Phase I clinical evaluation of a six-plasmid multiclade HIV-1 DNA candidate vaccine. Vaccine 25:4085–4092. doi: 10.1016/j.vaccine.2007.02.050. [DOI] [PubMed] [Google Scholar]
- 29.Barouch DH, Yang Z, Kong W, Korioth-Schmitz B, Sumida SM, Truitt DM, Kishko MG, Arthur JC, Miura A, Mascola JR, Letvin NL, Nabel GJ. 2005. A human T-cell leukemia virus type 1 regulatory element enhances the immunogenicity of human immunodeficiency virus type 1 DNA vaccines in mice and nonhuman primates. J Virol 79:8828–8834. doi: 10.1128/JVI.79.14.8828-8834.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cranenburgh RM, Hanak JA, Williams SG, Sherratt DJ. 2001. Escherichia coli strains that allow antibiotic-free plasmid selection and maintenance by repressor titration. Nucleic Acids Res 29:E26. doi: 10.1093/nar/29.5.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jacks T, Power MD, Masiarz FR, Luciw PA, Barr PJ, Varmus HE. 1988. Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature 331:280–283. doi: 10.1038/331280a0. [DOI] [PubMed] [Google Scholar]
- 32.Perdiguero B, Gómez CE, Cepeda V, Sánchez-Sampedro L, García-Arriaza J, Mejías-Pérez E, Jiménez V, Sánchez C, Sorzano CÓ Oliveros SJC, Delaloye J, Roger T, Calandra T, Asbach B, Wagner R, Kibler KV, Jacobs BL, Pantaleo G, Esteban M. 2015. Virological and immunological characterization of novel NYVAC-based HIV/AIDS vaccine candidates expressing clade C trimeric soluble gp140(ZM96) and Gag(ZM96)-Pol-Nef(CN54) as virus-like particles. J Virol 89:970–988. doi: 10.1128/JVI.02469-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 34.Raab D, Graf M, Notka F, Schödl T, Wagner R. 2010. The GeneOptimizer Algorithm: using a sliding window approach to cope with the vast sequence space in multiparameter DNA sequence optimization. Syst Synth Biol 4:215–225. doi: 10.1007/s11693-010-9062-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tartaglia J, Perkus ME, Taylor J, Norton EK, Audonnet JC, Cox WI, Davis SW, van der Hoeven J, Meignier B, Riviere M. 1992. NYVAC: a highly attenuated strain of vaccinia virus. Virology 188:217–232. doi: 10.1016/0042-6822(92)90752-B. [DOI] [PubMed] [Google Scholar]
- 36.Srivastava IK, Kan E, Sun Y, Sharma VA, Cisto J, Burke B, Lian Y, Hilt S, Biron Z, Hartog K, Stamatatos L, Diaz-Avalos R, Cheng RH, Ulmer JB, Barnett SW. 2008. Comparative evaluation of trimeric envelope glycoproteins derived from subtype C and B HIV-1 R5 isolates. Virology 372:273–290. doi: 10.1016/j.virol.2007.10.022. [DOI] [PubMed] [Google Scholar]
- 37.Donaldson MM, Kao S-F, Eslamizar L, Gee C, Koopman G, Lifton M, Schmitz JE, Sylwester AW, Wilson A, Hawkins N, Self SG, Roederer M, Foulds KE. 2012. Optimization and qualification of an 8-color intracellular cytokine staining assay for quantifying T cell responses in rhesus macaques for pre-clinical vaccine studies. J Immunol Methods 386:10–21. doi: 10.1016/j.jim.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tomaras GD, Yates NL, Liu P, Qin L, Fouda GG, Chavez LL, Decamp AC, Parks RJ, Ashley VC, Lucas JT, Cohen M, Eron J, Hicks CB, Liao H-X, Self SG, Landucci G, Forthal DN, Weinhold KJ, Keele BF, Hahn BH, Greenberg ML, Morris L, Karim SSA, Blattner WA, Montefiori DC, Shaw GM, Perelson AS, Haynes BF. 2008. Initial B-cell responses to transmitted human immunodeficiency virus type 1: virion-binding immunoglobulin M (IgM) and IgG antibodies followed by plasma anti-gp41 antibodies with ineffective control of initial viremia. J Virol 82:12449–12463. doi: 10.1128/JVI.01708-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zolla-Pazner S, deCamp A, Gilbert PB, Williams C, Yates NL, Williams WT, Howington R, Fong Y, Morris DE, Soderberg KA, Irene C, Reichman C, Pinter A, Parks R, Pitisuttithum P, Kaewkungwal J, Rerks-Ngarm S, Nitayaphan S, Andrews C, O'Connell RJ, Yang Z, Nabel GJ, Kim JH, Michael NL, Montefiori DC, Liao H-X, Haynes BF, Tomaras GD. 2014. Vaccine-induced IgG antibodies to V1V2 regions of multiple HIV-1 subtypes correlate with decreased risk of HIV-1 infection. PLoS One 9:e87572. doi: 10.1371/journal.pone.0087572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yates NL, Liao H-X, Fong Y, deCamp A, Vandergrift NA, Williams WT, Alam SM, Ferrari G, Yang Z, Seaton KE, Berman PW, Alpert MD, Evans DT, O'Connell RJ, Francis D, Sinangil F, Lee C, Nitayaphan S, Rerks-Ngarm S, Kaewkungwal J, Pitisuttithum P, Tartaglia J, Pinter A, Zolla-Pazner S, Gilbert PB, Nabel GJ, Michael NL, Kim JH, Montefiori DC, Haynes BF, Tomaras GD. 2014. Vaccine-induced Env V1-V2 IgG3 correlates with lower HIV-1 infection risk and declines soon after vaccination. Sci Transl Med 6:228ra39. doi: 10.1126/scitranslmed.3007730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu X, Gilbert PB, Hioe CE, Zolla-Pazner S, Self SG. 2012. Statistical approaches to analyzing HIV-1 neutralizing antibody assay data. Stat Biopharm Res 4:1–13. doi: 10.1080/19466315.2011.633860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gaschen B, Taylor J, Yusim K, Foley B, Gao F, Lang D, Novitsky V, Haynes B, Hahn BH, Bhattacharya T, Korber B. 2002. Diversity considerations in HIV-1 vaccine selection. Science 296:2354–2360. doi: 10.1126/science.1070441. [DOI] [PubMed] [Google Scholar]
- 43.Liao H-X, Sutherland LL, Xia S-M, Brock ME, Scearce RM, Vanleeuwen S, Alam SM, McAdams M, Weaver EA, Camacho Z, Ma B-J, Li Y, Decker JM, Nabel GJ, Montefiori DC, Hahn BH, Korber BT, Gao F, Haynes BF. 2006. A group M consensus envelope glycoprotein induces antibodies that neutralize subsets of subtype B and C HIV-1 primary viruses. Virology 353:268–282. doi: 10.1016/j.virol.2006.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Todd CA, Greene KM, Yu X, Ozaki DA, Gao H, Huang Y, Wang M, Li G, Brown R, Wood B, D'Souza MP, Gilbert P, Montefiori DC, Sarzotti-Kelsoe M. 2012. Development and implementation of an international proficiency testing program for a neutralizing antibody assay for HIV-1 in TZM-bl cells. J Immunol Methods 375:57–67. doi: 10.1016/j.jim.2011.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McLinden RJ, Labranche CC, Chenine A-L, Polonis VR, Eller MA, Wieczorek L, Ochsenbauer C, Kappes JC, Perfetto S, Montefiori DC, Michael NL, Kim JH. 2013. Detection of HIV-1 neutralizing antibodies in a human CD4+/CXCR4+/CCR5+ T-lymphoblastoid cell assay system. PLoS One 8:e77756. doi: 10.1371/journal.pone.0077756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pollara J, Hart L, Brewer F, Pickeral J, Packard BZ, Hoxie JA, Komoriya A, Ochsenbauer C, Kappes JC, Roederer M, Huang Y, Weinhold KJ, Tomaras GD, Haynes BF, Montefiori DC, Ferrari G. 2011. High-throughput quantitative analysis of HIV-1 and SIV-specific ADCC-mediating antibody responses. Cytometry A 79:603–612. doi: 10.1002/cyto.a.21084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bialuk I, Whitney S, Andresen V, Florese RH, Nacsa J, Cecchinato V, Valeri VW, Heraud J-M, Gordon S, Parks RW, Montefiori DC, Venzon D, Demberg T, Guroff MR, Landucci G, Forthal DN, Franchini G. 2011. Vaccine induced antibodies to the first variable loop of human immunodeficiency virus type 1 gp120, mediate antibody-dependent virus inhibition in macaques. Vaccine 30:78–94. doi: 10.1016/j.vaccine.2011.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seaman MS, Janes H, Hawkins N, Grandpre LE, Devoy C, Giri A, Coffey RT, Harris L, Wood B, Daniels MG, Bhattacharya T, Lapedes A, Polonis VR, McCutchan FE, Gilbert PB, Self SG, Korber BT, Montefiori DC, Mascola JR. 2010. Tiered categorization of a diverse panel of HIV-1 Env pseudoviruses for assessment of neutralizing antibodies. J Virol 84:1439–1452. doi: 10.1128/JVI.02108-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hovav A-H, Panas MW, Rahman S, Sircar P, Gillard G, Cayabyab MJ, Letvin NL. 2007. Duration of antigen expression in vivo following DNA immunization modifies the magnitude, contraction, and secondary responses of CD8+ T lymphocytes. J Immunol 179:6725–6733. doi: 10.4049/jimmunol.179.10.6725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang S, Farfan-Arribas DJ, Shen S, Chou T-HW, Hirsch A, He F, Lu S. 2006. Relative contributions of codon usage, promoter efficiency and leader sequence to the antigen expression and immunogenicity of HIV-1 Env DNA vaccine. Vaccine 24:4531–4540. doi: 10.1016/j.vaccine.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 51.Kibler KV, Gomez CE, Perdiguero B, Wong S, Huynh T, Holechek S, Arndt W, Jimenez V, Gonzalez-Sanz R, Denzler K, Haddad EK, Wagner R, Sékaly RP, Tartaglia J, Pantaleo G, Jacobs BL, Esteban M. 2011. Improved NYVAC-based vaccine vectors. PLoS One 6:e25674. doi: 10.1371/journal.pone.0025674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perdiguero B, Gómez CE, Di Pilato M, Sorzano COS, Delaloye J, Roger T, Calandra T, Pantaleo G, Esteban M. 2013. Deletion of the vaccinia virus gene A46R, encoding for an inhibitor of TLR signalling, is an effective approach to enhance the immunogenicity in mice of the HIV/AIDS vaccine candidate NYVAC-C. PLoS One 8:e74831. doi: 10.1371/journal.pone.0074831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gómez CE, Perdiguero B, Nájera JL, Sorzano COS, Jiménez V, González-Sanz R, Esteban M. 2012. Removal of vaccinia virus genes that block interferon type I and II pathways improves adaptive and memory responses of the HIV/AIDS vaccine candidate NYVAC-C in mice. J Virol 86:5026–5038. doi: 10.1128/JVI.06684-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harari A, Dutoit V, Cellerai C, Bart P-A, Du Pasquier RA, Pantaleo G. 2006. Functional signatures of protective antiviral T-cell immunity in human virus infections. Immunol Rev 211:236–254. doi: 10.1111/j.0105-2896.2006.00395.x. [DOI] [PubMed] [Google Scholar]
- 55.Haynes BF, Gilbert PB, McElrath MJ, Zolla-Pazner S, Tomaras GD, Alam SM, Evans DT, Montefiori DC, Karnasuta C, Sutthent R, Liao H- X, DeVico AL, Lewis GK, Williams C, Pinter A, Fong Y, Janes H, DeCamp A, Huang Y, Rao M, Billings E, Karasavvas N, Robb ML, Ngauy V, de Souza MS, Paris R, Ferrari G, Bailer RT, Soderberg KA, Andrews C, Berman PW, Frahm N, De Rosa SC, Alpert MD, Yates NL, Shen X, Koup RA, Pitisuttithum P, Kaewkungwal J, Nitayaphan S, Rerks-Ngarm S, Michael NL, Kim JH. 2012. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N Engl J Med 366:1275–1286. doi: 10.1056/NEJMoa1113425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chung AW, Ghebremichael M, Robinson H, Brown E, Choi I, Lane S, Dugast A-S, Schoen MK, Rolland M, Suscovich TJ, Mahan AE, Liao L, Streeck H, Andrews C, Rerks-Ngarm S, Nitayaphan S, de Souza MS, Kaewkungwal J, Pitisuttithum P, Francis D, Michael NL, Kim JH, Bailey-Kellogg C, Ackerman ME, Alter G. 2014. Polyfunctional Fc-effector profiles mediated by IgG subclass selection distinguish RV144 and VAX003 vaccines. Sci Transl Med 6:228ra38. doi: 10.1126/scitranslmed.3007736. [DOI] [PubMed] [Google Scholar]
- 57.Zolla-Pazner S. 2014. A critical question for HIV vaccine development: which antibodies to induce? Science 345:167–168. doi: 10.1126/science.1256526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim M, Qiao Z-S, Montefiori DC, Haynes BF, Reinherz EL, Liao H-X. 2005. Comparison of HIV type 1 ADA gp120 monomers versus gp140 trimers as immunogens for the induction of neutralizing antibodies. AIDS Res Hum Retroviruses 21:58–67. doi: 10.1089/aid.2005.21.58. [DOI] [PubMed] [Google Scholar]
- 59.Kovacs JM, Nkolola JP, Peng H, Cheung A, Perry J, Miller CA, Seaman MS, Barouch DH, Chen B. 2012. HIV-1 envelope trimer elicits more potent neutralizing antibody responses than monomeric gp120. Proc Natl Acad Sci U S A 109:12111–12116. doi: 10.1073/pnas.1204533109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kwong PD, Mascola JR, Nabel GJ. 2011. Rational design of vaccines to elicit broadly neutralizing antibodies to HIV-1. Cold Spring Harb Perspect Med 1:a007278. doi: 10.1101/cshperspect.a007278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Iyer SPN, Franti M, Krauchuk AA, Fisch DN, Ouattara AA, Roux KH, Krawiec L, Dey AK, Beddows S, Maddon PJ, Moore JP, Olson WC. 2007. Purified, proteolytically mature HIV type 1 SOSIP gp140 envelope trimers. AIDS Res Hum Retroviruses 23:817–828. doi: 10.1089/aid.2006.0261. [DOI] [PubMed] [Google Scholar]
- 62.Sanders RW, Derking R, Cupo A, Julien J-P, Yasmeen A, de Val N, Kim HJ, Blattner C, de la Peña AT, Korzun J, Golabek M, de Los Reyes K, Ketas TJ, van Gils MJ, King CR, Wilson IA, Ward AB, Klasse PJ, Moore JP. 2013. A next-generation cleaved, soluble HIV-1 Env trimer, BG505 SOSIP.664 gp140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies. PLoS Pathog 9:e1003618. doi: 10.1371/journal.ppat.1003618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jardine J, Julien J-P, Menis S, Ota T, Kalyuzhniy O, McGuire A, Sok D, Huang P-S, MacPherson S, Jones M, Nieusma T, Mathison J, Baker D, Ward AB, Burton DR, Stamatatos L, Nemazee D, Wilson IA, Schief WR. 2013. Rational HIV immunogen design to target specific germline B cell receptors. Science 340:711–716. doi: 10.1126/science.1234150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vargas-Inchaustegui DA, Robert-Guroff M. 2013. Fc receptor-mediated immune responses: new tools but increased complexity in HIV prevention. Curr HIV Res 11:407–420. doi: 10.2174/1570162X113116660063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hidajat R, Xiao P, Zhou Q, Venzon D, Summers LE, Kalyanaraman VS, Montefiori DC, Robert-Guroff M. 2009. Correlation of vaccine-elicited systemic and mucosal nonneutralizing antibody activities with reduced acute viremia following intrarectal simian immunodeficiency virus SIVmac251 challenge of rhesus macaques. J Virol 83:791–801. doi: 10.1128/JVI.01672-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Florese RH, Demberg T, Xiao P, Kuller L, Larsen K, Summers LE, Venzon D, Cafaro A, Ensoli B, Robert-Guroff M. 2009. Contribution of nonneutralizing vaccine-elicited antibody activities to improved protective efficacy in rhesus macaques immunized with Tat/Env compared with multigenic vaccines. J Immunol 182:3718–3727. doi: 10.4049/jimmunol.0803115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Robb ML, Rerks-Ngarm S, Nitayaphan S, Pitisuttithum P, Kaewkungwal J, Kunasol P, Khamboonruang C, Thongcharoen P, Morgan P, Benenson M, Paris RM, Chiu J, Adams E, Francis D, Gurunathan S, Tartaglia J, Gilbert P, Stablein D, Michael NL, Kim JH. 2012. Risk behaviour and time as covariates for efficacy of the HIV vaccine regimen ALVAC-HIV (vCP1521) and AIDSVAX B/E: a post-hoc analysis of the Thai phase 3 efficacy trial RV144. Lancet Infect Dis 12:531–537. doi: 10.1016/S1473-3099(12)70088-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Montefiori DC, Karnasuta C, Huang Y, Ahmed H, Gilbert P, de Souza MS, McLinden R, Tovanabutra S, Laurence-Chenine A, Sanders-Buell E, Moody MA, Bonsignori M, Ochsenbauer C, Kappes J, Tang H, Greene K, Gao H, LaBranche CC, Andrews C, Polonis VR, Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Self SG, Berman PW, Francis D, Sinangil F, Lee C, Tartaglia J, Robb ML, Haynes BF, Michael NL, Kim JH. 2012. Magnitude and breadth of the neutralizing antibody response in the RV144 and Vax003 HIV-1 vaccine efficacy trials. J Infect Dis 206:431–441. doi: 10.1093/infdis/jis367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jalah R, Kulkarni V, Patel V, Rosati M, Alicea C, Bear J, Yu L, Guan Y, Shen X, Tomaras GD, LaBranche C, Montefiori DC, Prattipati R, Pinter A, Bess J, Lifson JD, Reed SG, Sardesai NY, Venzon DJ, Valentin A, Pavlakis GN, Felber BK. 2014. DNA and protein co-immunization improves the magnitude and longevity of humoral immune responses in macaques. PLoS One 9:e91550. doi: 10.1371/journal.pone.0091550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li J, Valentin A, Kulkarni V, Rosati M, Beach RK, Alicea C, Hannaman D, Reed SG, Felber BK, Pavlakis GN. 2013. HIV/SIV DNA vaccine combined with protein in a co-immunization protocol elicits highest humoral responses to envelope in mice and macaques. Vaccine 31:3747–3755. doi: 10.1016/j.vaccine.2013.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]