

Abstract
Cholest-4-en-3-one, whether added exogenously or generated intracellularly from cholesterol, inhibits the growth of Mycobacterium tuberculosis when CYP125A1 and CYP142A1, the cytochrome P450 enzymes that initiate degradation of the sterol side chain, are disabled. Here we demonstrate that a 16-hydroxy derivative of cholesterol, which was previously reported to inhibit growth of M. tuberculosis, acts by preventing the oxidation of the sterol side chain even in the presence of the relevant cytochrome P450 enzymes. The finding that (25R)-cholest-5-en-3β,16β,26-triol (1) (and its 3-keto metabolite) inhibit growth suggests that cholesterol analogs with non-degradable side chains represent a novel class of anti-mycobacterial agents. In accord with this, two cholesterol analogs with truncated, fluorinated side chains have been synthesized and shown to similarly block the growth in culture of M. tuberculosis.
Keywords: cholesterol metabolism, cytochrome P450, dehydrogenase, enzyme inhibitor, Mycobacterium tuberculosis
Introduction
Mycobacterium tuberculosis (Mtb)2 is the causative agent of tuberculosis, a disease that the World Health Organization estimates in 2014 caused illness in 9.6 million individuals and killed 1.5 million people (1), with the bulk of this disease burden falling on third world countries. The scale of this threat, compounded by the emergence of increasingly drug-resistant strains of Mtb, makes it imperative to find new ways to combat tuberculosis (2).
Due to the absence of key enzymes, including squalene monooxygenase and oxidosqualene cyclase, cholesterol is not synthesized in Mtb, but its utilization is important for the infectivity and virulence of this pathogenic agent (3). In vitro, Mtb can grow on a defined medium that includes cholesterol as the sole source of carbon and energy (4–6). Furthermore, the ability of Mtb to infect macrophages after their membranes have been depleted of cholesterol is severely impaired (7). Early experiments also established that the igr locus is required for cholesterol metabolism, and its deletion impairs intracellular growth and virulence (8, 9).
Cholesterol degradation in Mtb is initiated by oxidation of a terminal methyl of the cholesterol side chain to a carboxylic acid by cytochrome P450 enzymes CYP125A1 (Rv3545c) and CYP142A1 (Rv3518c) (4, 6, 10). This oxidation reaction can occur with cholesterol itself, but does so more efficiently with cholest-4-en-3-one (4), the metabolite formed by oxidation of the 3-hydroxyl group to a ketone by a mycobacterial 3β-hydroxysterol dehydrogenase (3β-HSD, Rv1106c) (11–13). The β-oxidation of the side chain gives rise to 4-androstenedione, which can undergo further degradation (3). The acetyl- and propionoyl-CoA fragments obtained from the side chain are incorporated into cellular lipids and virulence factors (14, 15).
Mtb without functional CYP125A1 and CYP142A1 is unable to grow on cholesterol as a carbon source, but grows readily on carbon sources such as glycerol, glucose, or acetate (6, 14). In these mycobacteria, cholesterol accumulates in the form of cholest-4-en-3-one (14). We have shown that cholest-4-en-3-one can inhibit the growth of Mtb in defined media with glycerol as the carbon source, indicating that this steroid can act adversely on the utilization of glycerol by the mycobacterium (14). Cholest-4-en-3-one is not a useful growth inhibitor, however, because it is rapidly degraded by wild-type mycobacteria and only causes transient inhibition. A screen of various 3β-hydroxy sterols as Mtb growth inhibitors recently demonstrated that several, including (25R)-cholest-5-en-3β,16β,26-triol (1), significantly inhibited the growth of the H37Rv strain of Mtb, whereas they show no toxicity to the Vero mammalian cell line (16).
Here we demonstrate that cholest-4-en-3-one inhibits growth of Mtb on defined media with not only glycerol, as already reported (14), but also with acetate and glucose as the sole carbon source. We then establish that 1 is oxidized by the mycobacterial 3β-HSD enzyme to the 3-keto form, but it is not a substrate for CYP125A1 or CYP142A1, resulting in the accumulation of a non-degradable analog of cholest-4-en-3-one. This agent is a potent inhibitor of Mtb growth despite the normal presence of catalytically active CYP125A1 and CYP142A1. Furthermore, two cholesterol analogs with truncated, fluorinated side chains (2 and 3) that are resistant to degradation similarly inhibit Mtb growth.
Experimental Procedures
Chemicals
Diosgenin was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). All other chemicals were synthesized as described, or were purchased from Sigma or Fisher Scientific (Pittsburgh, PA), including: β-nicotinamide adenine dinucleotide phosphate, spinach ferredoxin, spinach ferredoxin-NADP+-reductase, bovine liver catalase, glucose 6-phosphate, glucose-6-phosphate dehydrogenase, N,O-bis(trimethylsilyl)trifluoro-BSTacetamide (BSTFA), and methyl-β-cyclodextrin (MβCD). All solvents used in synthetic procedures were distilled prior to use. Anhydrous solvents were dried according to standard procedures and distillation was conducted under vacuum or a N2 atmosphere. Melting points (m.p.) were determined on a SRS DigiMelt MPA161 (200 to 250 VAC) and are uncorrected. NMR spectra were recorded on a Bruker Advance 400 MHz spectrometer unless otherwise stated. The experiments were performed in CDCl3 unless otherwise stated and the residual solvent peak (CHCl3) was calibrated to δH 7.24 and the CDCl3 center peak at δC 77.0. Mass spectra (ESI-MS) were obtained with a Bruker Esquire HCT spectrometer. GC-MS spectra were recorded on a Shimadzu GC-MS QP2010-plus spectrometer, a ZB-5MS column, 30 m. Standard GC-MS program: column flow 1.7 ml/min; total flow 66.3 ml/min; injector 250 °C; detector 250 °C; oven at 200 °C held for 1 min, increased to 300 °C (20 °C/min), and held for 44 min; total program time was 50 min. HR-MS were obtained on a Bruker micrOTOFQ spectrometer in a positive ion mode.
(25R)-Cholest-5-en-3β,16β,26-triol (1)
The synthesis of 1 was carried out according to a previously reported method (17). The identity and purity of the final product were confirmed using NMR and GC-MS recorded on a Bruker AM-500 and an Agilent6850 series II 5988A, respectively.
(20R)-3β-(tert-Butyldimethylsilyloxy)-24-oxo-25,26,27-trisnorcholest-5-ene (4)
To a solution of (20R)-3β-(tert-butyldimethylsilyloxy)-24-hydroxy-25,26,27-trisnorcholest-5-ene (18) (271 mg, 0.57 mmol) and NaOAc (17 mg, 0.21 mmol) in anhydrous CH2Cl2 (13 ml) under a N2 atmosphere was added PCC (160 mg, 0.74 mmol) in one portion. The reaction mixture was stirred for 20 h at room temperature, diluted with CH2Cl2 (5 ml), and filtered through a pad of silica gel. The filtrate was concentrated in vacuo. The product was purified via silica column chromatography using petroleum ether/CH2Cl2 (1:3) to give the title compound as a white solid (201 mg, 74%) (m.p. 148–152 °C (literature (19); m.p. 137–139 °C)). 1H and 13C NMR spectral data matches those reported in the literature (19). GC-MS: 457 (3, M+ − CH3), 415 (100, M+ − tBu), 397 (5), 355 (1), 281 (6), 207 (18), 81 (7), 75 (35), 41 (6).
(20R)-3β-(tert-Butyldimethylsilyloxy)-24,24-difluoro-25,26,27-trisnorcholest-5-ene (5)
DeoxoFluor (117 μl, 0.64 mmol) was added to a solution of aldehyde 4 (100 mg, 0.21 mmol) in anhydrous CH2Cl2 (12 ml). The reaction mixture was heated under reflux for 18 h and then cooled and a saturated aqueous solution of NaHCO3 (6 ml) was added slowly. The organic phase was separated and the aqueous phase was extracted with CH2Cl2 (3 × 6 ml). The combined organic phases were washed with brine (15 ml), dried (MgSO4), and concentrated in vacuo. The product was purified via silica column chromatography using petroleum ether/CH2Cl2 (9:1) to give the title compound as a white solid (62 mg, 59%) (m.p. 144–146 °C). 1H NMR (400 MHz, CDCl3): δ 0.04 (s, 6H, (CH3)2Si), 0.66 (s, 3H, 18-CH3), 0.87–2.00 (m, 38H, including 0.87 (s, 9H, (CH3)3CSi), 0.92 (d, J = 6.6 Hz, 3H, 21-CH3), 0.98 (s, 3H, 19-CH3)), 2.12–2.28 (m, 2H), 3.46 (m, 1H, 3α-CH), 5.29 (m, 1H, 6-CH), 5.75 (tt, J = 57 and 4.5 Hz, 1H, CF2H). 13C NMR (100 MHz, CDCl3): δ −4.6 (2C, (CH3)2Si), 11.8, 18.3, 18.4, 19.4, 21.0, 24.2, 25.9 (3C, (CH3)3CSi), 27.9 (t, 3JCF = 5.4 Hz), 28.1, 30.8 (t, 2JCF = 20.4 Hz), 31.9, 32.1, 35.1, 36.6, 37.4, 39.8, 42.4, 42.8, 50.2, 55.7, 56.8, 72.6 (3-C), 117.9 (t, 1JCF = 238 Hz), 121.1 (6-C), 141.6 (5-C). GC-MS: 479 (2, M+ − CH3), 437 (100, M+ − tBu), 438 (33), 419 (2), 361 (4), 343 (4), 281 (6), 255 (1), 207 (35), 81 (10), 75 (54), 41 (21). HR-MS (C30H52F2OSiNa): found 517.3644; calculated 517.3648.
(20R)-24,24-Difluoro-25,26,27-trisnorcholest-5-en-3β-ol (2)
To a solution of 5 (61 mg, 0.13 mmol) in anhydrous THF (10 ml) under a N2 atmosphere at 0 °C was added anhydrous pyridine (2 ml) and HF/pyridine (2.5 ml). The reaction mixture was stirred at room temperature for 24 h and then filtered through a pad of silica gel and the filtrate concentrated in vacuo to give a white solid. The product was purified via silica column chromatography using petroleum ether/CH2Cl2 (1:1) to give the title compound as a white solid (32 mg, 68%) (m.p. 125–127 °C). 1H NMR (400 MHz, CDCl3): δ 0.67 (s, 3H, 18-CH3), 0.91–2.30 [m, 31H, including 0.92 (d, J = 6.6 Hz, 3H, 21-CH3), 0.99 (s, 3H, 19-CH3)), 3.50 (m, 1H, 3α-CH), 5.33 (m, 1H, 6-CH), 5.75 (tt, 1H, J = 57 and 4.5 Hz, 24-CF2H). 13C NMR (100 MHz, CDCl3): δ 11.9, 18.4, 19.4, 21.1, 24.2, 27.9 (t, 3JCF = 4.9 Hz), 28.1, 30.8 (t, 2JCF = 20.4 Hz), 31.6, 31.86, 31.87, 35.1, 36.5, 37.2, 39.7, 42.28, 42.34, 50.1, 55.6, 56.7, 71.8 (3-C), 117.9 (t, 1JCF = 237 Hz), 121.6 (6-C), 140.7 (5-C). GC-MS: 381 (26), 380 (100, M+), 365 (26, M+ − CH3), 362 (52, M+ − H2O), 347 (47), 295 (49), 269 (63), 255 (22), 213 (31), 207 (40), 107 (45), 81 (39), 41 (86). HR-MS (C24H38F2ONa): found 403.2773; calculated 403.2783.
Ethyl(20R,24E)-3β-(tert-butyl-dimethylsilyloxy)-27-norcholesta-5,24-dien-26-oate (6)
A solution of aldehyde 4 (140 mg, 0.30 mmol) and ethyl 2-(triphenylphosphoranylidene)acetate (20) (309 mg, 0.90 mmol) in anhydrous CH2Cl2 (9 ml) was heated under reflux under a N2 atmosphere for 24 h. The reaction mixture was then concentrated in vacuo to give a pale yellow solid. The product was purified via silica column chromatography using petroleum ether/CH2Cl2 (7:3) to give the title compound as a white solid (140 mg, 87%) (m.p. 116–118 °C). 1H NMR (400 MHz, CDCl3): δ 0.04 (s, 6H, (CH3)2Si), 0.65 (s, 3H, 18-CH3), 0.87–2.28 (m, 43H, including 0.87 (s, 9H, (CH3)3CSi), 0.92 (d, J = 6.6 Hz, 3H, 21-CH3), 0.98 (s, 3H, 19-CH3), 1.27 (t, J = 6.9 Hz, 3H, CO2CH2CH3)), 3.46 (m, 1H, 3α-CH), 4.16 (q, J = 7.1 Hz, 2H, CO2CH2CH3), 5.29 (m, 1H, 6-CH), 5.78 (dt, J = 15.6 and 1.5 Hz, 1H), 6.94 (m, 1H). 13C NMR (100 MHz, CDCl3): δ −4.6 (2C, (CH3)2Si), 11.9, 14.3, 18.3, 18.5, 19.4, 21.0, 24.2, 25.9 (3C, (CH3)3CSi), 28.2, 29.0, 31.9, 32.1, 34.3, 35.5, 36.6, 37.4, 39.8, 42.4, 42.8, 50.2, 55.9, 56.8, 60.1, 72.6 (3-C), 121.0 (25-C), 121.1 (6-C), 141.6 (5-C), 150.0 (24-C), 166.8 (26-C). GC-MS: 527 (2, M+ − CH3), 486 (38), 485 (100, M+ − tBu), 365 (5), 347 (1), 281 (4), 255(1), 207 (16), 81 (6), 75 (25), 41 (14). HR-MS (C34H58O3SiNa): found 565.4052; calculated 565.4047.
(20R)-3β-(Tert-Butyldimethyl-silyloxy)-26-hydroxy-27-norcholest-5-ene (7)
A mixture of 6 (140 mg, 0.26 mmol) and PtO2 (7 mg, 5 wt %) in 1,4-dioxane (3.1 ml) and AcOH (62 μl) was placed under a H2 atmosphere. The reaction mixture was then stirred for 6 min. When almost all of the starting material was consumed as indicated by GC-MS, the mixture was filtered through a pad of celite and washed thoroughly with ethyl acetate. The filtrate was concentrated in vacuo to give the crude ester as a white solid that was used for the next step. To a suspension of LiAlH4 (44 mg, 1.2 mmol) in anhydrous THF (7 ml) was added the crude ester (126 mg, 0.23 mmol). The reaction mixture was heated under reflux for 2 h. The excess hydride was quenched by dropwise addition of water (44 μl), NaOH aqueous solution (44 μl, 15% w/v), and water (132 μl) successively. The reaction mixture was then filtered and the filtrate was concentrated in vacuo. The product was purified via silica column chromatography using CH2Cl2 to give the title compound as a white solid (104 mg, 80% over 2 steps) (m.p. 167–169 °C). 1H NMR (400 MHz, CDCl3): δ 0.04 (s, 6H, (CH3)2Si), 0.65 (s, 3H, 18-CH3), 0.86–2.28 (m, 44H, including 0.87 (s, 9H, (CH3)3CSi), 0.89 (d, J = 6.4 Hz, 3H, 21-CH3), 0.98 (s, 3H, 19-CH3)), 3.47 (m, 1H, 3α-CH), 3.62 (t, J = 6.7 Hz, 2H, 26-CH2), 5.29 (m, 1H, 6-CH). 13C NMR (100 MHz, CDCl3): δ −4.6 (2C, (CH3)2Si), 11.8, 18.3, 18.7, 19.4, 21.1, 24.3, 25.86, 25.94 (3C, (CH3)3CSi), 28.2, 31.90, 31.93, 32.1, 32.9, 35.7, 35.9, 36.6, 37.4, 39.8, 42.3, 42.8, 50.2, 56.1, 56.8, 63.1, 72.6 (3-C), 121.1 (6-C), 141.6 (5-C). GC-MS: 487 (1, M+ − CH3), 446 (36), 445 (100, M+ - tBu), 427 (4), 415 (6), 371 (1, M+ − OTBS), 281 (16), 255 (1), 207 (80), 81 (8), 75 (33), 41 (9). HR-MS (C32H58O23SiNa): found 525.4094; calculated 525.4098.
(20R)-3β-(Tert-Butyldimethyl-silyloxy)-26-oxo-27-norcholest-5-ene (8)
To a solution of 7 (136 mg, 0.27 mmol) and NaOAc (8 mg, 0.10 mmol) in anhydrous CH2Cl2 (7 ml) under a N2 atmosphere was added PCC (76 mg, 0.35 mmol) in one portion. The reaction mixture was stirred for 20 h at room temperature, diluted with CH2Cl2 (3 ml), and filtered through a pad of silica gel. The filtrate was concentrated in vacuo. The product was purified via silica column chromatography using petroleum ether/CH2Cl2 (1:3) to give the title compound as a white solid (86 mg, 64%) (m.p. 145–147 °C). 1H NMR (400 MHz, CDCl3): δ 0.03 (s, 6H, (CH3)2Si), 0.65 (s, 3H, 18-CH3), 0.87–2.42 (m, 44H, including 0.87 (s, 9H, (CH3)3CSi), 0.89 (d, J = 6.6 Hz, 3H, 21-CH3), 0.98 (s, 3H, 19-CH3)), 3.46 (m, 1H, 3α-CH), 5.29 (m, 1H, 6-CH), 9.74 (t, J = 1.9 Hz, 1H, 26-CHO). 13C NMR (100 MHz, CDCl3): δ −4.6 (2C, (CH3)2Si), 11.8, 18.3, 18.6, 19.4, 21.0, 22.5, 24.3, 25.6, 25.9 (3C, (CH3)3CSi), 28.2, 29.7, 31.89, 31.92, 32.1, 35.57, 35.64, 36.6, 37.4, 39.8, 42.3, 42.8, 44.0, 50.2, 56.0, 56.8, 72.6 (3-C), 121.1 (6-C), 141.6 (5-C), 203.0 (CHO). GC-MS: 485 (2, M+ − CH3), 444 (35), 443 (100, M+ − tBu), 281 (20), 255 (2), 207 (95), 81 (6), 75 (28), 41 (5).
(20R)-3β-(tert-Butyldimethylsilyloxy)-26,26-difluoro-27-norcholest-5-ene (9)
To a solution of 8 (60 mg, 0.12 mmol) in anhydrous CH2Cl2 (10 ml) was added DeoxoFluor (66 μl, 0.36 mmol). The reaction mixture was heated under reflux for 18 h, cooled, and a saturated aqueous solution of NaHCO3 (5 ml) was added slowly. The organic phase was separated and the aqueous phase was extracted with CH2Cl2 (3 × 5 ml). The combined organic phases were washed with brine (10 ml), dried (MgSO4), and concentrated in vacuo. The product was purified via silica column chromatography using petroleum ether/CH2Cl2 (9:1) to give the title compound as a white solid (47 mg, 76%) (m.p. 143–145 °C). 1H NMR (400 MHz, CDCl3): δ 0.03 (s, 6H, (CH3)2Si), 0.65 (s, 3H, 18-CH3), 0.87–2.28 (m, 44H, including 0.87 (s, 9H, (CH3)3CSi), 0.89 (d, J = 6.6 Hz, 3H, 21-CH3), 0.98 (s, 3H, 19-CH3)), 3.46 (m, 1H, 3α-CH), 5.29 (m, 1H, 6-CH), 5.77 (tt, J = 57 & 4.5 Hz, 1H, 26-CF2H). 13C NMR (100 MHz, CDCl3): δ −4.6 (2C, (CH3)2Si), 11.8, 18.3, 18.7, 19.4, 21.0, 22.6 (t, 3JCF = 5.3 Hz), 24.3, 25.5, 25.9 (3C, (CH3)3CSi), 28.2, 31.89, 31.92, 32.1, 34.1 (t, 2JCF = 20.4 Hz), 35.6, 35.7, 36.6, 37.4, 39.8, 42.3, 42.8, 50.2, 56.0, 56.8, 72.6 (3-C), 117.5 (t, 1JCF = 237 Hz), 121.1 (6-C), 141.6 (5-C). GC-MS: 507 (2, M+ − CH3), 466 (35), 465 (100, M+ − tBu), 389 (4), 371 (7), 281 (5), 255 (2), 207 (17), 81 (4), 75 (20), 41 (3). HR-MS (C32H56F2OSiNa): found 545.3938; calculated 545.3961.
(20R)-26,26-Difluoro-27-norcholest-5-en-3β-ol (3)
To a solution of 9 (47 mg, 0.09 mmol) in anhydrous THF (7 ml) under a N2 atmosphere at 0 °C was added anhydrous pyridine (1.4 ml) and HF/pyridine (1.7 ml). The reaction mixture was stirred at room temperature for 36 h, filtered through a pad of silica gel, and the filtrate concentrated in vacuo to give a white solid. The product was purified via silica column chromatography using CH2Cl2 to give a mixture of 3 (95%) and its C5 saturated derivative, (20R)-26,26-difluoro-27-norcholestan-3β-ol, 3a (5%) as a white solid (27 mg, total yield 73%) (m.p. 123–125 °C). 1H NMR (400 MHz, CDCl3): δ 0.62–0.65 (2x s, 3H, including 0.62 (s, 18-CH3 of 3a), 0.65 (s, 18-CH3 of 3)), 0.78–2.30 (m, 35H, including 0.78 (s, 19-CH3 of 3a), 0.89 (d, J = 6.6 Hz, 3H, 21-CH3 of 3), 0.99 (s, 19-CH3 of 3)), 3.46–3.60 (m, including 3.50 (m, 0.9H, 3α-CH of 3), 3.56 (m, 0.1H, 3α-CH of 3a)), 5.33 (m, 0.9H, 6-CH), 5.75 (tt, 1H, J = 57 and 4.5 Hz, 26-CF2H). 13C NMR (100 MHz, CDCl3): δ 11.8, 18.60, 18.65, 19.4, 21.1, 22.6 (t, 3JCF = 5.5 Hz), 24.3, 25.6, 28.2, 31.7, 31.9, 34.1 (t, 2JCF = 20.4 Hz), 35.6, 35.7, 36.5, 37.2, 39.8, 42.30, 42.32, 50.1, 56.0, 56.7, 71.8 (3-C), 117.5 (t, 1JCF = 240 Hz), 121.7 (6-C), 140.8 (5-C). GC-MS (compound 3): 409 (33), 408 (100, M+), 393 (28, M+ − CH3), 390 (54, M+ − H2O), 375 (41), 323 (43), 297 (48), 269 (14), 255 (21), 207 (7), 81 (17), 41 (23). GC-MS (compound 3a): 411 (28), 410 (100, M+), 395 (48, M+ − CH3), 377 (22), 284 (1), 233 (84), 215 (53), 206 (14), 165 (57), 108 (5). HR-MS (C26H42F2ONa): found 431.3088; calculated 431.3096.
Cytochrome P450 Expression and Purification
CYP125A1 and CYP142A1 were expressed and purified as previously described (21). Briefly, Escherichia coli DH5α cells containing the pCWori expression vector with the gene of interest were grown at 37 °C and 250 rpm in TB medium containing ampicillin (100 μg ml−1) to A600 = 0.7–0.8. Expression was induced by the addition of 1 mm isopropyl β-d-1-thiogalactopyranoside and 0.5 mm δ-aminolevulinic acid, and the culture continued for 36 h at 25 °C and 180 rpm. The cultures were harvested by centrifugation and stored at −80 °C. Cell pellets were thawed on ice and resuspended with agitation in 50 mm Tris-HCl, pH 7.5, buffer containing 0.15 m NaCl and 1 mm phenylmethylsulfonyl fluoride, followed by the addition of lysozyme (0.5 mg ml−1) and DNase (0.1 mg ml−1). The cells were disrupted by sonication using a Branson sonicator (six cycles of 1 min followed by 30-s rests), the sonicates were clarified by centrifugation at 100,000 × g for 45 min at 4 °C, and the proteins were purified on a nickel-nitrilotriacetic acid column washed with 10 column volumes of resuspension buffer plus 20 mm imidazole, and then eluted with resuspension buffer plus 250 mm imidazole. The eluted fractions were concentrated, dialyzed against 50 mm Tris-HCl, pH 7.5, and the enzyme concentration was determined from difference spectra using the extinction coefficient 91,000 m−1 cm−1 (22). The fraction of P420 species never exceeded 5%.
UV-visible Spectroscopy
UV-visible absorption spectra were recorded on a Cary UV-visible scanning spectrophotometer (Varian) using a 1-cm path length quartz cuvette at ambient temperature in 50 mm potassium phosphate buffer, pH 7.4, containing 150 mm NaCl, 10 mm MgCl2, and 0.05% MβCD. Spectral titrations were performed using 2 μm P450 with the sequential addition of substrates from concentrated stocks in methanol, with the same solution added to the reference cuvette. Difference spectra were recorded from 350 to 700 nm at a scanning rate of 120 nm/min. Data were fitted to the quadratic equation (Equation 1) using GraphPad Prism, where Aobs is the observed absorption shift, Amax is the maximal shift, KS is the apparent dissociation constant, [Et] is the enzyme concentration, and [S] is the ligand concentration (23).
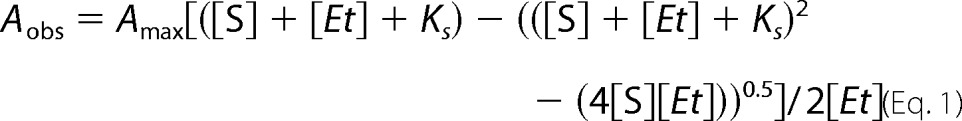 |
Cytochrome P450 Substrate Oxidation Assays
CYP125A1 and CYP142A1 (5 μm) were incubated at ambient temperature for 5 min with 0.1 mm substrate (compounds 1, 2, or 3) from a 10 mm stock dissolved in methanol in 50 mm potassium phosphate (pH 7.4) containing 0.05% (w/v) MβCD, 150 mm NaCl, and 10 mm MgCl2. Reactions were initiated by addition of an NADPH regeneration system consisting of 1 mm NADPH, 1 μm spinach ferredoxin, 0.2 unit ml−1 of spinach ferredoxin-NADP+ reductase, 0.1 mg ml−1 of bovine liver catalase, 0.4 unit ml−1 of glucose-6-phosphate dehydrogenase, and 5 mm glucose 6-phosphate. After 16 h the reactions were quenched with an equal volume of 1 n HCl, and the mixtures dried under N2 gas and then extracted five times with 0.5 ml of acetone. The combined extracts were dried under N2 gas, derivatized for 1 h at 37 °C with 75 μl of BSTFA (Sigma), and analyzed on an Agilent 6850 gas chromatograph fitted with an HP-5MS column (30 m, 0.25 mm internal diameter, Agilent) coupled to an Agilent 5973 network mass selective detector in the flame ionization mode operating at 70 eV. Helium was the carrier gas. The temperature program was 150 °C for 2 min, 150–300 °C at 10 °C/min, 300 °C for 18 min.
3β-HSD Expression and Purification
E. coli BL21(DE3)pLysS competent cells were transformed with a PET15b plasmid encoding M. tuberculosis 3β-HSD (Rv1106c) with a C-terminal His6 tag (11). Ampicillin-resistant colonies were picked and used to innoculate 100 ml of overnight small-scale LB cultures. A 15-ml aliquot of the overnight LB culture was transferred to 1 liter of YT medium containing 100 μg/ml of ampicillin. The cultures were grown at 37 °C and 220 rpm for ∼4 h until the optical density at 600 nm reached 0.8. Isopropyl β-d-1-thiogalactopyranoside was added at a final concentration of 0.4 mm to induce protein expression. The cultures were continued at 18 °C at 180 rpm for 20 h before harvesting. The cell cultures were harvested by centrifugation at 5,000 × g for 30 min and the cell pellets were frozen and stored at −80 °C. All purification steps were performed at 4 °C. The cell pellet (15 g) was suspended in 150 ml of buffer A (50 mm triethanolamine hydrochloride, pH 8.5) with one cOmpleteTM mini-protease inhibitor mixture tablet, EDTA-free (Roche), and lysed with sonication. Cell debris was removed by centrifugation at 35,000 rpm for 1 h. The supernatant was collected and 5 mm imidazole was added before loading onto a HisTrap HP column (GE Healthcare Life Sciences) that had been equilibrated with 10 column volumes of buffer A with 5 mm imidazole. After washing with 10 column volumes of buffer A with 5 mm imidazole, 10 column volumes of buffer A with 50 mm imidazole, and 10 column volumes of buffer A with 100 mm imidazole, the 3β-HSD protein was eluted with buffer A with 500 mm imidazole. The eluted 3β-HSD protein was pooled and assayed at 30 °C by monitoring NADH absorbance at 340 nm in buffer A in the presence of 120 μm cholesterol.
3β-HSD Oxidation of the 3β-Hydroxy Group in Cholesterol Analogs
The 3β-HSD catalyzed reactions contained 0.41 pm 3β-HSD, 1.4 mm NAD, and 120 μm triol or difluoro compounds in 50 mm triethanolamine hydrochloride buffer, pH 8.5. The reactions were terminated with the same volume of 1 n HCl. For HPLC analysis, the acidified samples were centrifuged and the supernatants were collected and analyzed on an Agilent 1200 series HPLC system with a reverse phase C18 column (Waters Xterra C18, 3.5 μm, 2.1 × 50 mm). The triol and difluoro compounds and their metabolites were monitored at 240 nm. For the reactions with difluoro compounds, the samples were eluted isocratically at a flow rate of 0.5 ml/min (solvent A, H2O + 0.1% formic acid; solvent B, CH3CN + 0.1% formic acid) with a gradient starting at 70% B for 1 min and then increasing to 100% B over 11 min. The elution was maintained at 100% B up to 14 min and then decreased to 70% B over 1 min, followed by equilibration at the same composition for 2 min before the next run. For the triol reactions, the samples were eluted isocratically at a flow rate of 0.5 ml/min (solvent A, H2O + 0.1% formic acid; solvent B, CH3CN + 0.1% formic acid) with a gradient starting at 50% B for 1 min and then increasing to 80% B over 11 min. The solvent was then increased to 100% B over 0.5 min and maintained at 100% B until 14 min and then decreased to 50% B within 1 min, followed by equilibration at the same composition for 2 min before the next run. Identification of the metabolites of the triol compound was accomplished by GC-MS analysis. The terminated reaction samples (0.5 ml) were extracted with hexane (2 × 2 ml). After vortexing, the mixture was centrifuged (2,000 rpm, 5 min) and the organic layer was separated and evaporated under nitrogen. The dried sample residue was then trimethylsilylated to give the trimethylsilyl derivatives by resuspension in 100 μl of BSTFA (Pierce) for 1 h at 37 °C. The reaction mixtures were analyzed using an HP5790 gas chromatography system fitted with a DB5-MS column (30 m × 0.25 mm × 0.25 μm). Separation was achieved by using a column temperature of 70 °C for 2 min, then increasing by 10 °C/min to 300 °C, and finally holding at 300 °C for 5 min. The reaction samples with difluoro compounds were analyzed by LC-MS. Identification of the metabolites of difluoro compounds was accomplished by LC-MS on a Waters Micromass ZQ (Waters, Milford, Massachusetts) coupled to a Waters Alliance HPLC equipped with a 2695 separation module, a Waters 2487 Dual λ Absorbance detector, and Waters Xterra C18 column (3.5 μm, 2.1 × 50 mm). The mass spectrometer was operated in the positive ion mode to monitor the product ion of m/z 379 for the oxidation product of compound 2 and m/z 407 for the oxidation product of compound 3. The mass spectrometer settings for metabolite detection were as follows: mode, ES+; capillary voltage, 2.0 kV; cone voltage, 45 V; extractor voltage, 10 V; RF voltage, 0.5 V; desolvation temperature, 300 °C; source temperature, 120 °C; desolvation flow, 350 liters/h; cone flow, 65 liters/h. The UV absorbance at 240 nm and MS ES+ spectra were recorded simultaneously.
M. tuberculosis in Vitro Culture
The wild-type CDC1551 strain was obtained from Colorado State University, and wild-type Erdmann strain from the Cox laboratory (University of California, San Francisco, CA). All experiments with M. tuberculosis cells were performed in a biosafety level-3 (BSL-3) facility. The CDC1551 Δcyp125A1 mutant strain was created by homologous recombination using specialized transducing phages as described previously (4). M. tuberculosis cells were cultured in 7H9 medium supplemented with 10% oleic acid-albumin-dextrose complex, 0.2% glycerol, and 0.05% Tween 80. For growth in carbon-restricted media, log-phase cells grown in 7H9 medium were used to inoculate a defined minimal medium (1 g/liter of KH2PO4, 2.5 g/liter of Na2HPO4, 0.5 g/liter of asparagine, 50 mg/liter of ferric ammonium citrate, 10 mg/liter of MgSO4·7H2O, 0.5 mg/liter of CaCl2, and 0.1 mg/liter of ZnSO4), and 0.1% glycerol at an initial A600 nm of 0.05. Then transferred to the same defined media without glycerol, with the addition of the appropriate carbon source (0.1% glycerol, 0.5% glucose, or 0.5% acetate), with or without 0.1 mm oxysterol supplements (4-cholesten-3-one, (25R)-cholest-5-en-3β, 16β, 26-triol (1), (20R)-24,24-difluoro-25,26,27-trisnorcholest-5-en-3β-ol (2), or (20R)-26,26-difluoro-27-norcholest-5-en-3β-ol (3)) at an initial A600 nm of 0.05. A stock solution of oxysterol (50 mg/ml) was prepared in tyloxapol/ethanol (1:1) and warmed at 65 °C for 20 min before addition.
Results
Growth Inhibition by Cholest-4-en-3-one
As already noted, CYP125A1 and CYP142A1 are required for the catabolism of cholesterol in Mtb. In the absence of these enzymes in an active form, cholest-4-en-3-one accumulates due to the action of a mycobacterial 3β-sterol dehydrogenase. In wild-type Mtb, growth on cholesterol is retarded in the presence of large amounts of cholesterol, or added cholest-4-en-3-one, with growth occurring as the cholest-4-en-3-one concentration decreases. Furthermore, cholest-4-en-3-one inhibits the growth of Mtb in defined media with glycerol as the sole carbon source (14). It is therefor able to interfere with the glycerol, as well as cholesterol, metabolic pathways. To determine whether cholest-4-en-3-one can inhibit growth on other carbon sources, we investigated its effects on cells growing on acetate or glucose. In effect, in the absence of CYP125A1 and CYP142A1, growth on both of these alternative carbon sources was also blocked by exogenously added cholest-4-en-3-one (Fig. 1, A, B, D, and E).
FIGURE 1.

Effect of cholest-4-en-3-one and 1 on growth of wild-type (A) or Δcyp125A1 (B, D, and E) strains of CDC1551, and Erdmann (C) Mtb on defined media with glycerol (A–C), acetate (D), or glucose (E) as the sole carbon source. Data shown are the average of three independent growths.
Interaction of (1) with CYP125A1 and CPY142A1
Triol 1 (the “triol”), an inhibitor of the growth of H37Rv Mtb in culture (16), is closely related in structural terms to 26-hydroxycholesterol, the first product generated by the action of CYP125A1 and CYP142A1 on cholesterol (4). To determine whether this cholesterol analog, like cholesterol, was a substrate for the subsequent conversion of the 26-hydroxy group to the aldehyde and carboxylic acid, we investigated the interaction of 1 with these two mycobacterial P450 enzymes. As shown in Fig. 2, triol (1) binds to CYP125A1 in a concentration-dependent manner with a Ks = 15.4 ± 1.1 μm. The reverse type-1 spectrum (Fig. 2, inset) that is observed suggests that binding of the sterol does not displace the distal water ligand from the heme iron atom, but rather may sterically, or through hydrogen bonding, trap the distal water, promoting its coordination as the distal ligand (24). In contrast, only minor spectroscopic changes were observed on attempted binding of 1 to CYP142A1. If the sterol binds to this P450 enzyme, it does so with very little perturbation of the active site environment.
FIGURE 2.

Reverse type I binding of 1 to CYP125A1 (2 μm) as determined by difference spectroscopy. The fit of the data to the quadratic binding equation gives Ks = 15.4 ± 1.1 μm. Data shown are the average of three independent titrations, error is the standard error. AU, absorbance units.
Unexpectedly, catalytic assays searching for the oxidation of 1 to the corresponding aldehyde or acid were unsuccessful under conditions where the conversion of cholesterol to the acid was readily observed (data not shown). Thus, although the triol binds to CYP125A1, it is not a detectable substrate of the enzyme, and the triol is neither a detectable ligand nor substrate of CYP142A1.
Inhibition of Mtb Growth by 1
The wild-type CDC1551 strain of Mtb has an active CYP125A1, but lacks a functional CYP142A1. Knock-out of the cyp125A1 gene therefore produces a strain that lacks both of the P450 enzymes that are able to effectively oxidize the C26-carbon of cholesterol and cholest-4-en-3-one (4). As shown in Fig. 1A, the CDC1551 strain, which grows normally on glycerol, is able to grow, perhaps even better, when 0.1 mm cholest-4-en-3-one is added to the medium. The Erdmann strain of Mtb, which retains active forms of both CYP125A1 and CYP142A1, also grows well on glycerol with or without the addition of cholest-4-en-3-one (Fig. 1C). However, as reported previously (14), the CDC1551 Δcyp125A1 mutant, which lacks both CYP125A1 and CYP142A1, is not able to grow on glycerol in the presence of cholest-4-en-3-one.
In striking contrast, wild-type CDC1551, which grew well in the presence of cholest-4-en-3-one because this compound can be catabolized, was completely unable to grow on glycerol in the presence of 1 (Fig. 1A). Interestingly, the triol completely inhibits the growth of the wild-type Erdmann strain, which has both active CYP125A1 and CYP142A1 (Fig. 1C). The triol thus inhibits growth on glycerol whether the two P450 enzymes that oxidize the cholesterol side chain are present or not. Furthermore, triol 1 blocks growth of the CDC1551 strain of Mtb not only on glycerol, but also on acetate or glucose (not shown).
Synthesis of 2 and 3
Difluoromethyl analogs 2 and 3 (Scheme 1) were synthesized from the corresponding protected aldehydes (4 and 8) by reaction with DeoxoFluor (25). The rationale for the choice of these two compounds is presented under “Discussion.” The side chain of all of the targets was constructed from the known precursor (20S)-3β-(tert-butyldimethylsilyloxy)-20-hydroxymethylpregn-5-ene, using a variety of standard transformations (Scheme 1) (18). The 5,6-double bond was not completely inert to the hydrogenation conditions utilized to remove the Δ16 alkene and consequently a small amount (<10%) of the chromatographically inseparable compounds lacking the C5 double bond was formed concomitantly with the desired analogs (2 and 3).
SCHEME 1.

Synthesis of difluoromethyl analogs 2 and 3. Reagents and conditions: (a) i, see Ref. 18; ii, PCC, NaOAc, CH2Cl2, 74%. (b) DeoxoFluor, CH2Cl2, reflux, 5: 59%, 9: 76%. (c) i, ethyl 2-(triphenylphosphoranylidene)acetate (20), CH2Cl2, reflux, 87%; ii, H2, PtO2, 1,4-dioxane/AcOH; iii, LiAlH4, THF, reflux (80% over 2 steps); iv, PCC, NaOAc, CH2Cl2, 64%. (d) HF/pyridine, THF, 2: 68%, 3: 73%. The structure of compound 1, independently synthesized by a literature procedure (17), is also shown.
Inhibition of CDC1551 Mtb by Side Chain Fluorinated Cholesterol Analogs 2 and 3
Incubation of CDC1551 Mtb in defined medium with glycerol as the carbon source results in normal growth of the mycobacteria. However, in the presence of a 0.1 mm concentration of either 2 or 3, growth is completely prevented (Fig. 3). The same is true if the Δcyp125a1 mutant strain of CDC1551 is substituted for the wild-type CDC1551 strain.
FIGURE 3.

Inhibition of the growth on 0.1% glycerol of wild-type CDC1551 Mtb and its Δcyp125A1 mutant by 0.1 mm compound 2 (A) and 0.1 mm compound 3 (B). Data shown are the average of three independent growths.
Oxidation of 1, 2, and 3 to the 3-Keto Derivatives by 3β-HSD
Earlier studies have shown that cholest-4-en-3-one is a better substrate for oxidation by CYP125A1 and CYP142A1 than cholesterol itself (4). We therefore examined whether 1 could be oxidized by the mycobacterial 3β-HSD to the 3-keto-4-ene form. As shown in (Fig. 4), triol 1 is readily converted to the corresponding keto form under conditions that similarly convert cholesterol to cholest-4-en-3-one. The structure of the metabolite was firmly established by the mass spectrum, which shows a loss of two hydrogen atoms, and by the acquisition of a UV-visible spectrum consistent with the presence of the 3-keto-4-ene chromophore. Similar experiments with fluorosteroids 2 and 3 (Fig. 4) establish that these compounds are also readily oxidized to the 3-keto-4-ene forms by 3β-HSD. Thus, the triol and the two fluorosteroids can be converted to the presumably more inhibitory 3-keto-4-ene metabolites.
FIGURE 4.

Oxidation of 1, 2, and 3 by 3β-HSD to the 3-keto derivatives. HPLC analysis of the oxidation of 1 by 3β-HSD (A) and cholesterol oxidase (B); and the oxidation of 2 (C) and 3 (D) by 3β-HSD.
Discussion
An important finding of this study is that cholest-4-en-3-one inhibits growth of Mtb on defined medium, not only when glycerol is the sole source of carbon, but also when the glycerol is replaced by acetate or glucose (Fig. 1). Cholest-4-en-3-one also inhibits growth on cholesterol, but inhibition in all these cases is transient, as cholest-4-en-3-one is catabolized by the mycobacteria if they retain the activity of either (or both) CYP125A1 or CYP142A1 (4). Cholest-4-en-3-one thus acts as an inhibitor at one or more crucial sites in the pathways that enable the utilization of all these carbon sources. The identities of these sites remain unknown, but they are clearly of interest as potential sites for antitubercular drug development.
Cholest-4-en-3-one, whether generated within the mycobacteria from cholesterol or added exogenously, is not of interest as an inhibitor of Mtb growth because it is rapidly catabolized and only has a transient existence. Nevertheless, the inhibitory properties of cholest-4-en-3-one become apparent when examined with strains of Mtb that lack both CYP125A1 and CYP142A1 (4, 14). These results suggest that analogs of cholesterol (or cholest-4-en-3-one) with degradation-resistant side chains might be inhibitors of wild-type Mtb growth, so long as the modifications of the sterol structure do not interfere with its interaction at the sites at which growth inhibition is imposed. We establish here that, unexpectedly, this can be achieved by placing a 16-hydroxyl group on the tetracyclic sterol nucleus, as shown by the fact that 1 is not a substrate for either CYP125A1 or CYP142A1. Although spectroscopic evidence shows that this sterol can bind to CYP125A1 (but possibly not to CYP142A1), it does so in a non-productive manner that prevents side chain oxidation. Triol 1 has two more hydroxyl groups than cholesterol, but the 26-hydroxyl group is equivalent to the 26-hydroxyl group that is added to cholesterol by the P450 enzymes in their first catalytic turnover and therefore cannot contribute to the resistance to side chain degradation. Furthermore, the mycobacterial 3β-HSD oxidizes the 3-hydroxyl group of 1 in the same way as it does the 3-hydroxyl group of cholesterol (Fig. 4). Thus, the key substitution in this molecule is the 16-hydroxyl group. The spectroscopic observation of a reverse type I binding spectrum with CYP125A1, together with the failure of the P450 enzymes to oxidize the triol, suggests that the 16-hydroxyl sterically interferes with internalization of the sterol side chain into the P450 active site. This is consistent with the observation of a reverse type I binding spectrum, which suggests that the water ligand on the heme iron atom has not been displaced, a necessary step for electron transfer, oxygen binding, and catalysis.
In effect, the 16-hydroxyl group of 1 makes it a degradation-resistant analog of cholesterol, and its 3-keto metabolite a similar analog of cholest-4-en-3-one. As a result, the triol is a potent inhibitor of the growth of Mtb, presumably acting at the same target sites as cholest-4-en-3-one.
To further test this hypothesis, we synthesized two cholesterol analogs with truncated, fluorinated side chains. In one case (3), one methyl of the terminal isopropyl function has been deleted and the other substituted with two fluorine atoms. In the other case, the entire isopropyl function has been deleted and two fluorines added to the terminus of the truncated side chain (2). In previous work (18), we have shown that 24-bromochol-5-en-3-ol, in which the side chain is truncated to the same extent as in 2, yields a sterol that binds to CYP125A1 and CYP142A1, but is not a detectable substrate. Removal of one methyl from the side chain terminus, as in 27-norcholesterol, gives a compound that also binds to the two P450 enzymes, but in this case is catalytically oxidized by the enzymes to give the terminal hydroxylation product with kcat rates 5–8 times slower than for cholesterol itself (18). As predicted from these earlier findings, 2 did not give a detectable product when incubated with either of the two P450 enzymes (data not shown). Again, as predicted by the earlier work, 3 was slowly oxidized by both enzymes to the carboxylic acid. HPLC and GC-MS analysis showed formation of the acid metabolite with a retention time of 26.5 min and the expected parent molecular ion of 546.5 Da for the di-trimethylsilylated derivative (Fig. 5). The acid is expected from hydroxylation of the terminal -CHF2 group, as it will spontaneously eliminate fluoride to give an acyl fluoride, which in turn could undergo spontaneous hydrolysis to the acid (26). A minor additional product with a GC-MS retention time of 24.5 min and a parent molecular mass of 488.5 Da was also observed. This metabolite corresponds to the same acid, but with the carboxyl group present as a methyl ester due to trapping of the acyl fluoride intermediate by methanol (Scheme 2). Trimethylsilylation is then only possible at the 3β-hydroxyl group (Fig. 5). Methanol is present in low amounts in the incubation as the sterol substrate is added as a methanol solution. Formation of the C26 methyl ester in itself confirms formation of the acyl fluoride in the incubation. A nucleophile on the P450 enzyme could also conceivably compete with water (or methanol) for addition to the acyl fluoride, resulting in covalent binding to the P450 enzyme, but we have no evidence in this regard. Most importantly, both 2 and 3 completely block growth of CDC1551 Mtb on media containing glycerol as the carbon source. This inhibition does not depend on the presence or absence of CYP125A1.
FIGURE 5.

GC-MS analysis of the oxidation of compound 3 (solid line) to the carboxylic acid (retention time 26.5 min) by CYP125A1 (small dashes) and CYP142A1 (large dashes). A second unidentified metabolite is seen at retention time 24.5 min.
SCHEME 2.

Oxidation of difluoromethyl analog 3 by CYP125A1 and CYP142A1 to an acyl fluoride that can be trapped by water or methanol.
Previous studies of the central carbon metabolism in Mtb note the importance of the isocitrate lyase genes for growth on cholesterol in vitro and for virulence during mouse infections (27). This toxicity was attributed to the buildup of toxic amounts of propionate, which would not be likely in our model as cholesterol serves as a precursor to propionate. Indeed, previous work noted that supplementation of propionate inhibited growth of Mtb on glycerol, but failed to inhibit growth on acetate (28), unlike our findings. The results presented here thus implicate a distinct and more robust mechanism of growth inhibition. They define a new paradigm for the development of agents that can, at least in culture, block the growth of M. tuberculosis. Further exploration of this paradigm is a promising route for the development of therapeutically useful inhibitors.
Author Contributions
D. J. F. did all the biological and analytical work on P450 enzymes, Y. Z. the work with the dehydrogenase, and S. H. W. and D. B. the synthetic work. J. J. D.V. directed the synthetic work and P. R. O. M. conceived the project and directed the biological studies. All the authors participated in writing the manuscript.
Acknowledgments
We thank Dr. Jeffrey Cox, and members of the P. O. M. and Cox laboratories for their technical assistance and useful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grant AI074824. The authors have no conflict of interest.
- Mtb
- Mycobacteriuam; tuberculosis
- 3β-HSD
- 3β-hydroxysteroid dehydrogenase
- BSTFA
- N,O-bis(trimethyl-silyl)trifluoroacetamide
- MβCD
- methyl-β-cyclodextrin
- THF
- tetrahydrofuran.
References
- 1. World Health Organization (2015) Tuberculosis, Fact Sheet No. 104, World Health Organization, http://www.who.int/mediacentre/factsheets/fs104/en/
- 2. Sharma S. K., and Mohan A. (2006) Multidrug-resistant tuberculosis: a menace that threatens to destabilize tuberculosis control. Chest 130, 261–272 [DOI] [PubMed] [Google Scholar]
- 3. Ouellet H., Johnston J. B., and de Montellano P. R. (2011) Cholesterol catabolism as a therapeutic target in Mycobacterium tuberculosis. Trends Microbiol. 19, 530–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Johnston J. B., Ouellet H., and Ortiz de Montellano P. R. (2010) Functional redundancy of steroid C26-monooxygenase activity in Mycobacterium tuberculosis revealed by biochemical and genetic analyses. J. Biol. Chem. 285, 36352–36360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brzostek A., Pawelczyk J., Rumijowska-Galewicz A., Dziadek B., and Dziadek J. (2009) Mycobacterium tuberculosis is able to accumulate and utilize cholesterol. J. Bacteriol. 191, 6584–6591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Capyk J. K., Kalscheuer R., Stewart G. R., Liu J., Kwon H., Zhao R., Okamoto S., Jacobs W. R. Jr., Eltis L. D., and Mohn W. W. (2009) Mycobacterial cytochrome P450 125 (CYP125) catalyzes the terminal hydroxylation of C27-steroids. J. Biol. Chem. 284, 35534–35542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gatfield J., and Pieters J. (2000) Essential role for cholesterol in entry of mycobacteria into macrophages. Science 288, 1647–1650 [DOI] [PubMed] [Google Scholar]
- 8. Chang J. C., Harik N. S., Liao R. P., and Sherman D. R. (2007) Identification of mycobacterial genes that alter growth and pathology in macrophages and in mice. J. Infect. Dis. 196, 788–795 [DOI] [PubMed] [Google Scholar]
- 9. Chang J. C., Miner M. D., Pandey A. K., Gill W. P., Harik N. S., Sassetti C. M., and Sherman D. R. (2009) igr Genes and Mycobacterium tuberculosis cholesterol metabolism. J. Bacteriol. 191, 5232–5239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Driscoll M. D., McLean K. J., Levy C., Mast N., Pikuleva I. A., Lafite P., Rigby S. E., Leys D., and Munro A. W. (2010) Structural and biochemical characterization of Mycobacterium tuberculosis CYP142. J. Biol. Chem. 285, 38270–38282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang X., Dubnau E., Smith I., and Sampson N. S. (2007) Rv1106c from Mycobacterium tuberculosis is a 3β-hydroxysteroid dehydrogenase. Biochemistry 46, 9058–9067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Klink M., Brzezinska M., Szulc I., Brzostek A., Kielbik M., Sulowska Z., and Dziadek J. (2013) Cholesterol oxidase is indispensable in the pathogenesis of Mycobacterium tuberculosis. PLOS One 8, e73333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Griffin J. E., Gawronski J. D., Dejesus M. A., Ioerger T. R., Akerley B. J., and Sassetti C. M. (2011) High-resolution phenotypic profiling defines genes essential for Mycobacterial growth and cholesterol catabolism. PloS Pathogens 7, e1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ouellet H., Guan S., Johnston J. B., Chow E. D., Kells P. M., Burlingame A. L., Cox J. S., Podust L. M., and de Montellano P. R. (2010) Mycobacterium tuberculosis CYP125A1, a steroid C27 monooxygenase that detoxifies intracellularly generated cholest-4-en-3-one. Mol. Microbiol. 77, 730–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pandey A. K., and Sassetti C. M. (2008) Mycobacterial persistence requires the utilization of host cholesterol. Proc. Natl. Acad. Sci. U.S.A. 105, 4376–4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schmidt A. W., Choi T. A., Theumer G., Franzblau S. G., and Knölker H. J. (2013) Inhibitory effect of oxygenated cholestan-3β-ol derivatives on the growth of Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 23, 6111–6113 [DOI] [PubMed] [Google Scholar]
- 17. Alessandrini L., Ciuffreda P., Santaniello E., and Terraneo G. (2004) Clemmensen reduction of diosgenin and kryptogenin: synthesis of [16,16,22,22,23,23-2H6]-(25R)-26-hydroxycholesterol. Steroids 69, 789–794 [DOI] [PubMed] [Google Scholar]
- 18. Johnston J. B., Singh A. A., Clary A. A., Chen C.-K., Hayes P. Y., Chow S., De Voss J. J., and Ortiz de Montellano P. R. (2012) Substrate analog studies of the ω-regiospecificity of Mycobacterium tuberculosis cholesterol metabolizing cytochrome P450 enzymes CYP124A1, CYP125A1 and CYP142A1. Bioorg. Med. Chem. 20, 4064–4081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Martin R., Entchev E. V., Däbritz F., Kurzchalia T. V., and Knölker H.-J. (2009) Synthesis and hormonal activity of the (25S)-cholesten-26-oic acids: potent ligands for the DAF-12 receptor in Caenorhabditis elegans. Eur. J. Org. Chem. 2009, 3703–3714 [DOI] [PubMed] [Google Scholar]
- 20. Sadhukhan S., Han Y., Zhang G.-F., Brunengraber H., and Tochtrop G. P. (2010) Using isotopic tools to dissect and quantitate parallel metabolic pathways. J. Am. Chem. Soc. 132, 6309–6311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ouellet H., Lang J., Couture M., and Ortiz de Montellano P. R. (2009) Reaction of Mycobacterium tuberculosis cytochrome P450 enzymes with nitric oxide. Biochemistry 48, 863–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Omura T., and Sato R. (1964) The carbon monoxide-binding pigment of liver microsomes: II. solubilization, purification, and properties. J. Biol. Chem. 239, 2379–2385 [PubMed] [Google Scholar]
- 23. Morrison J. F. (1969) Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim. Biophys. Acta 185, 269–286 [DOI] [PubMed] [Google Scholar]
- 24. Schenkman J. B., Cinti D. L., Orrenius S., Moldeus P., and Kraschnitz R. (1972) The nature of the reverse Type I (modified Type II) spectral changes in liver microsomes. Biochemistry 11, 4243–4251 [DOI] [PubMed] [Google Scholar]
- 25. Lal G. S., Pez G. P., Pesaresi R. J., Prozonic F. M., and Cheng H. (1999) Bis(2-methoxyethyl)aminosulfur trifluoride: a new broad-spectrum deoxofluorinating agent with enhanced thermal stability. J. Org. Chem. 64, 7048–7054 [Google Scholar]
- 26. Halpert J. R., and Stevens J. C. (1991) Dihalomethyl compounds as mechanism-based inactivators of cytochromes P450. Methods Enzymol. 206, 540–548 [DOI] [PubMed] [Google Scholar]
- 27. Muñoz-Elías E. J., and McKinney J. D. (2005) Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat. Med. 11, 638–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee W., VanderVen B. C., Fahey R. J., and Russell D. G., (2013) Intracellular Mycobacterium tuberculosis exploits host-derived fatty acids to limit metobolic stress. J. Biol. Chem. 288, 6788–6800 [DOI] [PMC free article] [PubMed] [Google Scholar]