

Abstract
AMPA receptors (AMPARs) mediate fast excitatory neurotransmission in the central nervous system. Functional AMPARs are tetrameric complexes with a highly modular structure, consisting of four evolutionarily distinct structural domains: an amino-terminal domain (ATD), a ligand-binding domain (LBD), a channel-forming transmembrane domain (TMD), and a carboxyl-terminal domain (CTD). Here we show that the isolated TMD of the GluA1 AMPAR is fully capable of tetramerization. Additionally, removal of the extracellular domains from the receptor did not affect membrane topology or surface delivery. Furthermore, whereas the ATD and CTD contribute positively to tetramerization, the LBD presents a barrier to the process by reducing the stability of the receptor complex. These experiments pinpoint the TMD as the “tetramerization domain” for AMPARs, with other domains playing modulatory roles. They also raise intriguing questions about the evolution of iGluRs as well as the mechanisms regulating the biogenesis of AMPAR complexes.
Keywords: endoplasmic reticulum (ER), ion channel, ionotropic glutamate receptor, protein assembly, synapse, amino-terminal domain, ligand-binding domain, native PAGE
Introduction
Ionotropic glutamate receptors (iGluRs),2 predominantly the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor subtype, mediate fast excitatory neurotransmission in the mammalian central nervous systems. Functional AMPARs are tetramers consisting of four identical (homomeric) or similar (heteromeric) subunits (1) arranged as a dimer-of-dimers (2–4). The process of AMPAR assembly determines the availability of AMPARs to be trafficked to the cell surface, and therefore represents a target for modulating glutamatergic signaling (5).
iGluR subunits are highly modular, being composed of four well defined structural domains (6, 7) possessing distinct evolutionary origins (8): an amino-terminal domain (ATD), a ligand-binding domain (LBD), a transmembrane (TMD) consisting of three hydrophobic segments (M1, M3 and M4) as well as a re-entrant loop (M2), and a carboxyl-terminal (CTD) domain attached to the intracellular end of M4 (9, 10). Previous studies have identified structural elements that contribute to the assembly of AMPARs in almost every domain (5, 11, 12). The ATD of AMPARs forms dimers in solution and drive the initial dimerization process (13–15). Alternative splicing in the LBD (flip/flop) influences subunit composition of AMPAR heteromers, potentially by affecting the tetramerization process (16). Residues in the TMD, especially those near the channel pore (e.g. the Q/R editing site), also impact tetramerization (3, 17). Recent studies pinpointed the M4 segment, which is unique to eukaryotic iGluRs, as a critical determinant of AMPAR tetramerization (18, 19). Despite the large number of studies investigating individual factors influencing AMPAR assembly, the relative contribution of each structural domain is unclear.
Here, we took advantage of the highly modular nature of the iGluR structure to address the assembly process by deleting entire domains from an AMPAR subunit, GluA1. We demonstrate that the TMD is the minimal structural unit required for AMPAR tetramerization, highlighting the central role of this domain to the assembly process. The three other domains modulate tetramerization of AMPARs but are not required. Surprisingly, whereas the ATD and CTD contribute positively to tetramerization, the LBD presents an unexpected hindrance to the process. Our findings hint at previously unknown mechanisms through which AMPAR biogenesis could be regulated. They may also help inspire novel therapeutic strategies that modulate glutamatergic signaling by targeting the assembly of AMPARs.
Experimental Procedures
Construct Design, Mutagenesis, and Expression
All constructs were designed based on the “flip” variant of the rat GluA1 (accession number P19490) subunit. Constructs lacking the amino-terminal domain (ΔATD) of GluA1 were tagged with hemagglutinin (HA) near the N termini. The cDNA encoding the HA-GluA1-ΔATD construct was generated by introducing restriction sites (EcoRV and AscI) at positions flanking the ATD (Ala-1 and Asp-376) and then excising the entire ATD-encoding region and replacing it with a segment encoding the HA tag. The GluA1-ΔCTD and HA-GluA1-ΔATD/ΔCTD constructs were created by substituting residue Ser-814 with a stop codon in full-length GluA1 and HA-GluA1-ΔATD, respectively. The HA-GluA1-ΔECD construct was ordered from GeneScript and contains all LBD-TMD linkers as well as a stretch of 5 Gly-Thr (GTs) between the M3 and M4 segments to replace the S2 segment of LBD. The full amino acid sequence is as follows: MPYIFAFFCTGFLGAVVGANFYPYDVPDYAPNNIQIGGLFPNKPQKSKPGVFSFLDPLAYEIWMCIVFAYIGVSVVLFLVSRFSPYEWHSEEFEEGRDQTTSDQSNEFGIFNSLWFSLGAFMQQGCDISPRSLSGRIVGGVWWFFTLIIISSYTANLAAFLTVERMVSPIEGTGTGTGTGTECGSKDSGSKDKTSALSLSNVAGVFYILIGGLGLAMLVALIEFCYKSRSESKRMKGFCLIPQQSINEAIRTSTLPRNSGAGASGGGGSGENGRVVSQDFPKSMQSIPCMSHSSGMPLGATGL. The M1, M2, M3, and M4 regions are marked as bold, whereas the 5(GT) linker is italicized. The sequence encoding for EGFP was introduced after the CTD region of this construct to generate the HA-GluA1-ΔECD-EGFP construct. Residue Ser-814 in the HA-GluA1-ΔECD construct was changed to a stop codon to create HA-GluA1-ΔECD/ΔCTD. Calculated molecular masses of these constructs are listed in Table 1. In all of our constructs the amino acid numbering is for the mature receptor (without the signal peptide). Point mutations were generated using PCR-based methods and confirmed by sequence analysis. Human embryonic kidney (HEK) 293 cells were transfected with the cDNAs using X-tremeGene 9 or X-tremeGene HP (Roche Applied Science) (20).
TABLE 1.
Calculated molecular masses of constructs used in experiments
Molecular masses of each construct as well as the total molecular mass of the oligomeric complexes formed by that construct (in kDa) are calculated and listed.
| Constructs | Monomer | Dimer | Trimer | Tetramer |
|---|---|---|---|---|
| GluA1 | 99.7 kDa | 199.4 kDa | 299.1 kDa | 398.8 kDa |
| HA-GluA1-ΔATD | 58.2 kDa | 116.4 kDa | 174.6 kDa | 232.8 kDa |
| GluA1-ΔCTD | 92.0 kDa | 184.0 kDa | 276.0 kDa | 368.0 kDa |
| HA-GluA1-ΔATD/ΔCTD | 50.5 kDa | 101.0 kDa | 151.5 kDa | 202.0 kDa |
| HA-GluA1-ΔECD | 30.7 kDa | 61.4 kDa | 92.1 kDa | 122.8 kDa |
| HA-GluA1-ΔECD-EGFP | 57.8 kDa | 115.6 kDa | 173. 4 kDa | 231.2 kDa |
| HA-GluA1-ΔECD/ΔCTD | 22.9 kDa | 45.8 kDa | 68.7 kDa | 91.6 kDa |
Immunocytochemistry (ICC)
HEK 293 cells were plated in 24-well plates at a density of 0.5 × 105 cells/well on coverslips pre-treated with nitric acid and coated with poly-d-lysine (25 μg/ml in PBS). Cells were maintained in 10% FBS at 37 °C and 95% O2/5% CO2. 36∼48 h after transfection, surface expression was determined as previously described (18).
To determine surface expression of HA-tagged AMPAR constructs, primary antibodies labeling either the N terminally tagged HA (Covance, MMS-101P, mouse monoclonal 16B12) or the GluA1 C terminus (Millipore, rabbit polyclonal AB1504) dissolved in conditioned medium were added directly onto the coverslip and incubated in a humidified chamber at 37 °C for 30 min. Cells were then fixed in 4% paraformaldehyde and blocked in 1% goat serum. Cells were rinsed with PBS and incubated with Alexa Fluor® 546 goat anti-mouse or Alexa Fluor® 488 goat anti-rabbit secondary antibodies (Invitrogen) at room temperature. For permeablized conditions, the cells were fixed in 4% paraformaldehyde and permeablized in 0.25% Triton X-100 before blocking and primary antibody incubation. Prepared coverslips were examined using an upright Fluoview® FV1000 confocal microscope (Olympus).
Harvest of Membrane Proteins
Cells were plated on 15 × 60-mm tissue culture dishes at roughly 3 × 105 cells/dish. Maintenance of culture and transfections were done as for ICC.
Whole cell lysates (including membrane proteins) were harvested as previously described (21). Briefly, a solubilization buffer is prepared by dissolving 20 mm (1%) n-dodecyl-α-d-maltopyranoside (DDM) (Affymetrix, D310HA) in PBS containing a protease inhibitor mixture (Thermo Scientific, 1861278) as well as 1 mm phenylmethylsulfonyl fluoride. Cells were rinsed in a washing buffer (PBS containing 0.5 mm N-ethylmaleimide) and then harvested in the solubilization buffer. The lysate was rotated for 1 h at 4 °C and centrifuged at 50,000 rpm on a Beckman TLA 120.2 rotor for 40 min at 4 °C. The supernatant containing solubilized membrane proteins was collected for Blue Native-PAGE.
For assaying the detergent or thermal stability of various constructs, a lysis buffer (20 mm Tris in PBS) was used to harvest cells after rinsing in the aforementioned washing buffer. We homogenized the suspended cells by passing them through a 25-gauge needle six times using a 1-ml syringe. The cell lysate was then centrifuged at 3,000 rpm on an Eppendorf 5417R microcentrifuge for 3 min at 4 °C. The supernatant containing the membrane fraction was then centrifuged at 50,000 rpm on a Beckman TLA 120.2 rotor for 10 min at 4 °C. The pellet was isolated and re-suspended in chilled PBS, and the suspension was centrifuged again. The final pellet containing the membrane fraction was re-suspended in an alternative solubilization buffer (20 mm Tris and 50 mm NaCl in PBS) and was sonicated 4 times, each for 15 s. 6 mm (0.3%) DDM along with various concentrations of SDS (0∼0.2%) were added to the suspension. The membrane lysate was then rotated for 1 h at 4 °C and centrifuged at 50,000 rpm on a Beckman TLA 120.2 rotor for 20 min at 4 °C. The resulting supernatant containing only solubilized membrane proteins was collected for analysis. For thermal stability assays, the supernatant (solubilized with 20 mm DDM and no SDS) was incubated at a particular temperature (0∼40 °C) for 20 min right before Blue Native-PAGE.
Blue Native-PAGE (BN-PAGE)
Membrane proteins contained in the supernatant were resolved using Blue Native-PAGE as previously described (21, 22). Briefly, protein samples mixed with 1× Native PAGE sample buffer, 0.05% Native PAGE G-250 additive and supplemented with 10 mm DDM were loaded onto Novex 4–16% BisTris gradient gels. NativeMarkTM Unstained Protein Standard (Life Technologies) and horse spleen apoferritin (Sigma) were also loaded as molecular mass markers. Proteins were separated at constant voltage (115 V for 1 h and then 215 V for 2 h) at 4 °C. Gels were transferred overnight (14∼16 h) at constant amperage (35 mA) to polyvinylidene fluoride (PVDF) membranes. Following transfer, membranes were briefly rinsed with methanol and proteins were fixed on the membrane by incubating in 8% acetic acid for 15 min. The presence of proteins was confirmed by Ponceau S staining. Membranes were then rinsed with water, rehydrated with methanol, and rinsed with TBS-T (0.05% Tween) to remove the stain. Membranes were blocked in 5% milk/TBS and then incubated with either anti-GluA1 (Millipore, MAB2269, mouse monoclonal RH95) or anti-HA (Covance, MMS-101P, mouse monoclonal 16B12). After rinsing in TBS-T, membranes were incubated with HRP-conjugated anti-mouse IgG secondary antibodies (Santa Cruz Biotechnologies, sc-2030). Blots were developed using luminol reagent (Santa Cruz Biotechnologies, sc-2048) before exposure to chemiluminescence blue-sensitive film (Crystalgen). In certain instances, lanes from the same gel are presented in a different order from the original gel (indicated by a thin space between lanes) for clarity of presentation. Each experiment with a particular construct is repeated at least n = 3 times to ensure reproducibility.
Surface Biotinylation Assay
Cells expressing constructs of interest were rinsed with PBS containing 100 μm CaCl2 and 1 mm MgCl2 (PBS/CM) 48 h post-transfection. Cells were then incubated in either PBS/CM or 5 mg/ml of sulfo-NHS-LC-biotin (Thermo) for 45 min on ice. The reaction was terminated by incubation in 100 mm glycine. After rinsing with PBS/CM, whole cell lysates were harvested as described above and subsequently rotated with strepavidin-agarose beads (Thermo) top-to-end for 2 h at room temperature. Beads were rinsed three times with PBS/CM and bound proteins were eluted by heating at 95 °C in SDS sample buffer containing 50 mm DTT. Whole cell lysates (“W”), eluted proteins (“E”), as well as unbound fractions (“U”) were subjected to SDS-PAGE and proteins of interest are detected by immunoblotting using antibodies described above. The percentages of loading for these samples were 2, 50, and 2%, respectively. GAPDH was selected as a cytosolic loading control and was detected using a mouse monoclonal anti-GAPDH antibody (Calbiochem, CB1001). Intensity of each band was quantified using ImageJ (see below) and surface expression was measured as the ratio between the eluted (“E”) and total protein (“W”) bands.
Densitometry and Quantification
The concentration of antibodies, duration of incubation, as well as time of exposure were optimized to ensure that the intensities of major bands fall within the linear response range of the film (21). Developed films were scanned into .tiff format and analyzed using ImageJ. The oligomeric state of each well resolved band was designated based on its position relative to the molecular weight markers. The mean intensity (I) as well as the area (A) of each band of interest (the area of interest was defined using the “freehand selection” tool because the shapes of the bands may be irregular on BN-PAGE) was measured (21). The background mean intensity (IB) was also measured on an area where there was no signal. The cumulative intensity (C) of each band was calculated as Equation 1.
For each construct, the fraction of tetramer (T) was calculated from the cumulative intensities of each band (CT, CTri, CD, and CM) (21),
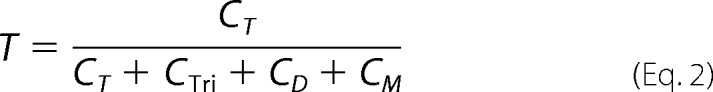 |
where T is defined as the fraction of subunits that are assembled into tetrameric complexes, assuming a linear relationship between the number of antibodies bound to a complex and the number of subunits it contains.
It should be noted that the intensity of each band is not necessarily proportional to the total amount of protein the band contains, because it could not be guaranteed that every single subunit in a given oligomeric complex is bound with an antibody. The fraction of tetramer intensity is therefore treated only as a relative measurement of tetramerization efficiency and cannot be used to calculate absolute physical quantities such as free energy of oligomerization, as was done in previous studies (23).
Data Analysis and Statistics
Stability assays, the percentage tetramer at a particular temperature or SDS concentration (Tn) was normalized to the baseline percentage at 0 °C or 0% SDS, respectively (T0).
 |
For each temperature or SDS condition, Student's t tests were performed between the Tn of each construct and the corresponding Tn of full-length GluA1.
To assess the relative stability (RS) of a particular mutant, we calculated the ratio between its normalized T and that of a wild type sample run on the same gel, either at a selected temperature (37 °C) or at a selected mid-point SDS concentration (0.11%).
 |
Paired Student's t tests were performed between the normalized T of each mutant and the normalized T of the corresponding wild type sample run on the same gel under the same condition.
Homology Modeling and Molecular Dynamics Simulation
The homology model of the GluA1 TMD is built based on the TMD of a full-length GluA2 crystal structure (PDB code 3KG2) (6) in MODELLER (24) by generating 50 models and selecting the one with the lowest score. The sequences of GluA2 and GluA1 TMD were aligned using Clustal Omega (25). To reduce the system size and speed up simulations, the intracellular M1-M2 linker (Tyr-545 to Asp-562) of each subunit was replaced with a GGG linker; otherwise, the TMD construct used in modeling and simulation is the same as that used in the experiments.
The GluA1 TMD structural model was first energy-minimized in vacuum with its Cα atoms fixed in position for 5000 steps, then it was embedded into a pre-equilibrated 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine bilayer obtained from CHARMM-GUI (26). Lipid and water molecules that overlapped with the protein were removed. The charge of the system was neutralized with NaCl at a salt concentration of about 150 mm. A second energy minimization was conducted with protein Cα atoms fixed in position for 5000 steps. MD simulation was then performed for 5 ns, in which only the Cα atoms of the transmembrane helices restrained using a harmonic potential with a gentle force constant of 1.0 kcal/(mol Å2). Finally, a 71.7-ns unbiased MD simulation was performed in which no restraints were applied to the system.
System preparation was done in VMD (27). All simulations were performed using NAMD version 2.9 (28) with CHARMM27 protein force field and CHARMM36 lipid force field (29). The simulation parameters were the same as those for the previous simulations (30, 31).
Results
AMPAR GluA1 Forms a Tetramer in the Absence of the ECD
Eukaryotic iGluRs consist of four structural domains: the ATD, LBD, TMD, and an intracellular CTD. The extracellular domain (ECD), which consists of the ATD and the LBD, accounts for 70∼80% of the molecular mass of an AMPAR complex (Fig. 1A). To investigate the significance of ECD in AMPAR tetrameric assembly, we used BN-PAGE to assess the oligomeric states of GluA1 with or without ECD. Consistent with previously published results (17, 19), full-length AMPAR subunits, in this case GluA1, expressed in HEK 293 cells readily formed homotetramers (Fig. 1B, left lane). A small dimer fraction was also detected, in keeping with the notion that AMPARs are dimer-of-dimers (2–4). When the ECD was deleted (HA-GluA1-ΔECD), the remaining domains of GluA1, including the TMD and CTD, still formed a homotetramer as estimated from its molecular mass (Fig. 1B, right lane). No dimer band was detected for the ECD-lacking construct, consistent with the pseudo-4-fold symmetry of the TMD (6) and the idea that the ATD is responsible for AMPAR dimerization (13).
FIGURE 1.
Assessing the oligomeric state of GluA1 with or without the ECD. A, left: crystal structure of the AMPA receptor GluA2 (Protein Data Bank code 3KG2) (6). The ECD, consisting of the ATD and LBD, is shaded gray. The dash line area indicates location and estimated size (∼26 Å) of the unresolved CTD. Purple shading represents the lipid bilayer, with in and out indicating the intracellular and extracellular faces. B, BN-PAGE of HA-GluA1 and HA-GluA1-ΔECD expressed in HEK 293 cells. Positions of molecular mass markers are labeled on the right (see “Experimental Procedures”). Oligomeric states of detected bands were estimated by molecular mass (see Table 1) and indicated as T and T′ (tetramers for HA-GluA1 and HA-GluA1-ΔECD, respectively) or D (dimer for HA-GluA1). C, BN-PAGE of HA-GluA1-ΔECD co-expressed with HA-GluA1-ΔECD-EGFP at various DNA ratios. Each construct formed a single tetramer band when expressed alone (leftmost and rightmost lanes). Four incremental upward shifts were observed above the HA-GluA1-ΔECD (open circles) tetramer band, each representing a distinct tetrameric stoichiometry with 1, 2, 3, or 4 EGFP-tagged subunit (green filled circles) incorporated, respectively.
To confirm that the single band observed with HA-GluA1-ΔECD represents a tetramer, we tagged the construct with EGFP at the C terminus, which caused an up-shift of its molecular mass (Fig. 1C, far right lane). When HA-GluA1-ΔECD and HA-GluA1-ΔECD-EGFP were co-expressed at various ratios, a total of four incremental up-shifts were observed (Fig. 1C, inner lanes), implying the incorporation of 1, 2, 3, and 4 EGFP-tagged subunits, respectively. Thus, the ECD-lacking GluA1 maintains a tetrameric stoichiometry.
Surface Trafficking, Membrane Topology, and Packing of Pore-lining Helices Are Maintained Despite the Lack of ECD
In full-length AMPARs, the N terminus is extracellular, whereas the C terminus is cytoplasmic (Fig. 1A). To test whether the tetramer formed by HA-GluA1-ΔECD maintained the same membrane topology, we performed ICC using two different primary antibodies: one against the N-terminal HA tag and the other against the CTD (Fig. 2A, left panel). Under non-permeabilizing conditions, we observed cell surface labeling only by the anti-HA antibody but not the anti-CTD antibody (Fig. 2A, right panel, left 3 columns). Under permeabilizing conditions, we observed labeling by both antibodies (Fig. 2A, right panel, rightmost column). Thus, HA-GluA1-ΔECD not only is capable of surface trafficking, but also has a membrane topology just like full-length GluA1, with the N terminus extracellular and the C terminus cytoplasmic.
FIGURE 2.

Surface trafficking, membrane topology, and helical packing of the ECD-lacking GluA1 construct. A, left, presumed membrane topology of the HA-GluA1-ΔECD construct. An anti-HA antibody (red) and an anti-GluA1-CTD antibody (blue) are used in ICC to detect the N terminus and the C terminus of the construct, respectively. Right, ICC of HEK 293 cells expressing HA-GluA1-ΔECD under non-permeabilizing (left 3 columns) or permeabilizing (right column) conditions. B and C, BN-PAGE of GluA1 and GluA1(A618T) (B) or HA-GluA1-ΔECD-EGFP and HA-GluA1(A618T)-ΔECD-EGFP (C). Estimated oligomeric states are indicated as T (tetramer), Tri (trimer), D (dimer), and M (monomer). D, surface biotinylation assay of GluA1 (left) or HA-GluA1-ΔECD (right) (see “Experimental Procedures” for details). Samples were immunoprecipitated (IP) with streptavidin-agarose beads. Proteins of interest were detected by immunoblot (IB) using either anti-GluA1 or anti-HA antibodies. Fractions are labeled as: W (total whole cell), E (elution), -Ctrl (biotin-free control), and U (unbound). The percentage of each sample that was loaded onto the SDS-PAGE gel is indicated below. The cytosolic protein GAPDH is used as a loading control. E, the ratio between the intensities of the E and W bands (elution/whole cell) for each protein was used as a quantification of surface expression. * indicates p value < 0.05 when compared with GAPDH in unpaired Student's t test.
To test whether the ECD-lacking GluA1 tetramer still contained an ion channel resembling that of the intact receptor, we used the lurcher mutation (A618T), which disrupts tetramerization of full-length AMPARs (3) possibly due to its location near the tightly packed activation gate of the fully formed tetrameric ion channel (6). Consistent with previous studies (3), A618T reduced the tetramer fraction for intact GluA1 (Fig. 2B) from 0.83 ± 0.04, n = 7 to 0.36 ± 0.09, n = 5 (mean ± S.E., n = number of samples). Similarly, the same mutation in the ECD-lacking GluA1 also reduced the tetramer fraction (Fig. 2C) from 1.00 ± 0.01, n = 5 to 0.70 ± 0.11, n = 5 (see “Experimental Procedures” for discussions on quantification). Thus, the general pattern of helical packing in the ion channel is retained even in the absence of the ECD. The reduction in tetramer fraction was accompanied by either an increase of dimers (Fig. 2B) or the emergence of trimers and dimers (Fig. 2C). No monomer band was observed. This might reflect that the positioning of Ala-618 in a non-tetrameric oligomer is different from that in a fully formed tetramer, resulting in A618T having no disruptive effect. Alternatively, interactions within the ATD (in the case of the full-length receptor), TMD (e.g. those mediated by the M4 segment), and/or CTD might compete with the lurcher mutation, thereby preventing the further breakdown of dimers.
Fig. 2A indicates that the ECD is not required for the surface expression of GluA1. To quantify the effect of ECD deletion on GluA1 trafficking, we performed a cell surface biotinylation assay. Both full-length GluA1 and HA-GluA1-ΔECD could be immunoprecipitated by streptavidin after surface biotinylation, in contrast to the cytosolic enzyme GAPDH (Fig. 2D). There was no significant difference between the immunoprecipitated fraction of GluA1 and HA-GluA1-ΔECD (Fig. 2E). These results, combined with the ICC experiment, suggest that the ECD does not contribute significantly to the efficiency of GluA1 surface trafficking.
The TMD Mediates Tetramerization While the CTD Plays a Modulatory Role
The ECD-lacking construct contains two domains, the TMD and CTD. To pinpoint which one of these is responsible for mediating tetramerization, we removed the CTD from this construct (HA-GluA1-ΔECD/ΔCTD) (see “Experimental Procedures”). This highly truncated construct was still able to form an oligomer, whose estimated molecular mass was consistent with that of a tetramer (91.6 kDa) (Fig. 3A, right lane). Given the homology between the TM region of iGluRs and K+ channels (8, 32), which still tetramerize even in the absence of their specialized tetramerization domains (33–35), it is highly likely that the oligomer formed by HA-GluA1-ΔECD/ΔCTD was also a tetramer. To probe the stoichiometry of this oligomer, we included 0.11% SDS in the solubilization buffer to disrupt inter-subunit interactions (see “Experimental Procedures” and the next section under “Results”). As a result, the single band seen in Fig. 3A (right lane) and Fig. 3B (left lane) was partially broken down into two bands, presumably representing the dimer and the monomer (Fig. 3B, right lane).
FIGURE 3.

Tetramerization of GluA1 with or without the CTD. A, BN-PAGE of HA-GluA1-ΔECD as well as the HA-GluA1-ΔECD/ΔCTD. For HA-GluA1-ΔECD/ΔCTD, a single band was detected (indicated as T′?) with an estimated molecular mass roughly consistent with that of a tetramer (91.6 kDa). B, BN-PAGE of HA-GluA1-ΔECD/ΔCTD harvested either without SDS or with 0.11% SDS in the solubilization buffer. C, homology modeling and MD simulation of an ECD/CTD-free GluA1 tetramer based on 3KG2 (see “Experimental Procedures”). R.m.s. deviation between the model and corresponding regions of Protein Data Bank 3KG2 over time is shown in purple (black dashed line shows averaged r.m.s. deviation between 50 and 76.7 ns). Initial conformation of the model after energy minimization (point 1 in r.m.s. deviation trace) was almost identical to 3KG2, whereas the final conformation after 76.7 ns of MD simulation (point 2 in trace) showed minimal changes (r.m.s. deviation <3.5 Å). D and E, BN-PAGE of GluA1 and GluA1-ΔCTD (D) or HA-GluA1-ΔATD and HA-GluA1-ΔATD/ΔCTD (E). Estimated oligomeric states are indicated as described in the legend to Fig. 2.
Unfortunately, we could not further verify the stoichiometry of HA-GluA1-ΔECD/ΔCTD using the experiment shown in Fig. 1C because attaching the EGFP tag directly to the N terminus of the M1 helix severely impaired protein folding (data not shown). We therefore performed homology modeling of a tetramer formed by this construct based on the GluA2 structure 3KG2 (6). The model after energy minimization was almost identical to the ion channel domain of 3KG2 (Fig. 3C, initial conformation), with an r.m.s. deviation of 0.54 Å. We also performed a total of 76.7 ns of constrained and unconstrained molecular dynamics simulations with the model and observed minimal changes in its structure (r.m.s. deviation <3.5 Å) (Fig. 3C, final conformation). Hence, the deletion of all extra-membrane domains (ECD and CTD) and the introduction of the GT linker (see “Experimental Procedures”) did not induce any notable structural changes in the TMD tetramer, lending further credence to our conclusion that the transmembrane segments alone, independent of other domains, are capable of tetramerizing.
In the absence of the ECD, the deletion of CTD caused no notable impairment to the tetramerization process (Fig. 3A). To test for any interaction between the ECD and CTD, we removed the CTD in either full-length GluA1 (GluA1-ΔCTD) or a construct that lacks the ATD (HA-GluA1-ΔATD/ΔCTD). Tetramerization of full-length GluA1 lacking the CTD still occurred (Fig. 3D, right lane) but in contrast to the TMD construct (Fig. 3A, right lane) the efficiency of tetramerization, assayed as the tetramer fraction, was significantly reduced from 0.83 ± 0.05, n = 7, to 0.39 ± 0.03, n = 7. Thus, the CTD positively modulates the efficiency of tetramer formation in full-length AMPARs. The ATD-lacking construct (HA-GluA1-ΔATD) still predominantly forms tetramers (Fig. 3E, left lane), as published previously (3, 36). The dimer band observed with full-length GluA1 is here replaced by a monomer, as expected from the idea that the ATD is responsible for the initial dimerization (13). When the CTD is removed in the absence of the ATD but still in the presence of the LBD, tetramerization was almost completely abolished (Fig. 3E, right lane), reducing the tetramer fraction from 0.77 ± 0.03, n = 5, to 0.16 ± 0.02, n = 3. This result is again consistent with the idea that CTD positively modulates tetramerization, but also suggests that the LBD presents a penalty to the tetramerization process, which we refer to here as the “LBD barrier.”
ATD and LBD Have Opposite Effects on Tetrameric Stability
To further address the energetic contributions of different domains to the assembly process, we assessed their influence on tetrameric stability by measuring changes in the tetramer fraction in response to various concentrations of SDS. SDS is a harsh anionic detergent that tends to disrupt non-covalent interactions between proteins, whereas at the same time partially mimicking the membrane environments required for TM helical contact (37). To improve the dynamic range of the assay, we also lowered the concentration of DDM in the solubilization buffer from 20 to 6 mm (see “Experimental Procedures”).
At 0% SDS, full-length GluA1, HA-GluA1-ΔATD, and HA-GluA1-ΔECD-EGFP largely remained as tetramers (Fig. 4A). As SDS concentration increased, the intensities of the tetramer bands decreased for all three constructs (Fig. 4A). At the same time, additional bands corresponding to lower number oligomers (dimers and monomers) began to appear. Pre-existing bands representing these lower oligomers also increased in intensity. These outcomes support the idea that incubation in SDS disrupts already formed tetramers, causing them to break down into lower number oligomers. We normalized the tetramer fraction at each SDS concentration to 0%, and fitted a sigmoidal SDS response curve for each construct (Fig. 4B). The curve for HA-GluA1-ΔATD (Fig. 4B, gray triangle and gray dash line) was significantly left-shifted compared with that of the full-length GluA1 (Fig. 4B, open diamond and black line), indicating that although the ATD is not required for tetramerization, it contributes to the stability of the tetramer once it is formed. Surprisingly, the SDS response curve of HA-GluA1-ΔECD-EGFP was right-shifted compared with that of HA-GluA1-ΔATD and was almost indistinguishable from that of full-length GluA1 (Fig. 4B, black square and black dash line). Thus, consistent with results shown in Fig. 3, the LBD seems to confer a penalty to tetrameric stability, effectively balancing out the stabilizing effect of the ATD.
FIGURE 4.

Resistance of GluA1 tetramers to SDS with or without the ECD. A, BN-PAGE of GluA1, HA-GluA1-ΔATD, or HA-GluA1-ΔECD-EGFP with various concentrations of SDS added to solubilization buffer (also containing 6 mm DDM). B, tetramer fractions of GluA1 (open diamond, black solid line), HA-GluA1-ΔATD (gray triangle, gray dash line), and HA-GluA1-ΔECD-EGFP (black square, black dash line) were normalized to those at 0% SDS (see “Experimental Procedures”). Normalized fractions are plotted against the SDS concentration and fitted with sigmoid functions. The midpoints of the sigmoid functions fitted for the three constructs are ∼0.11, 0.08, and 0.11%, respectively. C, tetramer fractions of the same three constructs (indicated as in B) at various temperatures are normalized to those at 0 °C (see “Experimental Procedures”). Normalized tetramer fractions are plotted against temperature and fitted with sigmoid functions.
As an alternative approach to measure tetramer stability, we incubated the membrane samples at progressively higher temperatures. Consistent with earlier studies (35) increasing temperatures led to progressively lower tetramer fractions for all three constructs (Fig. 4C). The derived curves (Fig. 4C) showed the exact same pattern as the SDS response curves (Fig. 4B), with HA-GluA1-ΔECD-EGFP being almost indistinguishable from the full-length GluA1, and HA-GluA1-ΔATD being left-shifted. The concordance between the SDS and the thermal approaches indicate that the results from both assays reflect the structural stability of the tetramers in a general sense, instead of being a fortuitous outcome associated with either treatment in particular.
As an additional validation of the SDS approach, we assayed the effect of the lurcher mutation (A618T) on tetrameric stability. For convenience, we selected as a reference a SDS concentration (0.11%) roughly at the mid-point of the dynamic range on the SDS response curves of the full-length and ECD-lacking constructs (Fig. 4B), where the tetramer fraction of these constructs would be most sensitive to mutations that affect their stability. We introduced A618T into both constructs and found that the mutation led to a significant decrease in normalized tetramer fraction at 0.11% SDS regardless of the presence of ECD (Fig. 5, A and B). This is consistent with the disruptive effect of A618T on tetramerization with or without the ECD (Fig. 2, B and C) and suggests that the effect of A618T resulted from reduced tetrameric stability.
FIGURE 5.

Effect of the “lurcher” mutation A618T on tetrameric stability in SDS. A, BN-PAGE of GluA1, GluA1(A618T), HA-GluA1-ΔECD-EGFP, or HA-GluA1-ΔECD-EGFP(A618T) with either 0 or 0.11% SDS. B, normalized tetramer fractions of the constructs in A at 0.11% SDS. * indicates p value < 0.05 in unpaired Student's t test.
Our stability assays on the ECD-lacking constructs exhibited a striking concordance with results shown in Fig. 3, D and E. Overall, these results indicate that the ATD and LBD modulate tetramerization in opposite directions, at least partially by influencing the stability of an already formed tetramer. The CTD also contributes positively to tetramerization (Fig. 3, D and E), although we did not measure its contribution to tetrameric stability, because the GluA1-ΔCTD showed a low baseline tetramer fraction, which would have limited the dynamic range of our assay.
The M4 Segment Is Critical to Tetramerization Only in the Presence of the LBD
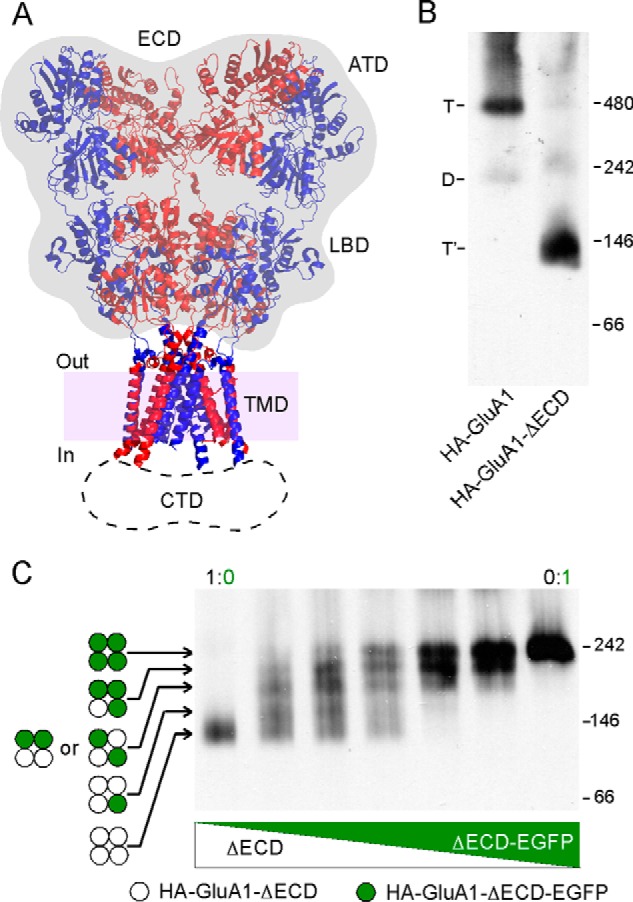
Previously we identified a face in the third transmembrane segment (M4) of AMPARs that is critical to tetramerization (18, 19). This face, dubbed “VLGAVE” after the amino acid residues comprising it, is closely aligned with the M1 and M3 segments of an adjacent subunit in the GluA2 structure (Fig. 6A), suggesting that interactions between M4 and the other TM segments play a critical role in AMPAR tetramerization. In support of this interpretation, mutating the residues within the VLGAVE face to tryptophan, which presumably disrupts molecular interactions between M4 and the rest of the TMD, prevents the formation of tetramers (Fig. 6B) (19). A similar outcome occurred for HA-GluA1-ΔATD (Fig. 6C), indicating that the TM interactions mediated by the VLGAVE face are still critical for tetramerization in the absence of the ATD. In contrast, the same mutations had no effect on tetramerization of the construct lacking ECD with or without the EGFP tag (Fig. 6D), suggesting a less important role of M4 in tetramerization when the LBD is absent. Thus, and again consistent with earlier results (Figs. 3 and 4), the LBD of GluA1 presents a barrier to receptor tetramerization that can be offset, at least in part, by interactions mediated by the M4 segment.
FIGURE 6.
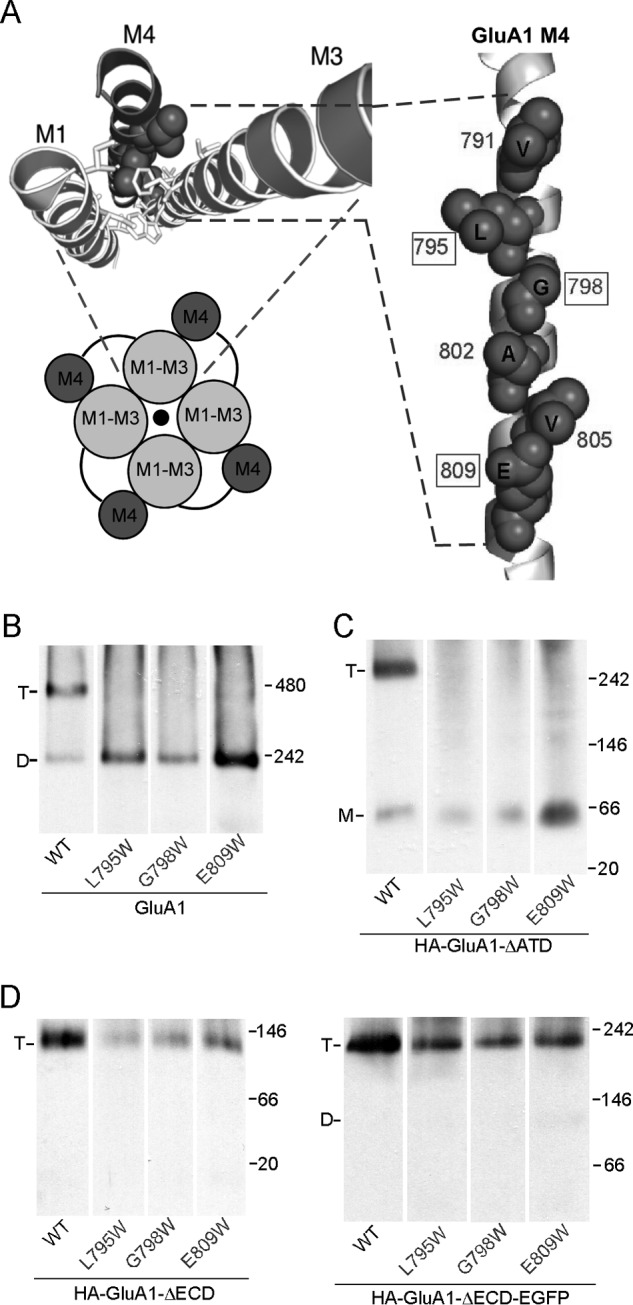
The effect of the M4 segment on tetramerization with or without the ECD. A, VLGAVE face (dark gray) of the M4 segment in the GluA2 structure of Protein Data Bank 3KG2 (6). Numbering is for GluA1. B, BN-PAGE of GluA1 and mutants with tryptophan (W) substitutions in the VLGAVE face (19). C and D, BN-PAGE of GluA1-ΔATD (C), HA-GluA1-ΔECD or HA-GluA1-ΔECD-EGFP (D) with Trp substitutions in the VLGAVE face.
An alternative explanation for the results in Fig. 6D is that in the absence of the LBD, the M4 helix gains more freedom of movement. As a result, inter-subunit interactions in the TMD are now mediated by a different set of residues instead of the VLGAVE face. We therefore tested the effect of a subset of M4 tryptophan substitutions both within and outside of the VLGAVE face (Fig. 7, gray and black residues, respectively) on the stability of the tetramer lacking ECD with the midpoint SDS concentration of 0.11% as a reference point, at which the assay would be most sensitive (see Fig. 4B). All of the tested mutations within the VLGAVE face (L795W, G798W, V805W, and E809W) significantly affected the stability of HA-GluA1-ΔECD-EGFP (Fig. 7B). In contrast, mutations outside of the VLGAVE face (G800W and L807W), which did not disrupt tetramerization in either full-length GluA1 or GluA2 (data not shown), showed either negligible or very slight effects on tetrameric stability (Fig. 7B). These results indicate that interactions mediated by the M4 VLGAVE face still occur in the absence of the LBD and contribute to the stability of the tetramer, despite not being necessary for its formation. Thus, in the full-length receptor, these interactions are at least partially responsible for offsetting the LBD barrier.
FIGURE 7.

Contribution of VLGAVE face to tetrameric stability in the absence of the ECD. A, example BN-PAGE of HA-GluA1-ΔECD-EGFP and mutants with tryptophan substitutions inside (gray) and outside of (black) the VLGAVE face. B, normalized tetramer fractions of HA-GluA1-ΔECD-EGFP with tryptophan substitutions relative to the wild type at 0.11% SDS. Dash line indicates normality. * indicates p value < 0.05 in paired Student's t test (comparing to wild type assayed on the same gel), whereas ** indicates p value < 0.01.
Restricting Quaternary Arrangement of the LBD Enhances Tetramerization
One possible mechanism underlying the LBD barrier involves steric clashes among the four subunits at the LBD level. These clashes could destabilize the quaternary arrangement of the subunits, thereby affecting the stability of the entire receptor complex. The LBD is arranged as a dimer-of-dimers. We therefore introduced a previously published disulfide bond located at the LBD dimer-dimer interface (Fig. 8A), which is known to stabilize that interface and restrict the quaternary movements of the LBD (38). The homologous cysteine substitution in GluA1, A661C, formed a disulfide cross-link as evidenced by redox-sensitive dimers in SDS-PAGE (Fig. 8B, upper and middle panels). To test the effect of the cross-link on GluA1 tetramerization, we used a mutation in the M4 VLGAVE face, G798A, which dramatically, but not completely, impeded tetramerization (Fig. 8B, lower panel) (19). The dimer-dimer cross-link was able to fully rescue the tetramerization deficit caused by G798A (from 0.46 ± 0.02, n = 9 to 0.92 ± 0.06, n = 2) (Fig. 8B, lower panel). Hence, the instability of the LBD quaternary arrangement is most likely the structural basis of the LBD barrier to tetramerization.
FIGURE 8.
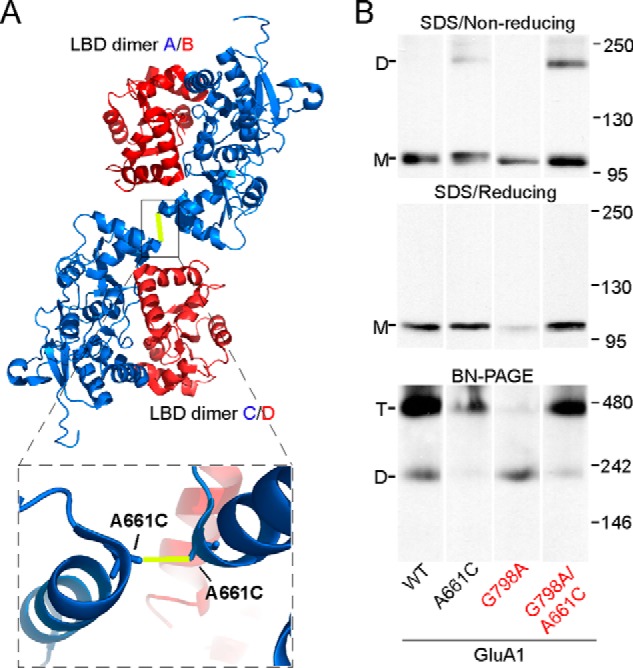
Effect of a disulfide cross-link at the LBD dimer-dimer interface on tetramerization. A, LBD tetramer in the GluA2 structure of Protein Data Bank 3KG2 (6) viewed from the N terminus. Subunits are colored blue (A/C conformation) and red (B/D conformation). The yellow line indicates the location of the disulfide bond between substituted cysteine A661C (numbering for GluA1). Box below shows a zoomed-in image of the dimer-dimer interface with the disulfide bond. B, upper and middle: SDS-PAGE of GluA1, GluA1(A661C), GluA1(G798A), and GluA1(G798A/A661C) under non-reducing (upper) or reducing (middle) conditions. Lower, BN-PAGE of the same constructs.
Discussion
Our experiments directly demonstrate that the TMD acts as the tetramerization domain in AMPARs (Fig. 3A). Neither the ECD, which encompasses the ATD and the LBD, nor the CTD was necessary for the tetrameric assembly of GluA1 (Figs. 1 and 3). Interestingly, the VLGAVE face of the M4 segment critical to the tetramerization of full-length GluA1 (19) is not required in the absence of the ECD (Fig. 6D), although it still contributes to tetrameric stability (Fig. 7). Together, our results suggest that the ion channel core (M1-M2-M3) of AMPARs alone is probably capable of tetramerization, with the peripheral M4 helix acting as a stabilizing “barrel hoop.” This scenario is similar to that of the KcsA channel, whose ion channel domain (homologous to the M1-M3 region of iGluRs) is fully capable of forming a functional tetramer, with the C-terminal “tetramerization domain” providing additional stability (35).
A surprising finding from our experiments was that the presence of the LBD reduces the tetramerization efficiency (Fig. 3, A and D). Indeed, multiple lines of evidence point to an energetic penalty introduced by the LBD to GluA1 tetrameric assembly, referred to here as the LBD barrier (Figs. 3, 4, and 6). The LBD barrier can be offset by some combination of the ATD, the M4 transmembrane segment, and the CTD (Figs. 3, 4, and 7).
One possible mechanism underlying the LBD barrier is that steric hindrance among the subunits at the LBD level destabilizes the tetrameric complex. Quaternary arrangements of the LBD are highly flexible (39–41). During the tetramerization process, quaternary rearrangements at the LBD level could lead to transient increases in steric hindrances. Consistent with this hypothesis, a disulfide cross-link introduced at the interface between two LBD dimers (Fig. 8A), which presumably prevents inter-dimer quaternary rearrangements (38), rescued the deficit in tetramerization caused by a mutation in the M4 VLGAVE face (Fig. 8B). In contrast, a mutation that restricts intra-dimer rearrangements in the LBD (L483Y in GluA2) has been found to negatively impact AMPAR assembly and maturation (42). This discrepancy seems to suggest that the LBD barrier arises from transient steric clashes between two LBD dimers but not within the dimers. Nevertheless, further work is required to pinpoint the exact structural determinants underlying the steric hindrance that destabilizes the LBD tetramer in the full-length receptor.
An alternative mechanism might involve the binding of glutamate at the LBD cleft. Biogenesis and assembly of nAChR are enhanced by the binding of nicotine (43, 44). Similarly, binding of glutamate, which exists in the endoplasmic reticulum at high concentrations (45), could influence AMPAR assembly as well, albeit negatively. Although no open state structure is yet available for iGluRs, significant displacements of TM helices could be induced by ligand binding at the LBD during iGluR gating (46–48), similar to K+ channels (49). Such displacements could interfere with the tight packing of the TMD tetramer, leading to its destabilization. Consistent with this idea, the lurcher mutation, which confers constitutive activity to the ion channel, also affects tetrameric stability (Figs. 2, B and C, 5). Interestingly, binding site mutations in the kainate receptor GluK2 that abolish its function actually increased the tetramer fraction (50). We therefore tested the tetrameric stability of the receptor using the SDS approach either with or without glutamate in the solubilization buffer (data not shown), but did not observe any difference, possibly due to limited sensitivity of the assay. Still, we cannot rule out that ligand binding and the gating motions it induces presents a barrier during tetramerization, but no longer poses a problem after the tetramer is formed.
Taken together, our observations seem to hint at a “checks and balances” scenario among multiple domains during the biogenesis of a functional AMPAR complex, with the LBD impeding tetramerization and the other domains collaborating to overcome the LBD barrier. The biological significance of such a scenario is unclear. One possibility is that a suboptimal efficiency of tetramerization provides an additional checkpoint for controlling the subunit composition of receptors available for surface expression. Another possibility is that the LBD barrier is an evolutionary by-product of the rapid gating kinetics characteristic of AMPARs. Consistent with this idea, the disulfide bond at the LBD dimer-dimer interface, in addition to enhancing tetramerization, also hampers gating efficiency (38). Furthermore, the prokaryotic GluR0, which lacks the ATD, the M4 segment, as well as the CTD, and is presumably more ancient than mammalian iGluRs, exhibit slow gating kinetics (51). We therefore speculate that the emergence of the ATD, the M4, and the CTD later in evolution conferred additional efficiency to receptor tetramerization, thus allowing the LBD to evolve structural features required for fast gating.
An intact and functional LBD capable of gating motions is essential for the maturation and forward trafficking of full-length AMPARs (52, 53). Surprisingly, the HA-GluA1-ΔECD construct, which lacks the LBD and is therefore incapable of agonist-induced gating motions, was expressed to the cell surface instead of being trapped in the ER (Fig. 2, A, D, and E). At least two possible explanations exist for this discrepancy: 1) certain ER chaperones (e.g. BiP) might bind specifically to the LBD and block forward trafficking until they sense the proper gating motion within the LBD. Thus, the HA-GluA1-ΔECD construct, which lacks the LBD, would be exempt from this ER checkpoint; 2) cytoplasmic trafficking machineries (e.g. the COPII complex) might bind to either the TMD or CTD and prevent the receptor from exiting the ER until they sense the proper conformational changes induced by LBD gating motions. The HA-GluA1-ΔECD construct might be more structurally flexible and therefore could take on those conformations without the help of the LBD. Again, much work is still needed to pinpoint the exact mechanism. Our findings have yielded novel insights into and provoked intriguing questions about the role of the LBD in AMPAR trafficking.
Based on our observations, we conclude that the TMD of AMPARs mediates its tetrameric assembly. The LBD constitutes a barrier to tetramer formation and stability, whereas the ATD, the M4 segment, and the CTD counteract the LBD barrier. This “checks and balances” scenario might have important implications on the regulation of AMPAR subunit composition and forward trafficking. Finally, in the context of neurons, the biogenesis of AMPARs might be modulated by auxiliary subunits and ER chaperone (54, 55) as well as other interacting partners.
Author Contributions
Q. G., J. D., H-X. Z., and L. P. W. designed research; Q. G. performed experiments; J. D. performed simulations; J. D., Q. G., and L. P. W. analyzed data; Q. G. and L. P. W. wrote the manuscript. All authors collaborated in editing the manuscript.
Acknowledgments
We thank Drs. Mark Bowen, Hiro Furukawa, Rashek Kazi, David McKinnon, and Catherine Salussolia for valuable comments, discussions, and suggestions.
This work was supported by an American Heart Association Predoctoral Fellowship (to Q. G.), National Institutes of Health RO1 Grants NS088479 (to L. P. W.) from the NINDS and GM0581879 (to H. Y. Z.) from the NIGMS, and a grant from the Thomas Hartman Center for Parkinson's Research (to L. P. W.). The authors declare no conflict of interest.
- iGluR
- ionotropic glutamate receptors
- AMPAR
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- ATD
- amino-terminal domain
- LBD
- ligand-binding domain
- TMD
- transmembrane domain
- CTD
- carboxyl-terminal domain
- ECD
- extracellular domain
- EGFP
- enhanced green fluorescent protein
- ICC
- immunocytochemistry
- DDM
- n-dodecyl-α-d-maltopyranoside
- BN-PAGE
- Blue Native-polyacrylamide gel electrophoresis
- ER
- endoplasmic reticulum
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. Dingledine R., Borges K., Bowie D., and Traynelis S. F. (1999) The glutamate receptor ion channels. Pharmacol. Rev. 51, 7–61 [PubMed] [Google Scholar]
- 2. Mansour M., Nagarajan N., Nehring R. B., Clements J. D., and Rosenmund C. (2001) Heteromeric AMPA receptors assemble with a preferred subunit stoichiometry and spatial arrangement. Neuron 32, 841–853 [DOI] [PubMed] [Google Scholar]
- 3. Kim K. S., Yan D., and Tomita S. (2010) Assembly and stoichiometry of the AMPA receptor and transmembrane AMPA receptor regulatory protein complex. J. Neurosci. 30, 1064–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tichelaar W., Safferling M., Keinänen K., Stark H., and Madden D. R. (2004) The three-dimensional structure of an ionotropic glutamate receptor reveals a dimer-of-dimers assembly. J. Mol. Biol. 344, 435–442 [DOI] [PubMed] [Google Scholar]
- 5. Gan Q., Salussolia C. L., and Wollmuth L. P. (2015) Assembly of AMPA receptors: mechanisms and regulation. J. Physiol. 593, 39–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sobolevsky A. I., Rosconi M. P., and Gouaux E. (2009) X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature 462, 745–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dürr K. L., Chen L., Stein R. A., De Zorzi R., Folea I. M., Walz T., Mchaourab H. S., and Gouaux E. (2014) Structure and dynamics of AMPA receptor GluA2 in resting, pre-open, and desensitized states. Cell 158, 778–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wo Z. G., and Oswald R. E. (1995) Unraveling the modular design of glutamate-gated ion channels. Trends Neurosci. 18, 161–168 [DOI] [PubMed] [Google Scholar]
- 9. Wollmuth L. P., and Sobolevsky A. I. (2004) Structure and gating of the glutamate receptor ion channel. Trends Neurosci. 27, 321–328 [DOI] [PubMed] [Google Scholar]
- 10. Traynelis S. F., Wollmuth L. P., McBain C. J., Menniti F. S., Vance K. M., Ogden K. K., Hansen K. B., Yuan H., Myers S. J., and Dingledine R. (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62, 405–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ayalon G., and Stern-Bach Y. (2001) Functional assembly of AMPA and kainate receptors is mediated by several discrete protein-protein interactions. Neuron 31, 103–113 [DOI] [PubMed] [Google Scholar]
- 12. Sukumaran M., Penn A. C., and Greger I. H. (2012) AMPA receptor assembly: atomic determinants and built-in modulators. Adv. Exp. Med. Biol. 970, 241–264 [DOI] [PubMed] [Google Scholar]
- 13. Jin R., Singh S. K., Gu S., Furukawa H., Sobolevsky A. I., Zhou J., Jin Y., and Gouaux E. (2009) Crystal structure and association behaviour of the GluR2 amino-terminal domain. EMBO J. 28, 1812–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rossmann M., Sukumaran M., Penn A. C., Veprintsev D. B., Babu M. M., and Greger I. H. (2011) Subunit-selective N-terminal domain associations organize the formation of AMPA receptor heteromers. EMBO J. 30, 959–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mayer M. L. (2011) Structure and mechanism of glutamate receptor ion channel assembly, activation and modulation. Curr. Opin. Neurobiol. 21, 283–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Penn A. C., Balik A., Wozny C., Cais O., and Greger I. H. (2012) Activity-mediated AMPA receptor remodeling, driven by alternative splicing in the ligand-binding domain. Neuron 76, 503–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Greger I. H., Khatri L., Kong X., and Ziff E. B. (2003) AMPA receptor tetramerization is mediated by Q/R editing. Neuron 40, 763–774 [DOI] [PubMed] [Google Scholar]
- 18. Salussolia C. L., Corrales A., Talukder I., Kazi R., Akgul G., Bowen M., and Wollmuth L. P. (2011) Interaction of the M4 segment with other transmembrane segments is required for surface expression of mammalian α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. J. Biol. Chem. 286, 40205–40218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Salussolia C. L., Gan Q., Kazi R., Singh P., Allopenna J., Furukawa H., and Wollmuth L. P. (2013) A eukaryotic specific transmembrane segment is required for tetramerization in AMPA receptors. J. Neurosci. 33, 9840–9845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yelshansky M. V., Sobolevsky A. I., Jatzke C., and Wollmuth L. P. (2004) Block of AMPA receptor desensitization by a point mutation outside the ligand-binding domain. J. Neurosci. 24, 4728–4736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Salussolia C. L., Gan Q., and Wollmuth L. P. (2015) Assaying AMPA receptor oligomerization. in Ionotropic Glutamate Receptor Technologies (Popescu G. K., ed), Vol. 106, pp. 3–14, Springer Science, New York [Google Scholar]
- 22. Schägger H., Cramer W. A., and von Jagow G. (1994) Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal. Biochem. 217, 220–230 [DOI] [PubMed] [Google Scholar]
- 23. Ojemalm K., Botelho S. C., Stüdle C., and von Heijne G. (2013) Quantitative analysis of SecYEG-mediated insertion of transmembrane α-helices into the bacterial inner membrane. J. Mol. Biol. 425, 2813–2822 [DOI] [PubMed] [Google Scholar]
- 24. Sali A., and Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 25. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jo S., Kim T., Iyer V. G., and Im W. (2008) CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comp. Chem. 29, 1859–1865 [DOI] [PubMed] [Google Scholar]
- 27. Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–38 [DOI] [PubMed] [Google Scholar]
- 28. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., and Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Klauda J. B., Venable R. M., Freites J. A., O'Connor J. W., Tobias D. J., Mondragon-Ramirez C., Vorobyov I., MacKerell A. D. Jr., and Pastor R. W. (2010) Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dai J., and Zhou H. X. (2013) An NMDA receptor gating mechanism developed from MD simulations reveals molecular details underlying subunit-specific contributions. Biophys. J. 104, 2170–2181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kazi R., Dai J., Sweeney C., Zhou H. X., and Wollmuth L. P. (2014) Mechanical coupling maintains the fidelity of NMDA receptor-mediated currents. Nat. Neurosci. 17, 914–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Janovjak H., Sandoz G., and Isacoff E. Y. (2011) A modern ionotropic glutamate receptor with a K+ selectivity signature sequence. Nat. Commun. 2, 232. [DOI] [PubMed] [Google Scholar]
- 33. Deutsch C. (2003) The birth of a channel. Neuron 40, 265–276 [DOI] [PubMed] [Google Scholar]
- 34. Isacoff E. Y., Jan L. Y., and Minor D. L. Jr. (2013) Conduits of life's spark: a perspective on ion channel research since the birth of neuron. Neuron 80, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Molina M. L., Encinar J. A., Barrera F. N., Fernández-Ballester G., Riquelme G., and González-Ros J. M. (2004) Influence of C-terminal protein domains and protein-lipid interactions on tetramerization and stability of the potassium channel KcsA. Biochemistry 43, 14924–14931 [DOI] [PubMed] [Google Scholar]
- 36. Pasternack A., Coleman S. K., Jouppila A., Mottershead D. G., Lindfors M., Pasternack M., and Keinänen K. (2002) α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor channels lacking the N-terminal domain. J. Biol. Chem. 277, 49662–49667 [DOI] [PubMed] [Google Scholar]
- 37. Tulumello D. V., and Deber C. M. (2009) SDS micelles as a membrane-mimetic environment for transmembrane segments. Biochemistry 48, 12096–12103 [DOI] [PubMed] [Google Scholar]
- 38. Lau A. Y., Salazar H., Blachowicz L., Ghisi V., Plested A. J., and Roux B. (2013) A conformational intermediate in glutamate receptor activation. Neuron 79, 492–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schauder D. M., Kuybeda O., Zhang J., Klymko K., Bartesaghi A., Borgnia M. J., Mayer M. L., and Subramaniam S. (2013) Glutamate receptor desensitization is mediated by changes in quaternary structure of the ligand binding domain. Proc. Natl. Acad. Sci. U.S.A. 110, 5921–5926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Meyerson J. R., Kumar J., Chittori S., Rao P., Pierson J., Bartesaghi A., Mayer M. L., and Subramaniam S. (2014) Structural mechanism of glutamate receptor activation and desensitization. Nature 514, 328–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gonzalez J., Du M., Parameshwaran K., Suppiramaniam V., and Jayaraman V. (2010) Role of dimer interface in activation and desensitization in AMPA receptors. Proc. Natl. Acad. Sci. U.S.A. 107, 9891–9896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shanks N. F., Maruo T., Farina A. N., Ellisman M. H., and Nakagawa T. (2010) Contribution of the global subunit structure and stargazin on the maturation of AMPA receptors. J. Neurosci. 30, 2728–2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sallette J., Pons S., Devillers-Thiery A., Soudant M., Prado de Carvalho L., Changeux J. P., and Corringer P. J. (2005) Nicotine upregulates its own receptors through enhanced intracellular maturation. Neuron 46, 595–607 [DOI] [PubMed] [Google Scholar]
- 44. Kuryatov A., Luo J., Cooper J., and Lindstrom J. (2005) Nicotine acts as a pharmacological chaperone to up-regulate human α4β2 acetylcholine receptors. Mol. Pharmacol. 68, 1839–1851 [DOI] [PubMed] [Google Scholar]
- 45. Meeker R. B., Swanson D. J., and Hayward J. N. (1989) Light and electron microscopic localization of glutamate immunoreactivity in the supraoptic nucleus of the rat hypothalamus. Neuroscience 33, 157–167 [DOI] [PubMed] [Google Scholar]
- 46. Dong H., and Zhou H. X. (2011) Atomistic mechanism for the activation and desensitization of an AMPA-subtype glutamate receptor. Nat. Commun. 2, 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kazi R., Gan Q., Talukder I., Markowitz M., Salussolia C. L., and Wollmuth L. P. (2013) Asynchronous movements prior to pore opening in NMDA receptors. J. Neurosci. 33, 12052–12066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sobolevsky A. I., Beck C., and Wollmuth L. P. (2002) Molecular rearrangements of the extracellular vestibule in NMDAR channels during gating. Neuron 33, 75–85 [DOI] [PubMed] [Google Scholar]
- 49. Long S. B., Campbell E. B., and Mackinnon R. (2005) Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 309, 897–903 [DOI] [PubMed] [Google Scholar]
- 50. Mah S. J., Cornell E., Mitchell N. A., and Fleck M. W. (2005) Glutamate receptor trafficking: endoplasmic reticulum quality control involves ligand binding and receptor function. J. Neurosci. 25, 2215–2225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen G. Q., Cui C., Mayer M. L., and Gouaux E. (1999) Functional characterization of a potassium-selective prokaryotic glutamate receptor. Nature 402, 817–821 [DOI] [PubMed] [Google Scholar]
- 52. Fleck M. W. (2006) Glutamate receptors and endoplasmic reticulum quality control: looking beneath the surface. Neuroscientist 12, 232–244 [DOI] [PubMed] [Google Scholar]
- 53. Penn A. C., Williams S. R., and Greger I. H. (2008) Gating motions underlie AMPA receptor secretion from the endoplasmic reticulum. EMBO J. 27, 3056–3068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Haering S. C., Tapken D., Pahl S., and Hollmann M. (2014) Auxiliary subunits: shepherding AMPA receptors to the plasma membrane. Membranes 4, 469–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Herring B. E., Shi Y., Suh Y. H., Zheng C. Y., Blankenship S. M., Roche K. W., and Nicoll R. A. (2013) Cornichon proteins determine the subunit composition of synaptic AMPA receptors. Neuron 77, 1083–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]