

Abstract
The DnaB-DnaC complex binds to the unwound DNA within the Escherichia coli replication origin in the helicase loading process, but the biochemical events that lead to its stable binding are uncertain. This study characterizes the function of specific C-terminal residues of DnaC. Genetic and biochemical characterization of proteins bearing F231S and W233L substitutions of DnaC reveals that their activity is thermolabile. Because the mutants remain able to form a complex with DnaB at 30 and 37 °C, their thermolability is not explained by an impaired interaction with DnaB. Photo-cross-linking experiments and biosensor analysis show an altered affinity of these mutants compared with wild type DnaC for single-stranded DNA, suggesting that the substitutions affect DNA binding. Despite this difference, their activity in DNA binding is not thermolabile. The substitutions also drastically reduce the affinity of DnaC for ATP as measured by the binding of a fluorescent ATP analogue (MANT-ATP) and by UV cross-linking of radiolabeled ATP. Experiments show that an elevated temperature substantially inhibits both mutants in their ability to load the DnaB-DnaC complex at a DnaA box. Because a decreased ATP concentration exacerbates their thermolabile behavior, we suggest that the F231S and W233L substitutions are thermolabile in ATP binding, which correlates with defective helicase loading at an elevated temperature.
Keywords: bacterial genetics, DNA replication, DNA-protein interaction, Escherichia coli (E. coli), nucleic acid enzymology
Introduction
DNA replication in Escherichia coli starts at the chromosomal replication origin (oriC) through a series of discrete biochemical events. First, DnaA bound to ATP recognizes specific DNA sequences within oriC to form a DnaA oligomer, which leads to the unwinding of an AT-rich region near the left border (reviewed in Ref. 1). At the step of helicase loading, DnaA then directs the binding of the DnaB-DnaC complex onto each unwound DNA strand. In vitro, the DnaB-DnaC complex not bound to DNA contains as many as six DnaC molecules per DnaB molecule, a hexamer of six identical subunits that in the absence of DnaC is a toroid (2–5). It acts as the replicative DNA helicase to unwind the parental duplex DNA so that each DNA strand can be copied by the cellular replicase, DNA polymerase III holoenzyme. Studies show that DnaB by itself is able to interact with single-stranded DNA (ssDNA)4 (6, 7). Other works indicate that one of two DNA strands of an artificial replication fork passes through the interior of the toroid (8, 9). Despite the ability of DnaB to interact with ssDNA, the conundrum is that loading of the helicase at oriC requires that it is complexed to DnaC. Considering that DnaC also interacts with ssDNA (see below), it is unclear whether this activity of DnaC participates during the events that lead to the stable binding of the DnaB-DnaC complex at oriC.
An AAA+ protein, DnaC, carries amino acid motifs implicated in ATP binding, in ATP hydrolysis, and in coordinating a conformational change with ATP hydrolysis (see Fig. 1). In those AAA+ motifs that have been examined, amino acid substitutions cause impaired function, indicating that these conserved residues are indispensable (10–13). Other residues near the C terminus (residue 245) are very highly conserved (14). This portion of DnaC is absent in the x-ray crystallographic structure of Aquifex aeolicus DnaC bound to ADP, but part of this region ending with the sensor 2 arginine forms a loop in the x-ray structure of this protein bound to ADP-BeF3, an ATP mimetic (12). Presumably, this C-terminal segment adopts this looped structure by the bonding of the sensor 2 arginine with the γ-phosphate of ATP that is represented by BeF3 but is flexible in its absence. These structures underpin a model of a DnaC oligomer assembled as a right-handed spiral with an open passageway along its long axis. The binding of ssDNA is suggested to occur within this central channel. In support of this model, amino acid substitutions at the proposed interface between adjacent DnaC molecules correlated with both impaired multimer formation as measured in a cross-linking assay and defective DNA binding (12). Complementing these studies, the x-ray crystallographic structures of the DnaB-DnaI complex of thermophilic bacteria for which DnaI may be considered the functional counterpart to DnaC and cryo-electron microscopy of the E. coli DnaB-DnaC complex reveal that the relative repositioning of DnaB protomers in the DnaB ring remodels the toroid structure to become a right-handed spiral by virtue of a single discontinuity between adjacent protomers of the DnaB hexamer (15, 16). The gap evidently permits passage of ssDNA into the interior of the spiral for DNA binding.
FIGURE 1.

Mutant DnaC proteins and their amino acid substitutions. Alignment of the primary amino acid sequence of E. coli DnaC with 29 DnaC homologues reveals conserved residues shown as uppercase letters relative to its secondary structure (hatched boxes, α helix; filled boxes, β strand) derived from the A. aeolicus DnaC x-ray crystallographic structure and from a secondary structure prediction method (PHDsec) (10, 12, 14). The numbers at the right are the coordinates for E. coli DnaC protein. The figure also shows the locations of single amino acid substitutions in mutant DnaCs. Those in green are from this work; substitutions in purple, yellow, pink, and red have been described in the respective publications (10, 11, 13, 17). AAA+ motifs shown as black bars are adapted from figures in Refs. 11 and 17. The residues shaded in gray represent segments that bind to DnaB (28). Those shaded in blue correspond to the initiator specific motif (ISM; residues 136–162) (12).
We previously developed a genetic method to identify mutations that impair E. coli dnaC function (17). With this method, which may be used to analyze other essential genes of E. coli and related bacteria, we isolated many more missense mutations of dnaC (see Fig. 1). We focused on mutant proteins bearing F231S and W233L substitutions near the C terminus (residue 245) because the substitutions are in strictly conserved residues, but their roles are unknown. We speculated that the W233L substitution affects ssDNA binding for the following reasoning. First, the binding of E. coli DnaC, which has three tryptophans, to ssDNA has been measured by an increase in tryptophan fluorescence (11). A W32G substitution in DnaC disrupts its interaction with DnaB (10). If this tryptophan is not also involved in DNA binding, this implicates the remaining tryptophans at residues 228 and/or 233. Second, many proteins that bind to ssDNA interpose aromatic residues between the stacked bases (18), so the fluorescence change of DnaC may be due to DNA binding by a neighboring aromatic residue that alters the environment of one or both of these tryptophans. Third, ATP-stimulated ssDNA binding by E. coli DnaC correlates with both the interaction of a conserved arginine of A. aeolicus DnaC (Arg226, equivalent to Arg236 of E. coli DnaC) with the ATP mimetic, ADP-BeF3, and the organization of nearby residues including those corresponding to Phe231 and Trp233 into a loop (11–13, 19). Hence, the interaction of this conserved arginine (proposed as the Sensor 2 arginine) with ATP may stabilize the loop to aid in DNA binding. In the study described herein, we determined that DnaC bearing an F231S or W233L substitution is thermolabile in DNA replication. Biochemical assays reveal that the mutants retain their ability to interact with DnaB but aberrantly bind to ssDNA. However, these activities are not thermolabile. Their drastically impaired ATP binding is apparently alleviated by higher concentrations of ATP so that the mutants are active in functions required for DNA replication at 30 °C. We also show that the mutants are defective in helicase loading at an elevated temperature.
Experimental Procedures
Reagents and Proteins
Replication proteins have been described (13, 20). Mutant DnaC proteins were purified essentially as described after their induced expression (13), but the host strain was E. coli Lemo21 (DE3). Briefly, this strain was transformed with a derivative of pET11a encoding the respective dnaC allele. Using the transformation mixture as an inoculum, cultures were grown overnight at 30 °C in M9 medium supplemented with 0.4% casamino acids, 0.2% glycerol, 1.0 mm CaCl2, 1.0 mm MgSO4, 0.02 mm FeSO4, 35 μg/ml chloramphenicol, and 100 μg/ml ampicillin. The cultures were then diluted into larger volumes of this medium followed by growth with shaking at room temperature. At a turbidity of 0.1–0.2 (A595 nm), IPTG was added to 0.4 mm with continued incubation with shaking for 20 min. The temperature was then adjusted to 16 °C before harvesting the cells after 17–20 h by centrifugation. Following cell lysis, purification of wild type DnaC, F231S, and W233L was as described in which the last step to obtain monomeric DnaC was by gel permeation chromatography (Superdex 75; GE Healthcare) (13).
Isolation and Genetic Analysis of dnaC Alleles
The genetic method to identify defective dnaC mutations relies on E. coli MF1061 (araD139 Δ(ara, leu)7697 ΔlacX74 galU galK rpsL hsdR2 (rK− mK+) mcrB1 ΔdnaC::cat recA635::kan) carrying the plasmid pAM34dnaCL1–4 that encodes the dnaC+ gene (17). This plasmid, which confers ampicillin resistance and complements the ΔdnaC::cat mutation of the host strain, requires IPTG in the culture medium for its maintenance. The function of a second plasmid that confers tetracycline resistance and encodes a dnaC mutation such as those characterized in this study was measured on LB plates supplemented with tetracycline (10 μg/ml) at 37 °C in the presence and absence of 0.5 mm IPTG. Transformants able to grow only in the presence of IPTG bear plasmids that encode nonfunctional dnaC mutations. To isolate novel dnaC mutations, a DNA fragment carrying dnaC was mutagenized by error-prone PCR amplification and then inserted into pACYC184 as described (17). After transformation into MF1061 carrying pAM34dnaCL1–4, individual transformants were screened at 37 °C on tetracycline-supplemented LB plates with or without IPTG. DNA sequence analysis was performed to identify alleles bearing single missense mutations. The derivatives of pACYC184 encoding these alleles were then retransformed into MF1061 (pAM34dnaCL1–4) followed by plating on tetracycline-containing media with and without IPTG to confirm their phenotypes.
In Vitro DNA Replication
Reactions to measure DNA replication with M13oriC2LB5 DNA (46 fmol; 200 ng) or M13 A site ssDNA (27 fmol; 80 ng), and the required purified replication proteins were performed as described (21, 22). The reactions were assembled at 0 °C and incubated at the indicated temperatures for 20 or 10 min, respectively. Total nucleotide incorporation of DNA (as pmol) was measured by liquid scintillation spectrometry after trichloroacetic acid precipitation onto glass fiber filters (Whatman GF/C).
UV Cross-linking to ssDNA
Reactions (10 μl) contained 20 mm HEPES-KOH, pH 8, 30 mm potassium chloride, 5 mm magnesium acetate, 1 mm EDTA, 4 mm dithiothreitol, 0.2% (v/v) Triton X-100, 100 μg/ml BSA, 2 mm ATP, 60 fmol of 5′ [α-32P]-CGAAAACAGTGATTTAGCAGGCATCGCGGAAATTATTATC, 0.4 μm DnaB (monomer), and 2 μm DnaC. After incubation at 30 or 37 °C for 10 min, duplicate samples were placed on a Parafilm-covered aluminum block equilibrated at the respective temperature and then irradiated for 15 min under a Westinghouse G15T8 germicidal lamp at a distance of 11 cm. Samples were denatured in a boiling water bath for 2 min, electrophoresed on 10% SDS-polyacrylamide gels, and stained with Coomassie Brilliant Blue to visualize the molecular weight standards. The dried gels were analyzed with a Molecular Dynamics Typhoon phosphorimaging device.
Fluorescence Measurement of the Binding of MANT-ATP, a Fluorescent ATP Analogue
Reactions (200 μl) contained 50 mm HEPES-KOH, pH 8.0, 30 mm KCl, 5 mm MgCl2, 5% glycerol, 0.002% Tween 20, 4 mm DTT, 1.5 μm DnaC, or the mutants and MANT-ATP at the indicated concentrations. Fluorescence emission spectra were measured at 25 °C from 400 to 550 nm after excitation at 356 nm using Cary Eclipse fluorescence spectrophotometer. Three independent measurements were taken for samples containing wild type DnaC or the mutants and also for those lacking protein. These values were corrected by subtracting measurements obtained with the buffer and an increasing ligand in absence of protein. The change in fluorescence (ΔF) at 440 nm (the emission maximum) for each measurement was plotted versus the concentration of MANT-ATP to obtain a binding curve and Fmax value. The dissociation constant (KD) was calculated from a double reciprocal plot of the binding curve. Linear regression of 1/(1 − (ΔF/Fmax) versus [MANT-ATP]/((ΔF/Fmax) yielded a slope of 1/KD.
Assembly, Isolation, and Analysis of the ABC Complex
An intermediate named the ABC complex, which forms by recruitment of the DnaB-DnaC complex by DnaA bound to a DnaA box in a hairpin structure, was assembled with M13 A site ssDNA (1.3 μg), single-stranded DNA-binding protein (600 ng), DnaA (400 ng), DnaB (2 μl), DnaC, F231S, or W233L each at 1.7 μg in 85 μl of ABC buffer (40 mm HEPES-KOH, pH 8.0, 40 mm potassium glutamate, 0.5 mm magnesium acetate, 4% (w/v) sucrose, 4 mm dithiothreitol, 100 μg/ml BSA, and 1 mm ATP), as described (23–26). After incubation for 10 min at 30 °C or 37 °C, the ABC complex 2 was isolated in the void volume of a Sepharose CL-4B (Pharmacia) gel filtration column equilibrated at room temperature in ABC buffer. To measure DNA replication, the indicated void volume fractions (20 μl) were supplemented with single-stranded DNA-binding protein, primase, DNA polymerase III holoenzyme, magnesium acetate (10 mm final concentration), and other required components at standard amounts for M13 A site ssDNA replication in a final volume of 25 μl, followed by incubation at 30 °C or 37 °C for 10 min, which showed that assembly of the ABC complex is thermolabile.5 In addition, the amounts of M13 A site ssDNA and DnaB were quantified in void volume fractions on ethidium bromide-stained agarose gels or by ELISA, respectively, using known amounts in duplicate or triplicate of the DNA or protein analyzed in parallel to prepare standard curves (25, 26).
Results
The F231S and W233L Substitutions Confer Thermolability in DNA Replication at 37 °C
Using a genetic assay that measures the function of plasmid-borne dnaC alleles to complement a null dnaC strain at 37 °C (17), we identified novel dnaC mutations (Fig. 1). For reasons described above, we focused on mutations encoding F231S and W233L substitutions and found that both were defective in this genetic assay at 37 °C but remained active at 30 °C (Table 1). Substantiating these in vivo results, the purified mutant proteins were comparable with wild type DnaC in their ability to support in vitro DNA replication of an oriC-containing plasmid at 30 °C but were impaired at 37 °C (Fig. 2, A and B). We also analyzed the mutants in DNA replication of a ssDNA (M13 A site) that contains a DnaA box in a hairpin structure (27). In this assay, DnaA binds to the DnaA box, which leads to the loading of the DnaB-DnaC complex onto the ssDNA. After activation of DnaB, the helicase moves on the ssDNA and transiently interacts with primase. This interaction is required for a primer synthesis, which is followed by extension of the primer and DNA synthesis by DNA polymerase III holoenzyme. In assays that contained different concentrations of ATP, W233L and F231S were also thermolabile, but the higher ATP concentration enhanced their activity (Fig. 2, C–F). The effect of ATP is discussed in more detail below. We conclude that the substitutions confer thermolabile activity to DnaC at 37 °C.
TABLE 1.
Mutant DnaCs are defective in DNA replication in vivo at 37 °C but not at 30 °C
| Plasmid | Relative plating efficiencya |
|
|---|---|---|
| 30 °C | 37 °C | |
| pACYC184 | 3.7 × 10−3 | 3.5 × 10−3 |
| pACYC184dnaC | 0.9 | 1 |
| pACYC184dnaCF231S | 1.2 | 1.6 × 10−3 |
| pACYC184dnaCW233L | 0.9 | 3.2 × 10−3 |
a The indicated plasmids were transformed into MF1061 (araD139 Δ(ara, leu)7697 ΔlacX74 galU galK rpsL hsdR2 (rK− mK+) mcrB1 ΔdnaC::cat recA635::kan) carrying the plasmid pAM34dnaCL1-4 (17). Dilutions of the transformation mixture were plated on LB medium augmented with kanamycin (40 μg/ml) and tetracycline (10 μg/ml) and lacking or supplemented with 0.5 mm IPTG followed by incubation at 30 or 37 °C for 24 or 16 h, respectively. The relative plating efficiency is the ratio of the number of colonies observed in the absence of IPTG to that in its presence. The efficiency of transformation in presence of IPTG was 4–6 × 107/μg of plasmid DNA.
FIGURE 2.
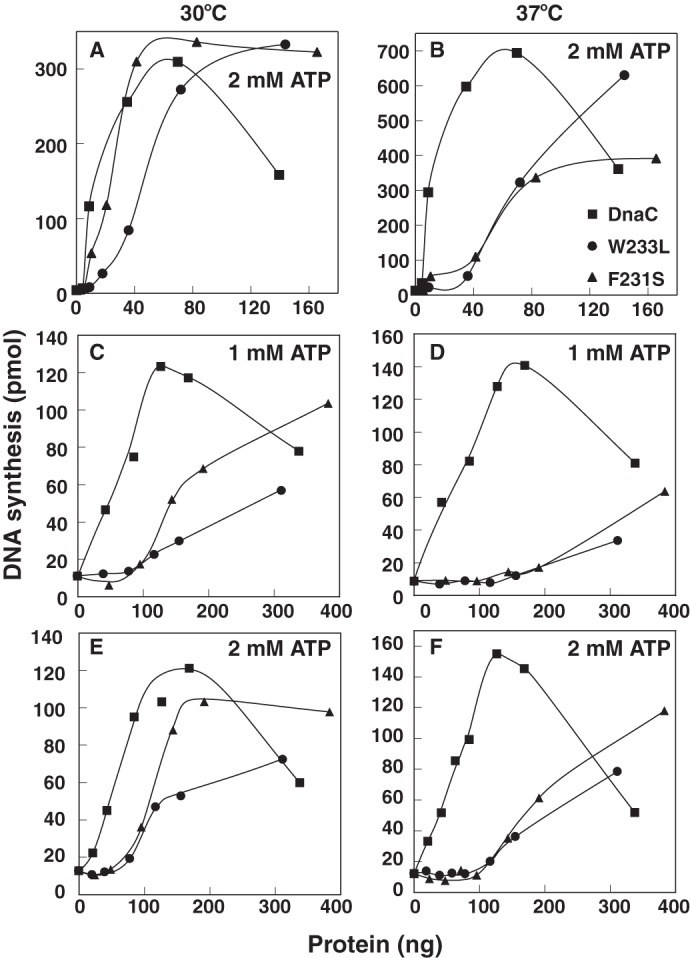
F231S and W233L are thermolabile in DNA replication of an oriC-containing plasmid and M13 A site ssDNA. Reactions containing 1 or 2 mm ATP, as indicated, were assembled with a supercoiled plasmid carrying oriC (M13oriC2LB5) (A and B), or a ssDNA bearing a DnaA box in a hairpin structure (M13 A site) (C–F), purified replication proteins, and the indicated amounts of DnaC, F231S, or W233L as described under “Experimental Procedures.” After incubation for 20 (A and B) or 10 min (C–F) at 30 or 37 °C, DNA synthesis (as pmol of acid-insoluble radioactivity) was measured by liquid scintillation spectrometry. Similar results to those presented were obtained in at least three essentially identical experiments.
The Mutant DnaCs Are Able to Interact with DnaB
Studies indicate that two sites near the N terminus of DnaC interact with DnaB in the DnaB-DnaC complex (10, 28). Hence, we expected that the substitutions would not cause impaired binding to DnaB because they do not reside in these sites. As a test, we immobilized the mutant proteins and wild type DnaC in the wells of a microtiter plate and measured their ability to interact with DnaB at 30 °C and 37 °C, by ELISA (Fig. 3). BSA served as a negative control. Supporting our expectation, the results indicate that the mutants remain able to interact with DnaB at both temperatures. Hence, these observations do not explain their thermolabile activity in DNA replication (Table 1 and Fig. 2).
FIGURE 3.
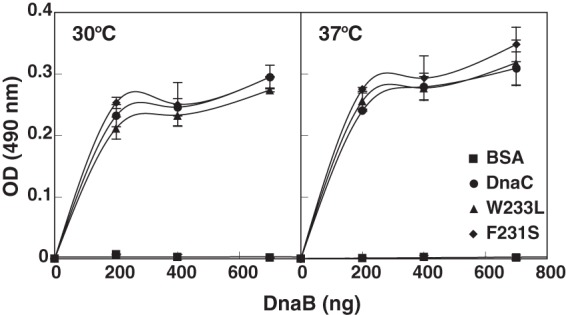
F231S and W233L are able to interact with DnaB. Enzyme-linked immunosorbent assays were performed essentially as described (20) with wild type DnaC, F231S, or W233L (200 ng in 100 μl of a buffer containing 10 mm Na2HPO4, 1.76 mm KH2PO4, 2.7 mm KCl, and 0.137 m NaCl) added per well of a microtiter plate. After incubation at room temperature for 1 h, unbound protein was removed. The indicated amounts of DnaB in triplicate were then added to each well in buffer (100 μl) containing 40 mm HEPES-KOH, pH 8.0, 20 mm Tris-HCl, pH 7.6, 10 mm MgCl2, 4 mm DTT, 1 mm ATP, and 0.1 mg/ml of BSA. After incubation for 1 h at 30 °C or 37 °C, the wells were washed with the same buffer containing 2% nonfat milk. A dilution of affinity-purified antibody that specifically recognizes DnaB was then added to each well followed by incubation at 4 °C overnight. After washing the plate as above, goat anti-rabbit antibody conjugated to horseradish peroxidase (ThermoScientific) was added, and immune complexes were detected colorimetrically at 490 nm. The average and standard deviation have been plotted.
The Mutant DnaCs Interact Aberrantly with ssDNA
The x-ray crystallographic structure of the AAA+ domain of A. aeolicus DnaC belongs to the hexagonal crystal family (12). Its P61 space group is the foundation of the model proposed by Berger and co-workers (12). They described that DnaC, like DnaA, bears two α helices between the Walker A and B boxes that compose the initiator specific motif (Fig. 1) wherein in the presence of ATP the first α helix abuts against the neighboring DnaC molecule of a DnaC oligomer assembled as a right-handed helicoid structure within which ssDNA binds. Their biochemical studies of mutant DnaCs add support, showing that substitution of hydrophobic residues located at the interface between DnaC molecules in the crystal structure led to their inability both to form multimers in a cross-linking assay and to bind to ssDNA.
As described above, we speculated that Phe231 and Trp233 of E. coli DnaC are involved in DNA binding. To test this idea, we performed biosensor assays with an immobilized oligonucleotide and observed reproducible concentration-dependent binding with wild type DnaC and the mutants (Fig. 4, A–C). Relative to wild type DnaC, the sensorgrams indicate an increased affinity of the mutant bearing the F231S substitution and weaker affinity of the other mutant (hereafter referred to as F231S and W233L, respectively) (Fig. 4D).
FIGURE 4.

Compared with wild type DnaC, F231S and W233L interact aberrantly with ssDNA. A–C, the binding of wild type DnaC (A) or each mutant (B and C) at 0.6, 1.25, 2.5, 5, 7.5, and 10 μm to an oligonucleotide (CATCCAATAAATCATACACAAGGC in which the 5′-end was biotinylated, 400 response units) immobilized onto a streptavidin-coated sensor chip was measured using a Biacore 2000 biosensor (GE Healthcare) in buffer containing 50 mm HEPES-KOH, pH 8.0, 30 mm KCl, 5 mm MgCl2, 5% (v/v) glycerol, 0.002% (v/v) Tween 20, 4 mm DTT, and 2 mm ATP. Representative sensorgrams from two essentially identical analyses are shown in which the background signal from a flow cell that lacks streptavidin was subtracted. D, using BIAevaluation 3.1 software, kinetic constants and the equilibrium dissociation constant (KD) of wild type DnaC were determined by fitting sensorgrams (A, dashed lines) to a global kinetic model. The sensorgrams obtained with F231S and W233L could not be fitted, so KD values were estimated by separately fitting association and dissociation curves. E and F, UV cross-linking of a 5′-labeled oligonucleotide (60 fmol) with 0.4 μm DnaB (monomer) where indicated, and 2 μm of either DnaC, F231S, or W233L was performed essentially as described (29), but with minor modifications (see “Experimental Procedures”). DnaB, wild type DnaC, and the mutants of this experiment were isolated as described under “Experimental Procedures” in which the final step of purification was by gel permeation chromatography on Superose 12 or Superdex 75 10/30 columns, respectively (GE Healthcare). DnaB eluted at the position expected for hexameric DnaB; DnaC and the mutants eluted at the position of monomeric DnaC. Following cross-linking at 30 or 37 °C, molecular weight markers (phosphorylase B (97 kDa), BSA (68 kDa), DnaB (52 kDa), ovalbumin (45 kDa), and DnaC (28 kDa)) separated by SDS-polyacrylamide gel electrophoresis were detected by Coomassie Brilliant Blue staining before autoradiography. The asterisk indicates the position of DnaB cross-linked to the oligonucleotide. In E, lanes 2–6 were rearranged by image editing for consistency with F. Results similar to those presented here were obtained in five essentially identical experiments.
The ability of DnaC to interact with ssDNA was first demonstrated by UV cross-linking of DnaC to a radiolabeled ssDNA in McMacken laboratory (29). Visualized by autoradiography after electrophoresis on an SDS-polyacrylamide gel, the formation of cross-linked complexes of ∼45 and 97 kDa ascribed to contain one and two DnaC monomers, respectively, was stimulated by DnaB and an adenine nucleotide. To compare with our biosensor experiments, we used this independent method to examine the ability of the mutants to become cross-linked to an oligonucleotide of the same length as that in the study described above. We also examined the influence of DnaB and temperature on formation of cross-linked complexes. With wild type DnaC under conditions that support formation of the DnaB-DnaC complex (Fig. 4, E and F, lane 3), we observed cross-linked complexes whose electrophoretic mobilities are similar to those described by McMacken and co-workers (29). If the complex near the position of the 45-kDa marker forms by a single cross-link to the oligonucleotide, additional cross-links between wild type DnaC and the oligonucleotide may explain the electrophoretic mobilities of the range of complexes that migrate more rapidly (see the vertical bars in Fig. 4, E and F).
With F231S and W233L, cross-linked complexes were observed of similar mobility as those seen with wild type DnaC, but their relative abundance differed (Fig. 4, E and F, lanes 4 and 5, respectively, compared with lane 3). In contrast with wild type DnaC, a range of complexes was also observed with F231S that migrated more slowly than the 97 kDa marker at 30 °C whose abundance was less at 37 °C.
We also observed a nominal level of oligonucleotide-cross-linked DnaB (see the symbol that denotes its position in Fig. 4, E and F, lane 2), which increased when ATPγS replaced ATP in the reaction (data not shown).5 The latter observation correlates with studies that AMP-PNP or ATPγS stabilizes DnaB bound to ssDNA (30–32). With oligo(dT)20, oligo(dT)56, or oligo(dT)70, a single protomer of the DnaB hexamer was determined to be cross-linked (31). In contrast, this complex is easily seen in reactions that also contain wild type DnaC, F231S, or W233L but not DnaCΔ51 (Fig. 4, E and F, lanes 3–6). The latter lacks the N-terminal 51 residues of wild type DnaC and amino acids that interact directly with DnaB to form the DnaB-DnaC complex. Together, these results indicate that the DnaB-DnaC complex leads to the elevated cross-linking of DnaB.
Compared with reactions supplemented with DnaB, its absence led to marginal cross-linking of wild type DnaC to the oligonucleotide (Fig. 4, E and F, lanes 3 and 7). This negligible photo-cross-linking affirms the weak affinity of DnaC for ssDNA (KD = 2.2 × 10−6; Fig. 4D; 3–4 × 10−6 m (13); 3–7 × 10−7 m (19)), and observations from the original UV cross-linking studies (29). In contrast with wild type DnaC, the greater abundance of oligonucleotide-cross-linked F231S at and slightly above the 97 kDa marker (Fig. 4, E and F, lane 8) confirms its stronger affinity for ssDNA as measured in biosensor assays (Fig. 4B) and suggests an altered interaction with the ssDNA. Of interest and despite the isolation of F231S as a monomer by gel permeation chromatography (“Experimental Procedures”), the increased relative abundance of this complex suggests that F231S is prone to self-oligomerize under the conditions of the assay. In comparison, the complex containing W233L migrated more rapidly than the 45-kDa marker (Fig. 4, E and F, lane 9). The greater abundance of this complex compared with wild type DnaC is not inconsistent with its weaker affinity; unlike our biosensor experiments, UV cross-linking does not necessarily reflect the affinity of a protein for DNA. Also, we note that this complex formed with either wild type DnaC or the mutants does not co-migrate with the complex above the 45-kDa marker produced in the presence of DnaB and thought to contain one DnaC molecule (Fig. 4, E and F, lanes 7–9 versus lanes 3–5). Presumably, the position of the cross-link in DnaC or the oligonucleotide is different in these complexes. To summarize, this set of experiments indicates that the respective substitutions cause aberrant ssDNA binding. In addition, DnaB greatly stimulates cross-linking of wild type DnaC, F231S, and W233L to ssDNA, but the complexes formed with the mutants differ in their relative abundance, which presumably arise from their aberrant ssDNA binding. The comparable results at 30 and 37 °C contrast with the thermolabile activity of the mutants in DNA replication.
Impaired ATP Binding by F231S and W233L
Studies show that DnaC specifically binds albeit weakly (KD = 6–9 μm) to adenine-containing nucleotides (11, 33, 34), which is required for DnaC function (12, 13). Because UV cross-linking showed a minimal effect of temperature on DNA binding by F231S and W233L, we considered the possibility that the mutants may be thermolabile in ATP binding to explain their impaired replication activity at 37 °C. UV cross-linking has been used to measure the weak ATP binding activity of DnaC (10, 13). With reactions incubated at 30 °C, we were able to cross-link [α-32P]ATP (0.17 μm) to wild type DnaC but not to the mutants (Fig. 5A). Control reactions showed that the inclusion of an excess of unlabeled ATP blocked the cross-linking of the radiolabeled ATP to wild type DnaC and that substitution of DnaC with BSA failed to cross-link it to [α-32P]ATP. Together, these results strongly suggest that the assay conditions measure ATP binding by DnaC and that the mutants are defective in ATP binding. Because ATP binding by DnaC is required for its function (12, 13), the quandary is the lack of an explanation for the DNA replication activity of the mutants at 30 °C (Table 1 and Fig. 2).
FIGURE 5.
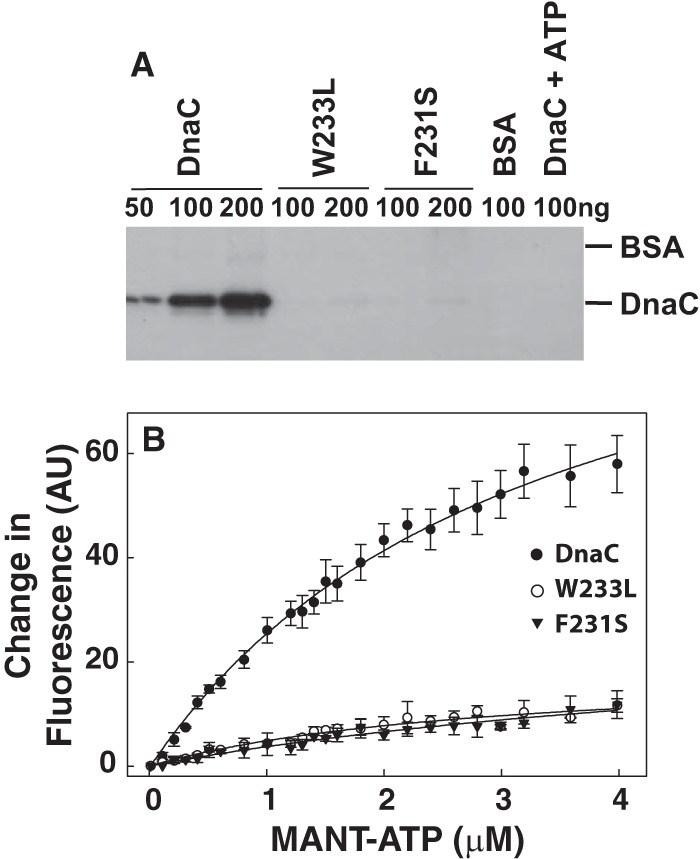
F231S and W233L are impaired in ATP-binding. A, duplicate samples of wild type DnaC, F231S, or W233L at the indicated amounts were incubated with [α-32P]ATP (10 μCi, 0.17 μm per reaction) for 2 min at 30 °C and then irradiated with UV light for 15 min on ice as described (10, 13). As negative controls, BSA was used in place of DnaC or DnaC was incubated in the presence of both [α-32P]ATP and 0.1 mm ATP. One of each pair of samples was then analyzed on separate 15% SDS-polyacrylamide gels. After staining the gels with Coomassie Brilliant Blue to determine the electrophoretic positions of DnaC and BSA, autoradiography of the dried gel was performed. Because the autoradiograms of each gel were essentially identical, only one is shown. Similar results to those presented were obtained in three essentially identical experiments. B, reactions (200 μl) containing 1.5 μm DnaC, F231S, or W233L and the indicated concentrations of MANT-ATP were incubated at 25 °C as described under “Experimental Procedures.” After excitation at 356 nm, emission from 400 to 550 nm revealed an emission maximum at 440 nm. The average change in fluorescence at 440 nm and standard error from three independent measurements were plotted versus the concentration of MANT-ATP. With the mutants, almost no binding was observed.
To attempt to resolve this dilemma, we measured nucleotide binding using a more sensitive method that relies on a fluorescent ATP analogue (MANT-ATP). As described by others (33), the binding of DnaC to MANT-ATP causes a fluorescence increase of ∼3-fold in concert with a shift of the emission wavelength from 450 to ∼440 nm. After confirming these observations in pilot experiments, we measured the change in fluorescence at 440 nm and 25 °C with increasing concentrations of MANT-ATP and a constant level of wild type DnaC (Fig. 5B). The calculated affinity of binding (KD) of 3.3 ± 0.24 μm is comparable with reported values (11, 33, 34). Under essentially identical conditions, we were unable to quantify the binding affinities of W233L and F231S because the fluorescence changes were marginal. Considering the negligible binding of the mutants to MANT-ATP at 25 °C together with their activity in DNA replication at 30 °C but not at 37 °C (Table 1 and Fig. 2), we did not expect that higher temperatures would lead to measurable binding of MANT-ATP.
Given that the mutants are active at 30 °C (Table 1 and Fig. 2), which contrasts with their feeble nucleotide binding at up to 4 μm MANT-ATP, the results suggest that a higher ATP concentration supports ATP binding to overcome this potentially disabling defect. In vivo, ATP is estimated at ∼1–3 mm (35); in reactions to measure DNA replication (Fig. 2), 1 or 2 mm ATP was included as indicated.
F231S and W233L Are Thermolabile at the Step of Loading of the DnaB-DnaC Complex at oriC or in the Subsequent Step of Helicase Activation
DnaA bound to oriC induces its localized unwinding and then assists in the loading of the DnaB-DnaC complex onto the unwound DNA. At this stage, ATP bound to DnaC is needed, after which ATP hydrolysis is required to release DnaC from DnaB (11, 13, 36). DnaB is then active as a DNA helicase, unwinding the parental duplex DNA so that each DNA strand can be copied by DNA polymerase III holoenzyme. We speculated that the mutants may be thermolabile at the step of loading of the DnaB-DnaC complex at oriC or in dissociating from DnaB, which is required for its activation. To address these possibilities, we examined the mutants in an assay that measures the production of a highly negatively supercoiled DNA named Form I* (37). Without other replication proteins required for DNA replication, the unwinding of a supercoiled plasmid carrying oriC by DnaB generates negative supercoils behind it and positive supercoils ahead of the translocating helicase. The action of DNA gyrase to remove the positive supercoils results in Form 1* DNA that can be separated from the naturally supercoiled oriC-containing plasmid by agarose gel electrophoresis and then quantified. In reactions at 30 °C containing 2 mm ATP, we found that F231S and W233L were similar in activity with wild type DnaC (Fig. 6A). In comparison (Fig. 6B), their lower activity at 1 mm ATP and 30 °C supports the idea that the higher ATP concentration partially alleviates their defective ATP binding activity. A higher ATP concentration (2 mm) similarly enhanced the activity of the mutants in DNA replication (Fig. 2, A and E versus C). Because the assay of Fig. 6 measures both the loading of the DnaB-DnaC complex and activation of the helicase, the results obtained by incubation at 37 °C show that the mutants are thermolabile in one or both of these activities.
FIGURE 6.

F231S and W233L are thermolabile in the formation of a highly negatively supercoiled DNA named Form I*. Reactions (25 μλ) contained 2 mm (A) or 1 mm (B) of ATP, DnaC, F231S, or W233L, a supercoiled plasmid carrying oriC (M13oriC2LB5, 200 ng), HU, single-stranded DNA-binding protein, DnaA, DnaB, and DNA gyrase as described (44). After incubation for 25 min at 30 or 37 °C, SDS and EDTA were added to final concentrations of 1% and 10 mm, respectively and then incubated for 4 min at 65 °C. After agarose gel electrophoresis, the electrophoretically separated DNAs were visualized by ethidium bromide staining. The reverse images are shown in which the leftmost lanes of each gel lack DnaC. In addition, the amounts of Form I* were quantified and normalized relative to the optimal level observed with wild type DnaC.
F231S and W233L Are Thermolabile in Loading the DnaB-DnaC Complex
With M13 A site ssDNA, DnaA directs the loading of a single DnaB-DnaC complex at a site near the DnaA box-containing hairpin to form an intermediate named the ABC complex (27). This intermediate can be isolated, and the proteins contained in it can be quantified by molecular methods (27). If the mutants are thermolabile in helicase loading, the expectation is that the stoichiometric ratio of DnaB to ssDNA will be less compared with the control in which the complex is assembled with wild type DnaC. If the mutants are thermolabile in helicase activation, the ratio of DnaB to ssDNA will be comparable when both mutants and wild type DnaC are used to assemble the complex at 37 °C. Experiments performed to discriminate between these possibilities show that the ratio of DnaB to ssDNA is comparable when the complex was assembled with the mutants or wild type DnaC in 1 mm ATP at 30 °C (Fig. 7). The values of 4–5 DnaB protomers support the presence of a DnaB hexamer in the ABC complex and the results of a previous study (27). In experiments performed in parallel but under conditions in which assembly was at 37 °C, the substantially reduced stoichiometry of DnaB when the complex was formed with the mutants but not with wild type DnaC indicates that the mutants are thermolabile at the step of loading the DnaB-DnaC complex.
FIGURE 7.
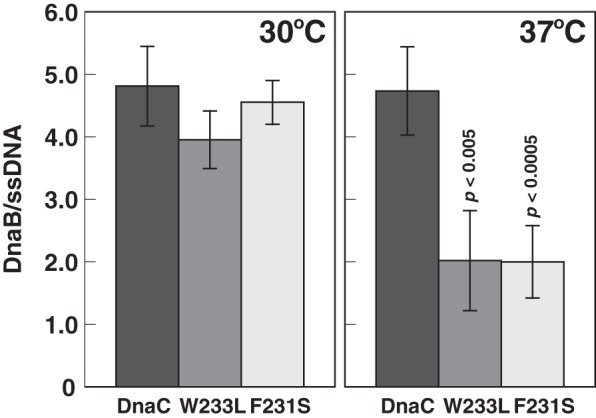
F231S and W233L are thermolabile in helicase loading. DnaA, DnaB, and DnaC were assembled onto M13 A site ssDNA at 30 or 37 °C. A portion of the sample equivalent to a standard assay to measure DNA replication was supplemented by the addition of primase, DNA polymerase III*, the β clamp and other required components at amounts used in a standard assay prior to isolation of the complex by gel filtration chromatography (Sepharose CL-4B; Pharmacia) as described under “Experimental Procedures.” The activity of the complex after isolation was also measured after adding required proteins and other components as described above to 20 μl of void volume fractions. The stoichiometry of DnaB, which represents the ratio of the DnaB-DnaC complex bound to the ssDNA, was then determined by quantitative analysis of the isolated complex. The mean and standard deviation from six or four independent analyses of isolated complexes from three or two experiments incubated at 37 or 30 °C, respectively, are shown in which the background ratios of nonspecifically bound DnaB per ssDNA in the absence of DnaA has been subtracted. These ratios ranged from 0.04 to 0.57. Student's t test analysis (two-tailed, two-sample equal variance) of the reduction at 37 °C compared with 30 °C yield p values of <0.005 for W233L and <0.0005 for F231S.
Discussion
Phe231 and Trp233 Contribute to the Native Structure of DnaC and Are Implicated in ssDNA Binding
In AAA+ proteins, a conserved arginine referred to as Sensor II is thought to make contact with the γ-PO4 of ATP (reviewed in Ref. 38). Mutational analysis shows that this residue in specific AAA+ proteins is involved in ATP binding or its hydrolysis and also may discriminate between ADP and ATP (reviewed in Ref. 38). The Berger laboratory found that Arg226, which is invariant among DnaC orthologues, contacts the BeF3 moiety in the x-ray crystallographic structure of the AAA+ domain of A. aeolicus DnaC perfused with ADP-BeF3 and proposed that this residue (Arg236 for E. coli DnaC) is the Sensor II arginine (12). Like the corresponding residue in DnaA (39, 40), for which DnaA and DnaC are considered paralogues (41), its substitution with alanine only modestly affected ATP binding (12). Of interest, residues that precede this arginine are in a loop in the x-ray structure of the AAA+ domain of A. aeolicus DnaC containing ADP-BeF3 but not ADP. Hence, the Sensor II arginine engaged with the γ-PO4 of ATP may stabilize the loop. ATP also increases the affinity of DnaC for ssDNA, but the loop is not an obligate requirement as the affinity of DnaC for ssDNA only increases 2–3-fold (13, 19). For E. coli DnaC, Arg236 corresponds to the Sensor II residue; Phe231 and Trp233 reside in the loop in a homology model of E. coli DnaC constructed using the above x-ray structure of A. aeolicus DnaC as a template (28).
Our experiments with MANT-ATP show that the F231S and W233L substitutions greatly reduce the affinity of DnaC for this fluorescent ATP analogue. Under the premise that the interaction of the Sensor II residue with ATP forms the loop, these substitutions ostensibly alter the conformation of the loop to affect ATP binding. The weak affinity of W233L for ATP also correlates with biosensor measurements of its reduced affinity for ssDNA, suggesting that ATP is only modestly effective in stimulating ssDNA binding by this mutant. Alternatively, if Trp233 interacts directly with ssDNA, the substitution may weaken the interaction of DnaC with ssDNA.
In contrast, biosensor assays measured an increased affinity of F231S for ssDNA, which correlates with results that this mutant in the absence of DnaB photo-cross-linked to ssDNA more readily than wild type DnaC. The greater abundance of cross-linked complexes of ≥97 kDa that likely contain more than one F231S molecule suggests that this mutant has an increased propensity to self-oligomerize, supporting other evidence that ssDNA binding by DnaC requires its self-oligomerization (12). In addition, the greater affinity of F231S for ssDNA suggests that Phe231 negatively regulates both self-oligomerization and ssDNA binding.
On the basis of the UV cross-linking assay, which reflects the relative proximity of an amino acid with the base of a nucleotide after formation of a free radical (reviewed in Ref. 42), F231S and W233L interact aberrantly with ssDNA compared with wild type DnaC. Evidently, the substituted residues lead to altered contacts between the mutant DnaCs and the ssDNA to affect the abundance of cross-linked complexes. We do not have a molecular understanding of how this occurs despite knowing their locations in a homology model of E. coli DnaC.
F231S and W233L Are Thermolabile in Helicase Loading
The assay that measures the formation of a highly underwound DNA named Form I* reflects the loading of the DnaB-DnaC complex at oriC and activation of DnaB. Presumably, DnaC is able to dissociate spontaneously from DnaB to unmask its helicase activity under these nonphysiological conditions that lack primase and DNA polymerase III in comparison with the primase-dependent mechanism (13). The mutants' inability to produce Form I* DNA at an elevated temperature indicates that they are thermolabile in either loading of the DnaB-DnaC complex or activating DnaB. To distinguish between these two possibilities, we compared the relative amounts of DnaB present in the ABC complex formed with the mutants or wild type DnaC. In contrast with the similar ratios of DnaB retained in this complex at 30 °C, the lower DnaB levels observed with the mutants at 37 °C indicate that they are thermolabile in supporting the loading of the DnaB-DnaC complex. These results explain their reduced activity in DNA replication of an oriC-containing plasmid and of the ssDNA bearing a DnaA box in a hairpin structure at the elevated temperature.
A mutant DnaC bearing a K112R substitution in the Walker A box is defective in ATP binding and fails to place the DnaB-DnaC complex at oriC (11). Hence, DnaC must bind ATP for its assembly as a complex with DnaB at oriC. However, this mutant, like F231S and W233L, retains its ability to interact with DnaB, confirming that ATP binding by DnaC is not required for it to form the DnaB-DnaC complex (11, 32, 43). For F231S and W233L, their ability to interact with DnaB or with ssDNA is not inhibited at an elevated temperature. Considering that DnaC in the DnaB-DnaC complex is thought to engage each unwound DNA strand within oriC (12), these observations taken together suggest that the mutants should be active in helicase loading. The improved activity of F231S and W233L in Form I* production at 2 mm ATP compared with 1 mm ATP suggests that the elevated ATP level at both temperatures mitigates their defective ATP binding activity. Despite this, the results also suggest that ATP binding is thermolabile. Apparently, their thermolability in ATP binding causes their defective activity in helicase loading at an elevated temperature, which may be exacerbated by their aberrant binding to ssDNA.
Author Contributions
M. M. F. purified the mutant proteins and performed the genetic and biochemical assays, J. M. S. and K. H.-K. isolated the dnaC mutations and characterized them in preliminary genetic experiments, S. A. assisted in biosensor experiments and analyzed the data, and M. M. F. and J. M. K. (who conceived the study) discussed and interpreted the results and wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Drs. Sundari Chodavarapu and Lisa Boughner (Michigan State University) for suggestions and for optimizing growth conditions for the induced expression of the mutant DnaCs. We also thank Charles Najt for help with fluorescence measurements.
This work was supported by Grant R01 GM090063 from the National Institutes of Health and by the USDA National Institute of Food and Agriculture, Hatch Project MICL02370, and Michigan AgBioResearch. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
M. M. Felczak and J. M. Kaguni, unpublished results.
- ssDNA
- single-stranded DNA
- IPTG
- isopropyl β-d-thiogalactopyranoside.
References
- 1. Bell S. P., and Kaguni J. M. (2013) Helicase loading at chromosomal origins of replication. Cold Spring Harb. Perspect. Biol. 5, a010124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Donate L. E., Llorca O., Bárcena M., Brown S. E., Dixon N. E., and Carazo J. M. (2000) pH-controlled quaternary states of hexameric DnaB helicase. J. Mol. Biol. 303, 383–393 [DOI] [PubMed] [Google Scholar]
- 3. Yang S., Yu X., VanLoock M. S., Jezewska M. J., Bujalowski W., and Egelman E. H. (2002) Flexibility of the rings: structural asymmetry in the DnaB hexameric helicase. J. Mol. Biol. 321, 839–849 [DOI] [PubMed] [Google Scholar]
- 4. Bailey S., Eliason W. K., and Steitz T. A. (2007) Structure of hexameric DnaB helicase and its complex with a domain of DnaG primase. Science 318, 459–463 [DOI] [PubMed] [Google Scholar]
- 5. Lo Y. H., Tsai K. L., Sun Y. J., Chen W. T., Huang C. Y., and Hsiao C. D. (2009) The crystal structure of a replicative hexameric helicase DnaC and its complex with single-stranded DNA. Nucleic Acids Res. 37, 804–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arai K., Low R. L., and Kornberg A. (1981) Movement and site selection for priming by the primosome in phage phi X174 DNA replication. Proc. Natl. Acad. Sci. U.S.A. 78, 707–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Biswas E. E., and Biswas S. B. (1999) Mechanism of DNA binding by the DnaB helicase of Escherichia coli: analysis of the roles of domain gamma in DNA binding. Biochemistry 38, 10929–10939 [DOI] [PubMed] [Google Scholar]
- 8. Jezewska M. J., Rajendran S., and Bujalowski W. (1997) Strand specificity in the interactions of Escherichia coli primary replicative helicase DnaB protein with a replication fork. Biochemistry 36, 10320–10326 [DOI] [PubMed] [Google Scholar]
- 9. Jezewska M. J., Rajendran S., Bujalowska D., and Bujalowski W. (1998) Does single-stranded DNA pass through the inner channel of the protein hexamer in the complex with the Escherichia coli DnaB helicase?: fluorescence energy transfer studies. J. Biol. Chem. 273, 10515–10529 [DOI] [PubMed] [Google Scholar]
- 10. Ludlam A. V., McNatt M. W., Carr K. M., and Kaguni J. M. (2001) Essential amino acids of Escherichia coli DnaC protein in an N-terminal domain interact with DnaB helicase. J. Biol. Chem. 276, 27345–27353 [DOI] [PubMed] [Google Scholar]
- 11. Davey M. J., Fang L., McInerney P., Georgescu R. E., and O'Donnell M. (2002) The DnaC helicase loader is a dual ATP/ADP switch protein. EMBO J. 21, 3148–3159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mott M. L., Erzberger J. P., Coons M. M., and Berger J. M. (2008) Structural synergy and molecular crosstalk between bacterial helicase loaders and replication initiators. Cell 135, 623–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Makowska-Grzyska M., and Kaguni J. M. (2010) Primase directs the release of DnaC from DnaB. Mol. Cell 37, 90–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hupert-Kocurek K., and Kaguni J. M. (2011) Understanding protein function: the disparity between bioinformatics and molecular methods. In Computational Biology and Applied Bioinformatics (Lopes H., ed) InTech, Rijeka, Croatia [Google Scholar]
- 15. Liu B., Eliason W. K., and Steitz T. A. (2013) Structure of a helicase-helicase loader complex reveals insights into the mechanism of bacterial primosome assembly. Nat. Commun. 4, 2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arias-Palomo E., O'Shea V. L., Hood I. V., and Berger J. M. (2013) The bacterial DnaC helicase loader is a DnaB ring breaker. Cell 153, 438–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hupert-Kocurek K., Sage J. M., Makowska-Grzyska M., and Kaguni J. M. (2007) Genetic method to analyze essential genes of Escherichia coli. Appl. Environ. Microbiol. 73, 7075–7082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Theobald D. L., Mitton-Fry R. M., and Wuttke D. S. (2003) Nucleic acid recognition by OB-fold proteins. Annu. Rev. Biophys. Biomol. Struct. 32, 115–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Biswas S. B., Flowers S., and Biswas-Fiss E. E. (2004) Quantitative analysis of nucleotide modulation of DNA binding by the DnaC protein of Escherichia coli. Biochem. J. 379, 553–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chodavarapu S., Gomez R., Vicente M., and Kaguni J. M. (2008) Escherichia coli Dps interacts with DnaA protein to impede initiation: a model of adaptive mutation. Mol. Microbiol. 67, 1331–1346 [DOI] [PubMed] [Google Scholar]
- 21. Marszalek J., and Kaguni J. M. (1992) Defective replication activity of a dominant-lethal dnaB gene product from Escherichia coli. J. Biol. Chem. 267, 19334–19340 [PubMed] [Google Scholar]
- 22. Chodavarapu S., Felczak M. M., and Kaguni J. M. (2011) Two forms of ribosomal protein L2 of Escherichia coli that inhibit DnaA in DNA replication. Nucleic Acids Res. 39, 4180–4191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carr K. M., and Kaguni J. M. (2001) Stoichiometry of DnaA and DnaB protein in initiation at the Escherichia coli chromosomal origin. J. Biol. Chem. 276, 44919–44925 [DOI] [PubMed] [Google Scholar]
- 24. Felczak M. M., and Kaguni J. M. (2004) The box VII motif of Escherichia coli DnaA protein is required for DnaA oligomerization at the E. coli replication origin. J. Biol. Chem. 279, 51156–51162 [DOI] [PubMed] [Google Scholar]
- 25. Felczak M. M., Simmons L. A., and Kaguni J. M. (2005) An essential tryptophan of Escherichia coli DnaA protein functions in oligomerization at the E. coli replication origin. J. Biol. Chem. 280, 24627–24633 [DOI] [PubMed] [Google Scholar]
- 26. Chodavarapu S., Felczak M. M., Yaniv J. R., and Kaguni J. M. (2008) Escherichia coli DnaA interacts with HU in initiation at the E. coli replication origin. Mol. Microbiol. 67, 781–792 [DOI] [PubMed] [Google Scholar]
- 27. Carr K. M., and Kaguni J. M. (2002) Escherichia coli DnaA protein loads a single DnaB helicase at a DnaA box hairpin. J. Biol. Chem. 277, 39815–39822 [DOI] [PubMed] [Google Scholar]
- 28. Chodavarapu S., Jones A. D., Feig M., and Kaguni J. M. (2016) DnaC traps DnaB as an open ring and remodels the domain that binds primase. Nucleic Acids Res. 44, 210–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Learn B. A., Um S. J., Huang L., and McMacken R. (1997) Cryptic single-stranded-DNA binding activities of the phage lambda P and Escherichia coli DnaC replication initiation proteins facilitate the transfer of E. coli DnaB helicase onto DNA. Proc. Natl. Acad. Sci. U.S.A. 94, 1154–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Arai K., and Kornberg A. (1981) Mechanism of dnaB protein action: III. allosteric role of ATP in the alteration of DNA structure by dnaB protein in priming replication. J. Biol. Chem. 256, 5260–5266 [PubMed] [Google Scholar]
- 31. Jezewska M. J., Kim U. S., and Bujalowski W. (1996) Binding of Escherichia coli primary replicative helicase DnaB protein to single-stranded DNA. Long-range allosteric conformational changes within the protein hexamer. Biochemistry 35, 2129–2145 [DOI] [PubMed] [Google Scholar]
- 32. Biswas S. B., and Biswas-Fiss E. E. (2006) Quantitative analysis of binding of single-stranded DNA by Escherichia coli DnaB helicase and the DnaB-DnaC complex. Biochemistry 45, 11505–11513 [DOI] [PubMed] [Google Scholar]
- 33. Galletto R., Rajendran S., and Bujalowski W. (2000) Interactions of nucleotide cofactors with the Escherichia coli replication factor DnaC protein. Biochemistry 39, 12959–12969 [DOI] [PubMed] [Google Scholar]
- 34. Wahle E., Lasken R. S., and Kornberg A. (1989) The dnaB-dnaC replication protein complex of Escherichia coli. I. Formation and properties. J. Biol. Chem. 264, 2463–2468 [PubMed] [Google Scholar]
- 35. Neuhard J., and Nygaard P. (1987) Purines and pyrimidines. In Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology (Neidhardt F. C., Ingraham J. L., Low K. B., Magasanik B., Schaechter M., and Umbarger H. E., eds) pp. 445–473, ASM Press, Washington, D.C. [Google Scholar]
- 36. Wahle E., Lasken R. S., and Kornberg A. (1989) The dnaB-dnaC replication protein complex of Escherichia coli: II. role of the complex in mobilizing dnaB functions. J. Biol. Chem. 264, 2469–2475 [PubMed] [Google Scholar]
- 37. Baker T. A., Sekimizu K., Funnell B. E., and Kornberg A. (1986) Extensive unwinding of the plasmid template during staged enzymatic initiation of DNA replication from the origin of the Escherichia coli chromosome. Cell 45, 53–64 [DOI] [PubMed] [Google Scholar]
- 38. Ogura T., Whiteheart S. W., and Wilkinson A. J. (2004) Conserved arginine residues implicated in ATP hydrolysis, nucleotide-sensing, and inter-subunit interactions in AAA and AAA+ ATPases. J. Struct. Biol. 146, 106–112 [DOI] [PubMed] [Google Scholar]
- 39. Nishida S., Fujimitsu K., Sekimizu K., Ohmura T., Ueda T., and Katayama T. (2002) A nucleotide switch in the Escherichia coli DnaA protein initiates chromosomal replication: evidnece from a mutant DnaA protein defective in regulatory ATP hydrolysis in vitro and in vivo. J. Biol. Chem. 277, 14986–14995 [DOI] [PubMed] [Google Scholar]
- 40. Su'etsugu M., Shimuta T. R., Ishida T., Kawakami H., and Katayama T. (2005) Protein associations in DnaA-ATP hydrolysis mediated by the Hda-replicase clamp complex. J. Biol. Chem. 280, 6528–6536 [DOI] [PubMed] [Google Scholar]
- 41. Koonin E. V. (1992) DnaC protein contains a modified ATP-binding motif and belongs to a novel family of ATPases including also DnaA. Nucleic Acids Res. 20, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Meisenheimer K. M., and Koch T. H. (1997) Photocross-linking of nucleic acids to associated proteins. Crit. Rev. Biochem. Mol. Biol. 32, 101–140 [DOI] [PubMed] [Google Scholar]
- 43. Galletto R., Jezewska M. J., and Bujalowski W. (2003) Interactions of the Escherichia coli DnaB helicase hexamer with the replication factor the DnaC protein: effect of nucleotide cofactors and the ssDNA on protein-protein interactions and the topology of the complex. J. Mol. Biol. 329, 441–465 [DOI] [PubMed] [Google Scholar]
- 44. Marszalek J., Zhang W., Hupp T. R., Margulies C., Carr K. M., Cherry S., and Kaguni J. M. (1996) Domains of DnaA protein involved in interaction with DnaB protein, and in unwinding the Escherichia coli chromosomal origin. J. Biol. Chem. 271, 18535–18542 [DOI] [PubMed] [Google Scholar]