

ABSTRACT
Monocyte infiltration into the CNS is a hallmark of several viral infections of the central nervous system (CNS), including retrovirus infection. Understanding the factors that mediate monocyte migration in the CNS is essential for the development of therapeutics that can alter the disease process. In the current study, we found that neuropeptide Y (NPY) suppressed monocyte recruitment to the CNS in a mouse model of polytropic retrovirus infection. NPY−/− mice had increased incidence and kinetics of retrovirus-induced neurological disease, which correlated with a significant increase in monocytes in the CNS compared to wild-type mice. Both Ly6Chi inflammatory and Ly6Clo alternatively activated monocytes were increased in the CNS of NPY−/− mice following virus infection, suggesting that NPY suppresses the infiltration of both cell types. Ex vivo analysis of myeloid cells from brain tissue demonstrated that infiltrating monocytes expressed high levels of the NPY receptor Y2R. Correlating with the expression of Y2R on monocytes, treatment of NPY−/− mice with a truncated, Y2R-specific NPY peptide suppressed the incidence of retrovirus-induced neurological disease. These data demonstrate a clear role for NPY as a negative regulator of monocyte recruitment into the CNS and provide a new mechanism for suppression of retrovirus-induced neurological disease.
IMPORTANCE Monocyte recruitment to the brain is associated with multiple neurological diseases. However, the factors that influence the recruitment of these cells to the brain are still not well understood. In the current study, we found that neuropeptide Y, a protein produced by neurons, affected monocyte recruitment to the brain during retrovirus infection. We show that mice deficient in NPY have increased influx of monocytes into the brain and that this increase in monocytes correlates with neurological-disease development. These studies provide a mechanism by which the nervous system, through the production of NPY, can suppress monocyte trafficking to the brain and reduce retrovirus-induced neurological disease.
INTRODUCTION
The recruitment of monocytes into the central nervous system (CNS) is associated with a number of neurological diseases. Monocyte recruitment to the CNS has been reported for traumatic brain injury (TBI), misfolded-protein diseases, and multiple sclerosis (MS), as well as many different viral infections, including West Nile virus (WNV), herpes simplex virus (HSV), and retroviruses, such as HIV (1–4). Monocyte trafficking is particularly important for retrovirus-induced neurological diseases, as monocytes are susceptible to retrovirus infection (5, 6).
Monocytes recruited to the CNS may contribute to pathogenesis in these neurological diseases. Decreased cognitive performance correlated with increased monocyte infiltration in HIV-associated neurocognitive disorders (HAND) (1). Additionally, infiltration of monocytes was found to be important for seizure development in a mouse model of Theiler's murine encephalitis (7). Recent studies in a mouse model of experimental autoimmune encephalitis (EAE) demonstrated that CCR2+ infiltrating monocytes, but not CX3CR1+ microglia, were responsible for axonal demyelination (8). Understanding the mediators of monocyte trafficking into the CNS is therefore important for inhibiting immune-mediated damage within the CNS.
Infiltrating monocytes can contribute to neurological disease induced by polytropic retrovirus infection in mice (9, 10). In this model, infection of newborn mice with neurovirulent murine retrovirus Fr98 or BE, a chimeric retrovirus encoding a neurovirulent epitope of the Fr98 envelope protein (11), results in the development of severe neurological disease characterized by repeated seizures, progressive ataxia, and ultimately death (12). The mechanism of neuronal damage is indirect, as these viruses do not productively infect neurons but rather primarily infect microglia in the CNS (13, 14). Interestingly, the histological changes associated with Fr98 infection are substantially different from those observed with ecotropic retroviruses, as neither spongiform degeneration nor intracerebral hemorrhages are associated with disease development (15). Instead, clinical signs of neurological disease are associated with upregulation of proinflammatory cytokines and chemokines, including CCL2 and tumor necrosis factor (TNF), and the recruitment of monocytes to the CNS (10, 12, 16). The expression of CCL2 correlates with the infiltration of monocytes, as these cells are recruited from the bone marrow to the blood and then to the brain through the CCL2 receptor, CCR2, expressed by monocytes (17). Indeed, mice deficient in CCR2 have reduced susceptibility to Fr98-induced disease (9), indicating that monocyte recruitment to the brain contributes to Fr98 neuropathogenesis. Deficiency in TNF, which is produced primarily by infiltrating monocytes in the brain, also inhibited Fr98-induced neurological disease (10), further supporting a contributing role for monocytes in disease pathogenesis.
We recently found that retrovirus infection in the CNS results in increased production of neuropeptide Y (NPY) by neurons (14). Further, deficiency in NPY significantly increased the incidence and kinetics of retrovirus-mediated neurological disease (14). However, we were unable to determine a specific mechanism for this inhibition, as we saw no significant differences in cell tropism of virus infection, localization of virus within the CNS, astrocyte and microglion activation, or neuronal damage. Additionally, we did not observe a clear change in histopathology of the CNS between wild-type and NPY-deficient mice that correlated with clinical-disease development. NPY is also upregulated in the cerebrospinal fluid of patients with HIV encephalopathy, although the effect of this upregulation remains unknown (18). Recent studies with macrophage-like cell lines or isolated macrophages have demonstrated that NPY can influence macrophage activation (19–24). Additionally, NPY has been found to inhibit interleukin 1β (IL-1β)-induced activation of microglia (25, 26), the primary resident myeloid cell type in the CNS. Thus, we hypothesized that NPY might influence retrovirus-induced neurological disease by affecting the recruitment or activation of monocytes and/or microglia in the brain.
In the current study, we focused on the potential effect of NPY on monocyte recruitment in the CNS. We found that NPY-deficient mice had increased numbers of inflammatory monocytes in the CNS compared to wild-type controls and that this increased influx of inflammatory cells correlated with the development of neurological disease. Analysis of the infiltrating monocyte population indicated that NPY suppressed the recruitment of both inflammatory M1 and alternatively activated M2 monocytes. Thus, NPY has a negative regulatory role on monocyte trafficking into the CNS during Fr98 infection.
MATERIALS AND METHODS
Mice.
129S1 (129SvImJ) and 129S1 NPY−/− mice (27) were purchased from Jackson Laboratories, and colonies were maintained at Rocky Mountain Laboratories. All animal procedures were conducted in accord with the Rocky Mountain Laboratories Animal Care and Use Committee guidelines under approved protocols 2008-45, 2011-64, 2012-13, 2012-16, and 2015-050.
Virus infection and NPY treatment of mice.
The construction of chimeric virus clones for Fr98 has been previously described (15). Virus stocks were prepared from the supernatants of confluent cultures of infected Mus dunni fibroblasts. Virus titers were determined by focus-forming assays using the envelope-specific monoclonal antibodies 514 and 720 (28, 29). Mice were infected within 24 h of birth by intraperitoneal injection with 100 μl of cell culture supernatant containing 103 focus-forming units (FFU) of virus. The mice were observed daily for clinical signs, which were characterized by hyperexcitability, followed by the development of multiple severe seizures and/or ataxia, which precedes death by 1 to 2 days. When mice developed multiple severe seizures or ataxia, they were scored as clinical and their tissues were removed. No severe seizures or ataxia was observed in uninfected wild-type or uninfected NPY−/− mice. For truncated NPY (NPY 13-36) treatment of mice, NPY−/− mice at 14 and 18 days postinfection (dpi) with Fr98 were inoculated intracerebrally with 15 nmol/kg of body weight of NPY 13-36 (R&D Systems) in a 10-μl volume diluted in saline. Vehicle-control-only animals received injections of saline.
Immunohistochemistry analysis of virus infection and glial activation in wild-type and NPY−/− mice.
At set time points, mice were perfused transcardially with heparin saline (100 U/ml), followed by 10% neutral buffer formalin. Whole brains were serially sectioned at 5 μm. The sections were blocked with block solution (2% donkey serum, 1% bovine serum albumin, 0.1% cold fish skin gelatin, 0.1% Triton X-100, 0.05% Tween 20 in phosphate-buffered saline [PBS], pH 7.2) at room temperature (RT) for 1 h. Primary antibodies against virus (1:200; goat anti-gp70), rabbit anti-glial fibrillary acidic protein (anti-GFAP) (1:1,000; Dako), and rabbit anti-Iba1 (1:200; Waco) were incubated overnight at 4°C in PBS. Secondary antibodies (donkey anti-goat AF488 and donkey anti-rabbit AF594; Invitrogen) were incubated for 1 h at RT. Slides were coverslipped with Prolong Gold mounting medium containing DAPI (4′,6-diamidino-2-phenylindole) and imaged using an Aperio ScanScope FL (fluorescent) slide scanner (Leica Biosystems) with a UPLSAPO 20× objective (numerical aperture [NA] 0.75) controlled by ScanScope software or an epifluorescence Nikon Eclipse 55i clinical microscope (Nikon) with a Plan Fluor 40× objective (NA 0.75) mounted with a Nikon DS-Ri1 digital camera operated by NIS Elementals v3.2 software. Representative images were exported to TIFF format. All figures were built using Canvas 14/16 (ACD Systems).
Isolation and staining of myeloid cells from the CNS.
Animals were anesthetized by inhalation of isoflurane, followed by perfusion through the left ventricle of the heart with 1× PBS or heparin-treated saline. Whole brains were removed and sliced into several pieces prior to homogenization with a Dounce homogenizer. Samples were then further dissociated using a 70-μm cell strainer (BioExpress). Cells were isolated from spleen tissue by homogenization through a 70-μm cell strainer. To isolate the myeloid and immune cells, cells were resuspended in 70% Percoll-PBS and fractionated using a Percoll gradient as previously described (30). Cells at the 30%-70% interface of the Percoll gradient were isolated and washed in PBS. The cells were then fixed in 2% paraformaldehyde and permeabilized in 0.1% saponin-2% bovine serum albumin (BSA)-1× PBS. Samples were incubated in an Fc-blocking solution containing rat anti-mouse CD16/CD32 Fcγ III/II antibody (BD catalog no. 553142) in 2% donkey serum-0.1% saponin-2% BSA-1× PBS. The cells were incubated with fluorochrome-conjugated antibodies at room temperature. The antibodies used were F4/80-allophycocyanin (APC) (eBiosciences; clone BM8), Ly6G-APC/Cy7 (Biolegend; clone 1A8), CD45-phycoerythrin (PE) (BD; clone 30-F11), Ly6C-AF700 (BD; clone AL-21), CD11b-BV510 (Biolegend; M1/70), CD11c-PE/Cy7 (BD; clone HL3), rabbit anti-gp70 and goat anti-rabbit AF488 (Invitrogen), and NPY2R-APC (LifeSpan Biosciences; polyclonal). After washing twice, the cells were resuspended in PBS and analyzed on an LSRII flow cytometer (BD Biosciences) or sorted using a FACSAria flow cytometer (BD Biosciences) controlled by FACSDiva software (BD Biosciences). Live cells were gated using forward and side scatter, and appropriate doublet discrimination was used.
Identification and analysis of brain cells by flow cytometry.
Myeloid and other immune cells from the brains of wild-type and NPY−/− mice were isolated and stained with antibodies as described above. T cell, dendritic cell, NK cell, or B cell infiltrate was not consistently observed in the isolated cell populations. Myeloid cells were identified by F4/80 expression and were approximately 75 to 90% of the cells recovered at the 30%-70% interface of the Percoll gradient from brain tissue. Myeloid cells were then analyzed for CD45 expression to distinguish microglia (F4/80+ and CD45lo) and brain monocytes (F4/80+ and CD45hi). In some experiments, the myeloid cell population was determined by staining and gating on CD11bhi Ly6G− cells rather than F4/80. All microglia and monocytes were CD11b+, CD11c−, and Ly6G−. These cells were subsequently analyzed for CD45 expression to delineate microglia (F4/80+ CD45lo) and monocytes (F4/80+ CD45hi), which were further subdivided into M1 monocytes (F4/80+ CD45hi Ly6Chi) or M2 monocytes (F4/80+ CD45hi Ly6Clo). Differences in axis are due to differences in gain settings, which can vary between individual flow cytometry runs. This was due to cells being analyzed at different times, which was necessary due to litters being born on different days and the inoculation of mice as newborns.
Because of the variability in the total numbers of cells isolated per animal by Percoll gradient, we used a ratio of monocytes to total myeloid cells (including microglia) as determined by F4/80 staining or CD11b+ Ly6G− staining. Microglia can be infected by Fr98, but the overall numbers of microglia in the brain did not change significantly during infection and were comparable between wild-type and NPY−/− mice (data not shown). This allows comparison of an infiltrating population (brain monocytes) versus a stable population (microglia) in the CNS and controls for any variability in the isolation process between animals.
For sorting, unfixed, live cells from the brain were stained as described above but kept on ice during the staining process. Live cells were then gated for microglia (F4/80+ CD45lo) or brain monocytes (F4/80+ CD45hi). For comparison purposes, we also gated on monocytes/macrophages from the spleen (CD11c− F4/80+ CD45hi). Following sorting, cells were lysed in ZR RNA buffer (Zymo) and analyzed for RNA expression.
RNA extraction, reverse transcription, and real-time PCR.
Total RNA was isolated from brain tissue using TRIzol reagent (Invitrogen, Carlsbad, CA) or from cells using a Zymo RNA extraction kit (Zymo Research, Irvine, CA). Total RNA was treated with DNase I (Ambion, Austin, TX) for 30 min at 37°C and purified over RNA cleanup columns (Zymo Research) following the manufacturer's instructions. RNA samples were transcribed to cDNA using the iScript reverse transcription kit (Bio-Rad) as previously described (14). Samples that did not undergo reverse transcription were used to verify the lack of DNA contamination. cDNA samples were diluted 5-fold in RNase-free water prior to analysis by quantitative real-time PCR using SYBR green Supermix with ROX. All primers were used at a concentration of 0.5 μM. Untranscribed controls and water were used as negative controls. All samples were run in triplicate on a 384-well plate using an Applied Biosystems ViiA7 machine with an automatically set baseline and a manually set CT (threshold cycle) of 0.19, which intersects the mid-log phase of curves for all of the PCR pairs. Dissociation curves were used to confirm amplification of a single product for each primer pair per sample. The data for each sample were calculated as the percent difference in the CT value with the housekeeping gene Gapdh (ΔCT = CT Gapdh − CT gene of interest) and then as the fold expression relative to mock-infected controls.
RESULTS
NPY deficiency increases neurological disease and myeloid cell activation factors in the CNS.
To determine whether NPY affected the development of Fr98-induced neurological disease, wild-type and NPY−/− mice were infected with 103 FFU of virus and analyzed for signs of neurological disease. NPY−/− mice developed signs of severe seizures and ataxia at a higher rate and higher incidence than wild-type mice (Fig. 1A), similar to studies with another murine retrovirus, BE (14). These clinical signs were not observed in mock-infected NPY−/− controls (data not shown) and occurred at approximately 2 to 4 weeks postinfection in infected mice. Analysis of viral RNA demonstrated comparable viral RNA levels between 1 and 2 weeks postinfection (7, 10, and 14 dpi), with a slight increase of approximately 1 log unit in viral RNA at 3 weeks postinfection (21 dpi) in NPY−/− mice compared to wild-type controls (Fig. 1B).
FIG 1.

NPY deficiency is associated with increased neurological disease, as well as increased expression of monocyte-related mRNAs in the CNS. (A) Wild-type (129S1) and NPY−/− (129S1 Npy−/−) mice were infected within 24 h of birth with 103 FFU of Fr98. The mice were then followed for clinical signs of neurological disease, including severe seizures and ataxia. The data are presented as percent nonclinical for 28 wild-type and 32 NPY−/− mice. Statistical analysis was completed by Mantel-Cox analysis. P = 0.0013. (B) RNA was isolated from brain tissue of wild-type and NPY−/− mice at 7, 10, 14, and 21 dpi and analyzed by real-time PCR for expression of viral RNA. The data are the means ± standard errors of the mean (SEM) for 3 to 6 samples per group per time point. Statistical analysis was completed by two-way analysis of variance (ANOVA) with Sidak's multiple-comparison test.
Histological comparison of virus infections in wild-type and NPY−/− mice.
To examine the potential effects of NPY deficiency on Fr98 infection in the CNS, we examined brain tissue from 10, 14, and 21 dpi for virus infection by staining for gp70 envelope protein (Fig. 2 and 3, green fluorescence), microglion activation/monocyte recruitment using Iba1 staining (Fig. 2 and 3, red fluorescence), and astrocyte activation using GFAP staining (Fig. 4, red fluorescence). Virus infection was observed in focal areas of the thalamus, cortex, ventricles, and corpus callosum in wild-type and NPY−/− mice as early as 10 dpi (Fig. 2A and B), spreading to the midbrain, cerebellum, and brainstem by 14 dpi (Fig. 2C and D) with increased virus expression at 21 dpi (Fig. 2E and F). There was no obvious or consistent difference in virus staining in the brain between wild-type and NPY−/− mice at any of the time points (Fig. 2). Staining for the microglion/monocyte/macrophage marker Iba1 (Fig. 2, red fluorescence) showed no distinguishable difference in localization or intensity by examination of whole-brain tissue.
FIG 2.

Analysis of Fr98 infection in brain tissue from wild-type and NPY−/− mice. Brain tissues from Fr98-infected wild-type (A, C, and E) and NPY−/− (B, D, and F) mice at 10, 14, and 21 dpi were stained for the viral envelope protein gp70 (green fluorescence) and the myeloid cell marker Iba1 (red fluorescence) and imaged on a ScanScope to compare areas of virus infection. Comparisons were made for 3 or 4 brain tissues per mouse strain per time point. Although individual mice varied slightly in areas of virus infection, similar infection of the ventricles, thalamus, cortex, and cerebellum was observed at 10 dpi (A and B), with spread to the midbrain and brainstem by 14 dpi (C and D) and patchy infection in multiple regions of the brain by 21 dpi (E and F). The images were generated using ScanScope software with ×1.5 to ×1.7 digital objective magnification.
FIG 3.

Fr98 infection of Iba1+ cells in brain tissue from wild-type and NPY−/− mice. Representative images from Fr98-infected wild-type (A and B) and NPY−/− (C to E) mice at 21 dpi stained for viral envelope protein gp70 (green fluorescence) and the myeloid cell marker Iba1 (red fluorescence) were analyzed for colocalization of virus (yellow fluorescence). Virus infection of Iba1+ (white arrows) and non-Iba1+ (yellow arrows) cells was observed in both wild-type and NPY−/− mice. Additionally, Iba1+ cells in and around blood vessels (bv) (blue arrows) were observed, suggesting infiltrating monocytes. The images were taken using an epifluorescence Nikon Eclipse 55i clinical microscope (Nikon) with a Plan Fluor 40× objective (NA 0.75) mounted with a Nikon DS-Ri1 digital camera operated by NIS Elementals v3.2 software.
FIG 4.

GFAP expression increases with Fr98 infection in both wild-type and NPY−/− mice. Brain tissues from Fr98-infected wild-type (A, C, and E) and NPY−/− (B, D, and F) mice at 10 (A and B), 14 (C and D), and 21 (E and F) dpi were stained for GFAP (red fluorescence) and counterstained with DAPI. Comparisons were made for 3 or 4 brain tissues per mouse strain per time point. Although individual mice varied slightly in areas of virus infection, similar increases in staining of GFAP was observed in both wild-type (A, C, and E) and NPY−/− (B, D, and F) mice. GFAP went from minimal staining at 10 dpi (A and B) to stronger staining in the cortex, corpus callosum, thalamus, midbrain, and brain stem by 21 dpi (E and F). The images were generated using ScanScope software using ×1.5 to ×1.7 digital objective magnification.
Our previous studies on infection of NPY−/− mice with the polytropic retrovirus BE showed no clear changes in the type of cell infected by polytropic retroviruses, with infection of endothelial cells, oligodendrocytes, microglia, and monocytes in both wild-type and NPY−/− mice (14). Similarly, in the current study, we saw no obvious difference in the type of cell infected between wild-type (Fig. 3A and B) and NPY−/− (Fig. 3C to E) mice, with myeloid (red fluorescence; white arrows) and endothelial (yellow arrows) cells infected by Fr98 (green fluorescence). There did appear to be an increase in Iba1+ monocytes in and around blood vessels (bv) in NPY−/− mice (Fig. 3D and E, blue arrows); however, it was difficult to quantify due to our inability to clearly distinguish these cells as monocytes versus activated microglia.
Since astrocyte activation is also associated with polytropic retrovirus infection, we analyzed whether astrocyte activation was altered during Fr98 infection in NPY-deficient mice. Staining with GFAP increased over the time course of Fr98 infection, with a substantial increase by 21 dpi (Fig. 4, red fluorescence). However, no observable difference in either general intensity or localization was found between wild-type and NPY−/− mice at either early or late time points (Fig. 4). Furthermore, no significant difference was observed in Gfap mRNA expression (data not shown). Similarly to previously published results (15), Fr98 did not infect astrocytes in either wild-type or NPY−/− mice (data not shown). Thus, the effect of NPY deficiency on Fr98-induced neurological disease did not appear to be mediated by an effect on astrocytes.
Treatment with NPY peptide induces protection in NPY-deficient mice.
To determine if we could reverse the effects of NPY deficiency by treatment with NPY, we directly treated NPY−/− mice intracerebrally with NPY 13-36, a cleaved peptide of NPY that binds the NPY 2 receptor (Y2R) that is expressed by neurons and immune cells (31, 32). NPY−/− mice were treated intracerebrally with NPY 13-36 at 14 and 18 dpi, after the establishment of virus in the brain (Fig. 1B). Treatment with the NPY 13-36 fragment significantly (P < 0.05) inhibited neurological disease, with less than 50% of treated animals developing neurological disease compared to approximately 90% of control animals (Fig. 5A). RNA analysis and immunohistochemistry analysis of PBS- versus NPY 13-36-treated mice showed no difference in virus infection in the CNS (Fig. 5B and C). Thus, treatment with NPY 13-36 at the late stages of disease, after virus was already present in the CNS, inhibited the development of neurological disease without altering the level of virus infection in the brain. This suggests that the mechanism of NPY suppression is not due to a direct effect on virus replication in the brain.
FIG 5.

Treatment with Y2R-specific NPY peptide protects against retrovirus-induced disease. NPY−/− mice were infected with 103 FFU of Fr98 within 24 h of birth. Mice were randomly assigned to two treatment groups; one received vehicle control and the other 15 nmol/kg of NPY 13-36. (A) Mice were given intracerebral inoculations at 14 and 18 dpi and followed for signs of severe neurological disease. The data are the percentages of animals that were not clinical for 10 vehicle control- and 11 NPY 13-36-treated mice. Statistical analysis was completed using a Mantel-Cox analysis. P < 0.05. (B) RNA was isolated from brain tissues of vehicle control- and NPY 13-36-treated mice at 3 weeks postinfection and analyzed by real-time PCR for expression of viral RNA. The data are the means and SEM for 3 or 4 samples per group. No statistical difference was observed between groups. (C and D) Immunohistochemistry of Iba1 (red fluorescence) and virus gp70 (green fluorescence) of brain tissue from 3 weeks of age showing similar infection of microglia in vehicle control- (C) and NPY 13-36-treated (D) mice. The images are representative of 3 or 4 mice per group. The images were taken using an epifluorescence Nikon Eclipse 55i clinical microscope (Nikon) with a Plan Fluor 40× objective (NA 0.75) mounted with a Nikon DS-Ri1 digital camera operated by NIS Elementals v3.2 software.
NPY deficiency does not affect virus infection or activation of microglia or monocytes in the brain.
One of the strongest associations with Fr98-induced neurological disease is the proinflammatory response that is associated with neurovirulent polytropic retrovirus infection, but not avirulent polytropic retrovirus infection (9, 11, 13, 33). Our previous studies have shown that the majority of this inflammatory response is due to the CNS myeloid cell population (16). This myeloid cell population is composed of resident microglia (Iba1+ CD11b+ F4/80+ Ly6G− CD45lo), as well as infiltrating monocytes (Iba1+ CD11b+ F4/80+ Ly6G− CD45hi) (Fig. 6A and B). To look at the responses of these cells, we sorted the cell populations from the brains of infected mice at 21 dpi, the time point when NPY-deficient mice started developing neurological disease, and compared them to peripheral monocytes/macrophages from the spleen. Viral RNA was highest in the brain monocyte population compared to microglia or spleen monocytes; however, there were no significant differences between cell types or between mouse strains (Fig. 6C). Nos2 mRNA expression, an indicator of inflammatory responses, was also higher in the brain monocyte population than in the microglia or spleen macrophages (Fig. 6D); however, on a per cell basis, there was no difference between wild-type and NPY−/− mice. Thus, the difference between wild-type and NPY−/− mice does not appear to be due to the level of either virus infection or immune cell activation of microglia or monocytes.
FIG 6.
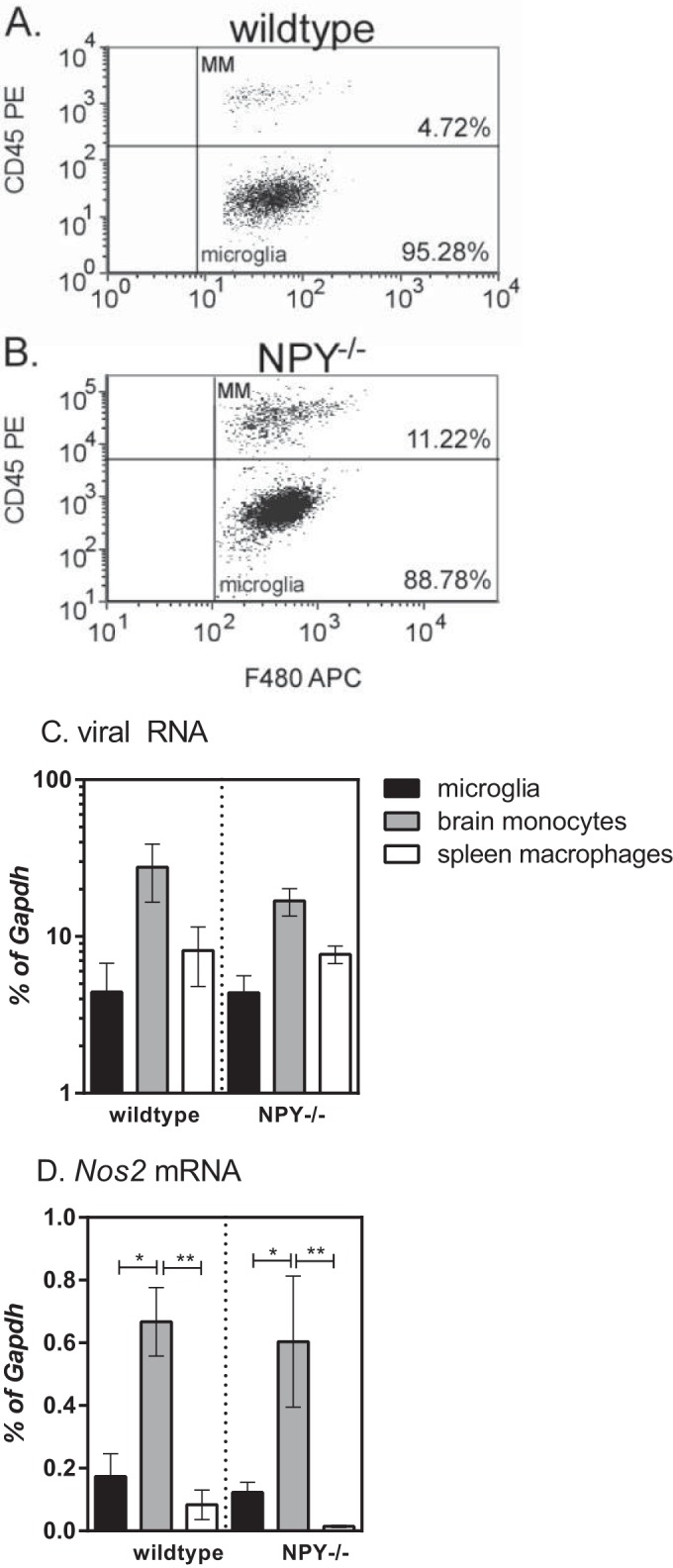
Analysis of myeloid cells in the CNS following retrovirus infection in wild-type and NPY−/− mice. Brain tissue from mock- or Fr98-infected wild-type mice and Fr98-infected mice was removed at 21 dpi and homogenized into single-cell suspensions, and myeloid cells were isolated by Percoll gradient centrifugation. Individual cells were separated from the cell debris and doublets using forward and side scatter. Whole-cell populations were then gated for F4/80 expression. (A and B) F4/80-positive cells were analyzed for CD45 expression to identify brain monocytes (CD45hi) and microglia (CD45lo). Representative plots for Fr98-infected wild-type (A) and NPY−/− (B) mice are shown. (C and D) CD45lo F4/80+ microglia, CD45hi F4/80+ brain monocytes, and CD45hi F4/80+ spleen monocytes were isolated by sorting from the brains and spleens of Fr98-infected wild-type and NPY−/− mice at 3 weeks of age. RNA was isolated from each cell population and analyzed by real-time PCR for viral RNA (C) and Nos2 mRNA (D) expression. Gene expression was normalized to Gapdh mRNA expression for each sample. The data are from 8 samples per group, isolated in duplicate experiments. The data were analyzed by two-way ANOVA with Sidak's posttest. *, P < 0.05; **, P < 0.01. The error bars indicate SEM.
NPY deficiency results in increased recruitment of monocytes into the CNS following Fr98 infection.
One of the notable differences in the sorting of myeloid cells was the higher percentage of monocytes in NPY−/− mice than in wild-type mice (Fig. 6A). Direct comparison showed that Fr98-infected NPY−/− mice had significantly higher percentages of infiltrating monocytes than Fr98-infected wild-type mice at 3 weeks postinfection (Fig. 7A). A slight, but not statistically significant, increase in the percentage of infiltrating monocytes was also observed in uninfected NPY−/− mice compared to uninfected controls (Fig. 7A). Time course analysis showed that the peak difference in monocyte infiltration between wild-type and NPY−/− mice was at 3 weeks postinfection, with a slight increase at other time points (Fig. 7B). Thus, NPY deficiency resulted in increased monocytes in the brain, primarily at 3 weeks postinfection, which correlates with the onset of clinical disease in NPY−/− mice.
FIG 7.
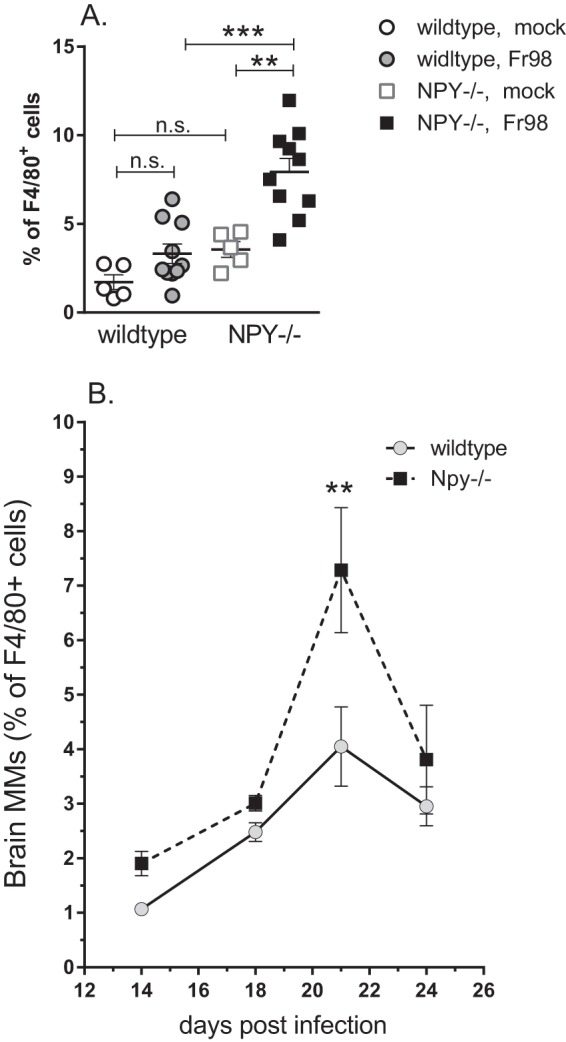
Increased monocytes in brain tissue from NPY−/− mice following Fr98 infection. (A) Analysis of the monocyte population at 3 weeks postinfection as a percentage of the total F4/80 population. The symbols represent individual mice and are from multiple experiments per group. Cells were isolated and analyzed as described in the legend to Fig. 6. Statistical analysis was completed by Mann-Whitney test. **, P < 0.01; ***, P < 0.001; n.s., not significant. (B) Monocytes in the CNS as a percentage of the total F4/80 population from 14 to 24 dpi. The data are the means ± SEM for 3 to 7 mice per mouse strain per time point. All time points represent combined data from at least 2 experiments. Two-way ANOVA analysis showed significant differences for both time (P < 0.001) and strain (P < 0.01).
NPY suppresses both M1 and M2 monocyte populations.
Infiltrating brain monocytes can be separated into two populations depending on their expression of Ly6C (34, 35). Inflammatory M1 monocytes are Ly6Chi, are dependent on CCR2 for recruitment to the CNS, and produce TNF and nitric oxide. In contrast, alternatively activated M2 monocytes/macrophages are Ly6Clo CCR2− and produce anti-inflammatory cytokines (34, 35). In addition, the M2 cell population may include infiltrating M1 cells that downregulate Ly6C and CCR2 after entry into the brain, as well as the small number of resident brain macrophages. Ly6C analysis of brain monocytes indicated that approximately 40% of these cells were M1 cells in Fr98-infected wild-type mice at 3 weeks postinfection (Fig. 8A). A similar ratio was observed in Fr98-infected NPY−/− mice (Fig. 8B). This ratio was also not affected by virus infection, as uninfected animals had a similar percentage of M1 cells relative to the monocyte population (Fig. 8C). Thus, NPY appears to affect both inflammatory M1 monocytes and alternatively activated monocytes.
FIG 8.
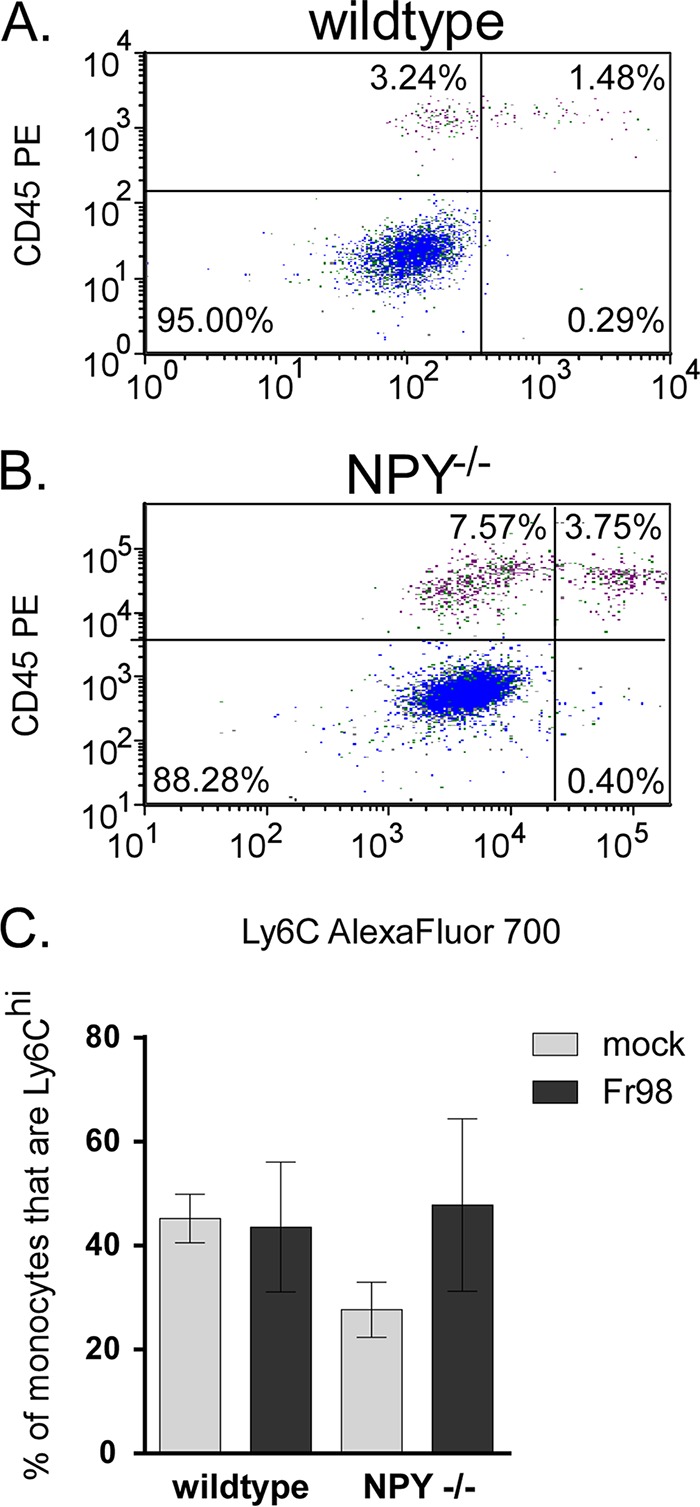
NPY deficiency results in increased numbers of monocytes in the brain, but with phenotypes similar to those of monocytes from wild-type mice. (A and B) Brain tissue from wild-type and NPY−/− mice infected with Fr98 was removed at 3 weeks postinfection as described for Fig. 2. F4/80-positive cells were also gated on CD45 (y axes) by Ly6C (x axes) to identify M2 noninflammatory monocytes (upper left quadrants) and M1 inflammatory monocytes (upper right quadrants). The data are representative of multiple animals. Representative data from Fr98-infected wild-type (A) and NPY−/− (B) mice are shown. (C) The percentage of monocytes that were Ly6Chi was not statistically different in any group. The data are the means ± SEM for 4 mice per group and represent a minimum of two replicate experiments.
Recent studies have shown that NPY 13-36, which specifically binds Y2R, can inhibit monocyte recruitment in other tissues (36, 37). Since the effects of NPY deficiency on Fr98-induced neurological disease could be reversed by treatment with NPY 13-36, we examined monocytes and microglia for their expression of Y2R. Y2R was detected on all 3 cell types (Fig. 9A to E). M2 monocytes had the highest level of Y2R expression (Fig. 9D), with M1 monocytes having an intermediate level of Y2R expression (Fig. 9C). Thus, by flow cytometric analysis, M2 monocytes expressed Y2R while M1 monocytes had lower expression levels. Similar results were observed between cell populations in Fr98-infected NPY−/− mice (data not shown).
FIG 9.

Y2R expression on all three myeloid cell types in the CNS. (A) Microglia, M1 monocytes, and M2 monocytes were isolated by CD45 and Ly6C staining on CD11b+ Ly6G− myeloid cells isolated from Fr98-infected wild-type mice at 3 weeks postinfection. The cells were analyzed for Y2R using directly conjugated antibodies. (B to E) Comparison of relative expression of Y2R on microglia (B), M1 monocytes (C), M2 monocytes (D), and all three cell populations (E). The gray line in each plot indicates no primary controls (antibody cocktail minus Y2R antibody).
DISCUSSION
The regulation of monocyte levels in the CNS is an important mediator of viral diseases, both in the recruitment of virus-infected cells to the CNS and in the mediation of damage within the brain (1, 2, 4). In the current study, we found that NPY, a small peptide that is produced by neurons of the central and peripheral nervous systems (23, 38), was an important inhibitor of monocyte recruitment to the brain during retrovirus infection. Mice deficient in NPY were more susceptible to retrovirus-induced neurological disease. This increase in disease was associated with an approximately 2-fold increase in the number of monocytes in the CNS, although it did not detectably alter the phenotype of the infiltrating monocytes. Thus, NPY appears to suppress retrovirus disease, at least in part, by inhibiting monocyte recruitment to the CNS. The ability of NPY, produced by neurons, to suppress monocyte recruitment to the CNS during retrovirus infection suggests that the nervous system can respond to damage or stress in the CNS and influence the immune response to that damage.
Monocyte trafficking to the CNS has been reported to be an important component in the development of seizures, including temporal lobe epilepsy in humans and in animal models (39). Monocyte recruitment to the CNS is also a contributing factor to Theiler's murine encephalomyelitis virus (TMEV)-induced seizures (40–42), as demonstrated by correlative experiments, monocyte transfers, and minocycline treatment. Our previous studies with polytropic retrovirus infection have also shown an important role for monocytes, as demonstrated by the effect of CCR2 deficiency on Fr98-induced neurological disease (9). Identifying how the nervous system regulates the recruitment of these cells to the CNS, such as through the production of NPY, will be important for developing methods for the treatment of seizures and other neurological disorders.
Treatment with NPY has been shown to have therapeutic effects for both kainate- and trimethyltin-induced seizure models of temporal lobe epilepsy (43, 44). The major protective effect of NPY in these models is thought to be through direct stimulation of neurons. However, since monocytes contribute to seizure development (39–42), NPY may also limit seizure induction by limiting monocyte recruitment. Thus, NPY may inhibit seizures through multiple mechanisms, including both direct and indirect effects on the nervous system.
The current study is the first to demonstrate that NPY suppresses monocyte recruitment to the CNS but is not the first study to show an effect of neurotransmitters like NPY on monocyte or macrophage responses. In vitro, NPY was found to stimulate macrophage functions, including chemotaxis, in some studies but inhibited these responses in others (19, 23, 45, 46). NPY was also found to inhibit the expression of costimulatory molecules on monocytes in a rat model of experimental autoimmune encephalitis (47). However, in the current study, we did not see any significant effect of NPY on CD80 or CD86 expression by monocytes or microglia following retrovirus infection (data not shown). This difference in the effect of NPY on the response of monocytes may be highly dependent on NPY receptor expression or the activation state of the cell when stimulated by NPY. Additionally, there may be some differences in monocyte activation or recruitment of cells to specific areas of the brain. The flow cytometry analysis of whole-brain tissue used in the current study would not detect specific changes in areas of virus infection versus areas without virus infection. However, it is expected that most monocytes would be recruited to areas of virus infection and therefore would be stimulated/recruited by similar stimuli.
The peak of monocyte recruitment to the brain was 21 dpi for both wild-type and NPY−/− mice, with a decrease a few days later. This peak of monocyte recruitment correlates with the onset of clinical disease in most animals. Cytokine expression also peaks just prior to the onset of clinical disease for Fr98 infection (9, 12). Thus, the reduction in monocyte recruitment at 24 dpi may be reflective of decreased CCL2 and other proinflammatory cytokines that recruit monocytes into the CNS.
Stimulation of immune cells using receptor-specific peptides indicates that the activating effects of NPY are mediated primarily by Y1R, while the inhibitory effects are mediated by Y2R and Y5R (20, 48). In agreement with this, the expression of DPP4, which cleaves NPY to a form that preferentially binds Y2R over Y1R, by monocytes also contributes to the anti-inflammatory effects of NPY (49). The ability of Y2R-specific NPY to inhibit retrovirus-induced neurological disease correlates with Y2R expression on monocytes. These data suggest that NPY may influence monocyte recruitment and/or accumulation in the CNS through Y2R.
One aspect of NPY inhibition that was not analyzed in this study was the ability of NPY to directly inhibit neurons. Although Fr98 does not infect neurons, virus infection in the CNS results in seizures (10, 15), suggesting an indirect mechanism of neuronal damage. Furthermore, XPR1, the receptor for polytropic retroviruses, is expressed by neurons, and binding of this receptor by virus proteins induces neurotoxicity (50). Neurons do express Y1R, Y2R, and Y5R, and stimulation through these receptors has been shown to limit seizures in animal models of epilepsy, possibly by regulation of glutamate release (51). Preliminary studies with glutamate inhibitors have not demonstrated a role for this pathway in Fr98-induced pathogenesis (unpublished observations). We cannot rule out the possibility that one of the effects of NPY on Fr98-induced disease is through direct stimulation of neurons and a direct neuroprotective effect. However, as monocyte recruitment is also important for Fr98-mediated disease (9, 10), the ability of NPY to suppress the recruitment of these cells is a critical factor in Fr98-mediated pathogenesis. Furthermore, as monocytes are important mediators of several neurological disorders, NPY- or Y2R-specific agonists may be potential therapeutics for suppressing or reducing monocyte-mediated damage in these diseases.
ACKNOWLEDGMENTS
This study was performed at Rocky Mountain Laboratories (RML) and was funded in part by the Division of Intramural Research (DIR) as part of the National Institute of Allergy and Infectious Diseases (NIAID) within the National Institutes of Health (NIH) and in part by National Natural Science Foundation of China grant 31270917 and Chinese Scholarship Council program grant 201404910139 to Min Du.
We thank Clayton Winkler, Bruce Chesebro, Lara Myers, Leonard Evans, Paul Policastro, and Byron Caughey for critical readings of the manuscript.
REFERENCES
- 1.Burdo TH, Lackner A, Williams KC. 2013. Monocyte/macrophages and their role in HIV neuropathogenesis. Immunol Rev 254:102–113. doi: 10.1111/imr.12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Getts DR, Terry RL, Getts MT, Muller M, Rana S, Shrestha B, Radford J, Van Rooijen N, Campbell IL, King NJ. 2008. Ly6c+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J Exp Med 205:2319–2337. doi: 10.1084/jem.20080421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Semple BD, Bye N, Rancan M, Ziebell JM, Morganti-Kossmann MC. 2010. Role of CCL2 (MCP-1) in traumatic brain injury (TBI): evidence from severe TBI patients and CCL2−/− mice. J Cereb Blood Flow Metab 30:769–782. doi: 10.1038/jcbfm.2009.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Terry RL, Getts DR, Deffrasnes C, van Vreden C, Campbell IL, King NJ. 2012. Inflammatory monocytes and the pathogenesis of viral encephalitis. J Neuroinflamm 9:270. doi: 10.1186/1742-2094-9-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams DW, Eugenin EA, Calderon TM, Berman JW. 2012. Monocyte maturation, HIV susceptibility, and transmigration across the blood brain barrier are critical in HIV neuropathogenesis. J Leukoc Biol 91:401–415. doi: 10.1189/jlb.0811394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Williams K, Burdo TH. 2012. Monocyte mobilization, activation markers, and unique macrophage populations in the brain: observations from SIV infected monkeys are informative with regard to pathogenic mechanisms of HIV infection in humans. J Neuroimmune Pharmacol 7:363–371. doi: 10.1007/s11481-011-9330-3. [DOI] [PubMed] [Google Scholar]
- 7.Cusick MF, Libbey JE, Patel DC, Doty DJ, Fujinami RS. 2013. Infiltrating macrophages are key to the development of seizures following virus infection. J Virol 87:1849–1860. doi: 10.1128/JVI.02747-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R, Wu PM, Doykan CE, Lin J, Cotleur AC, Kidd G, Zorlu MM, Sun N, Hu W, Liu L, Lee JC, Taylor SE, Uehlein L, Dixon D, Gu J, Floruta CM, Zhu M, Charo IF, Weiner HL, Ransohoff RM. 2014. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med 211:1533–1549. doi: 10.1084/jem.20132477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peterson KE, Errett JS, Wei T, Dimcheff DE, Ransohoff R, Kuziel WA, Evans L, Chesebro B. 2004. MCP-1 and CCR2 contribute to non-lymphocyte-mediated brain disease induced by Fr98 polytropic retrovirus infection in mice: role for astrocytes in retroviral neuropathogenesis. J Virol 78:6449–6458. doi: 10.1128/JVI.78.12.6449-6458.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peterson KE, Hughes S, Dimcheff DE, Wehrly K, Chesebro B. 2004. Separate sequences in a murine retroviral envelope protein mediate neuropathogenesis by complementary mechanisms with differing requirements for tumor necrosis factor alpha. J Virol 78:13104–13112. doi: 10.1128/JVI.78.23.13104-13112.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peterson KE, Pourciau S, Du M, Lacasse R, Pathmajeyan M, Poulsen D, Agbandje-McKenna M, Wehrly K, Chesebro B. 2008. Neurovirulence of polytropic murine retrovirus is influenced by two separate regions on opposite sides of the envelope protein receptor binding domain. J Virol 82:8906–8910. doi: 10.1128/JVI.02134-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peterson KE, Robertson SJ, Portis JL, Chesebro B. 2001. Differences in cytokine and chemokine responses during neurological disease induced by polytropic murine retroviruses map to separate regions of the viral envelope gene. J Virol 75:2848–2856. doi: 10.1128/JVI.75.6.2848-2856.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peterson KE, Chesebro B. 2006. Influence of proinflammatory cytokines and chemokines on the neuropathogenesis of oncornavirus and immunosuppressive lentivirus infections. Curr Top Microbiol Immunol 303:67–95. [DOI] [PubMed] [Google Scholar]
- 14.Du M, Butchi NB, Woods T, Morgan TW, Peterson KE. 2010. Neuropeptide Y has a protective role during murine retrovirus-induced neurological disease. J Virol 84:11076–11088. doi: 10.1128/JVI.01022-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robertson SJ, Hasenkrug KJ, Chesebro B, Portis JL. 1997. Neurologic disease induced by polytropic murine retroviruses: neurovirulence determined by efficiency of spread to microglial cells. J Virol 71:5287–5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peterson KE, Evans LH, Wehrly K, Chesebro B. 2006. Increased proinflammatory cytokine and chemokine responses and microglial infection following inoculation with neural stem cells infected with polytropic murine retroviruses. Virology 354:143–153. doi: 10.1016/j.virol.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 17.Lim JK, Obara CJ, Rivollier A, Pletnev AG, Kelsall BL, Murphy PM. 2011. Chemokine receptor Ccr2 is critical for monocyte accumulation and survival in West Nile virus encephalitis. J Immunol 186:471–478. doi: 10.4049/jimmunol.1003003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malessa R, Heimbach M, Brockmeyer NH, Hengge U, Rascher W, Michel MC. 1996. Increased neuropeptide Y-like immunoreactivity in cerebrospinal fluid and plasma of human immunodeficiency virus-infected patients: relationship to HIV encephalopathy. J Neurol Sci 136:154–158. doi: 10.1016/0022-510X(95)00305-L. [DOI] [PubMed] [Google Scholar]
- 19.Ahmed AA, Wahbi AH, Nordlin K. 2001. Neuropeptides modulate a murine monocyte/macrophage cell line capacity for phagocytosis and killing of Leishmania major parasites. Immunopharmacol Immunotoxicol 23:397–409. doi: 10.1081/IPH-100107339. [DOI] [PubMed] [Google Scholar]
- 20.Dimitrijevic M, Stanojevic S, Vujic V, Beck-Sickinger A, von Horsten S. 2005. Neuropeptide Y and its receptor subtypes specifically modulate rat peritoneal macrophage functions in vitro: counter regulation through Y1 and Y2/5 receptors. Regul Pept 124:163–172. doi: 10.1016/j.regpep.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 21.Phan TA, Taylor AW. 2013. The neuropeptides alpha-MSH and NPY modulate phagocytosis and phagolysosome activation in RAW 264.7 cells. J Neuroimmunol 260:9–16. doi: 10.1016/j.jneuroim.2013.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singer K, Morris DL, Oatmen KE, Wang T, DelProposto J, Mergian T, Cho KW, Lumeng CN. 2013. Neuropeptide Y is produced by adipose tissue macrophages and regulates obesity-induced inflammation. PLoS One 8:e57929. doi: 10.1371/journal.pone.0057929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Straub RH, Mayer M, Kreutz M, Leeb S, Scholmerich J, Falk W. 2000. Neurotransmitters of the sympathetic nerve terminal are powerful chemoattractants for monocytes. J Leukoc Biol 67:553–558. [DOI] [PubMed] [Google Scholar]
- 24.Zhou JR, Zhang LD, Wei HF, Wang X, Ni HL, Yang F, Zhang T, Jiang CL. 2013. Neuropeptide Y induces secretion of high-mobility group box 1 protein in mouse macrophage via PKC/ERK dependent pathway. J Neuroimmunol 260:55–59. doi: 10.1016/j.jneuroim.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 25.Ferreira R, Santos T, Cortes L, Cochaud S, Agasse F, Silva AP, Xapelli S, Malva JO. 2012. Neuropeptide Y inhibits interleukin-1 beta-induced microglia motility. J Neurochem 120:93–105. doi: 10.1111/j.1471-4159.2011.07541.x. [DOI] [PubMed] [Google Scholar]
- 26.Ferreira R, Xapelli S, Santos T, Silva AP, Cristovao A, Cortes L, Malva JO. 2010. Neuropeptide Y modulation of interleukin-1{beta} (IL-1{beta})-induced nitric oxide production in microglia. J Biol Chem 285:41921–41934. doi: 10.1074/jbc.M110.164020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Erickson JC, Clegg KE, Palmiter RD. 1996. Sensitivity to leptin and susceptibility to seizures of mice lacking neuropeptide Y. Nature 381:415–421. doi: 10.1038/381415a0. [DOI] [PubMed] [Google Scholar]
- 28.Robertson MN, Miyazawa M, Mori S, Caughey B, Evans LH, Hayes SF, Chesebro B. 1991. Production of monoclonal antibodies reactive with a denatured form of the Friend murine leukemia virus gp70 envelope protein: use in a focal infectivity assay, immunohistochemical studies, electron microscopy and western blotting. J Virol Methods 34:255–271. doi: 10.1016/0166-0934(91)90105-9. [DOI] [PubMed] [Google Scholar]
- 29.Sitbon M, Nishio J, Wehrly K, Lodmell D, Chesebro B. 1985. Use of a focal immunofluorescence assay on live cells for quantitation of retroviruses: distinction of host range classes in virus mixtures and biological cloning of dual-tropic murine leukemia viruses. Virology 141:110–118. doi: 10.1016/0042-6822(85)90187-4. [DOI] [PubMed] [Google Scholar]
- 30.Butchi NB, Du M, Peterson KE. 2010. Interactions between TLR7 and TLR9 agonists and receptors regulate innate immune responses by astrocytes and microglia. Glia 58:650–664. doi: 10.1002/glia.20952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sheikh SP, Hakanson R, Schwartz TW. 1989. Y1 and Y2 receptors for neuropeptide Y. FEBS Lett 245:209–214. doi: 10.1016/0014-5793(89)80223-6. [DOI] [PubMed] [Google Scholar]
- 32.Brothers SP, Wahlestedt C. 2010. Therapeutic potential of neuropeptide Y (NPY) receptor ligands. EMBO Mol Med 2:429–439. doi: 10.1002/emmm.201000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peterson KE, Du M. 2009. Innate immunity in the pathogenesis of polytropic retrovirus infection in the central nervous system. Immunol Res 43:149–159. doi: 10.1007/s12026-008-8060-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang J, Zhang L, Yu C, Yang XF, Wang H. 2014. Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark Res 2:1. doi: 10.1186/2050-7771-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geissmann F, Jung S, Littman DR. 2003. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19:71–82. doi: 10.1016/S1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 36.Stadler J, Le TP, Haas P, Nave H. 2011. Distinct effects of NPY13-36, a specific NPY Y2 agonist, in a model of rodent endotoxemia on leukocyte subsets and cytokine levels. Ann Anat 193:486–493. doi: 10.1016/j.aanat.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 37.Nave H, Bedoui S, Moenter F, Steffens J, Felies M, Gebhardt T, Straub RH, Pabst R, Dimitrijevic M, Stanojevic S, von Hörsten S. 2004. Reduced tissue immigration of monocytes by neuropeptide Y during endotoxemia is associated with Y2 receptor activation. J Neuroimmunol 155:1–12. doi: 10.1016/j.jneuroim.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 38.Straub RH, Schaller T, Miller LE, von Horsten S, Jessop DS, Falk W, Schölmerich J. 2000. Neuropeptide Y cotransmission with norepinephrine in the sympathetic nerve-macrophage interplay. J Neurochem 75:2464–2471. [DOI] [PubMed] [Google Scholar]
- 39.Ravizza T, Gagliardi B, Noe F, Boer K, Aronica E, Vezzani A. 2008. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis 29:142–160. doi: 10.1016/j.nbd.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 40.Kirkman NJ, Libbey JE, Wilcox KS, White HS, Fujinami RS. 2010. Innate but not adaptive immune responses contribute to behavioral seizures following viral infection. Epilepsia 51:454–464. doi: 10.1111/j.1528-1167.2009.02390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Libbey JE, Kennett NJ, Wilcox KS, White HS, Fujinami RS. 2011. Interleukin-6, produced by resident cells of the central nervous system and infiltrating cells, contributes to the development of seizures following viral infection. J Virol 85:6913–6922. doi: 10.1128/JVI.00458-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Howe CL, Lafrance-Corey RG, Sundsbak RS, Sauer BM, Lafrance SJ, Buenz EJ, Schmalstieg WF. 2012. Hippocampal protection in mice with an attenuated inflammatory monocyte response to acute CNS picornavirus infection. Sci Rep 2:545. doi: 10.1038/srep00545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Corvino V, Marchese E, Giannetti S, Lattanzi W, Bonvissuto D, Biamonte F, Mongiovì AM, Michetti F, Geloso MC. 2012. The neuroprotective and neurogenic effects of neuropeptide Y administration in an animal model of hippocampal neurodegeneration and temporal lobe epilepsy induced by trimethyltin. J Neurochem 122:415–426. doi: 10.1111/j.1471-4159.2012.07770.x. [DOI] [PubMed] [Google Scholar]
- 44.Elbrond-Bek H, Olling JD, Gotzsche CR, Waterfield A, Wortwein G, Woldbye DP. 2014. Neuropeptide Y-stimulated [35S]GTPgammas functional binding is reduced in the hippocampus after kainate-induced seizures in mice. Synapse 68:427–436. doi: 10.1002/syn.21762. [DOI] [PubMed] [Google Scholar]
- 45.De la Fuente M, Bernaez I, Del RM, Hernanz A. 1993. Stimulation of murine peritoneal macrophage functions by neuropeptide Y and peptide YY. Involvement of protein kinase C. Immunology 80:259–265. [PMC free article] [PubMed] [Google Scholar]
- 46.De la Fuente M, Del RM, Medina S. 2001. Changes with aging in the modulation by neuropeptide Y of murine peritoneal macrophage functions. J Neuroimmunol 116:156–167. doi: 10.1016/S0165-5728(01)00297-1. [DOI] [PubMed] [Google Scholar]
- 47.Dimitrijevic M, Mitic K, Kustrimovic N, Vujic V, Stanojevic S. 2012. NPY suppressed development of experimental autoimmune encephalomyelitis in Dark Agouti rats by disrupting costimulatory molecule interactions. J Neuroimmunol 245:23–31. doi: 10.1016/j.jneuroim.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 48.Mitic K, Stanojevic S, Kustrimovic N, Vujic V, Dimitrijevic M. 2011. Neuropeptide Y modulates functions of inflammatory cells in the rat: distinct role for Y1, Y2 and Y5 receptors. Peptides 32:1626–1633. doi: 10.1016/j.peptides.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 49.Dimitrijevic M, Stanojevic S, Mitic K, Kustrimovic N, Vujic V, Miletic T, Kovacević-Jovanović V. 2008. The anti-inflammatory effect of neuropeptide Y (NPY) in rats is dependent on dipeptidyl peptidase 4 (DP4) activity and age. Peptides 29:2179–2187. doi: 10.1016/j.peptides.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 50.Vaughan AE, Mendoza R, Aranda R, Battini JL, Miller AD. 2012. Xpr1 is an atypical G-protein-coupled receptor that mediates xenotropic and polytropic murine retrovirus neurotoxicity. J Virol 86:1661–1669. doi: 10.1128/JVI.06073-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Decressac M, Barker RA. 2012. Neuropeptide Y and its role in CNS disease and repair. Exp Neurol 238:265–272. doi: 10.1016/j.expneurol.2012.09.004. [DOI] [PubMed] [Google Scholar]