

ABSTRACT
The promoter most strongly induced upon activation of the Cpx two-component envelope stress response is the cpxP promoter. The 3′ untranscribed region (UTR) of the cpxP transcript is shown to produce a small RNA (sRNA), CpxQ. We investigated the role of CpxQ in combating envelope stress. Remarkably, the two effectors specified by the transcript are deployed to combat distinct stresses in different cellular compartments. CpxP acts in both a regulatory negative-feedback loop and as an effector that combats periplasmic protein misfolding. We find that CpxQ combats toxicity at the inner membrane (IM) by downregulating the synthesis of the periplasmic chaperone Skp. Our data indicate that this regulation prevents Skp from inserting β-barrel outer membrane proteins (OMPs) into the IM, a lethal event that likely collapses the proton motive force. Our findings suggest that Skp can fold and directly insert OMPs into a lipid bilayer in vivo without the aid of the Bam complex.
IMPORTANCE
Skp is a well-characterized periplasmic chaperone that binds unfolded OMPs. Surprisingly, we find that Skp can catalyze the folding and mistargeting of OMPs into the inner membrane without the aid of the other cellular proteins that normally assemble OMPs. Several OMPs function as diffusion pores. Accordingly, their mistargeting is lethal because it depolarizes the inner membrane. We show that the most highly expressed transcript of the Cpx stress response produces an sRNA from the 3′ UTR, CpxQ, which combats this potential toxicity by downregulating Skp production. Defects in OMP assembly trigger the σE response to upregulate factors, including Skp, that promote OMP folding. The Cpx response downregulates σE. Our findings reveal that this heretofore puzzling hierarchy exists to protect the inner membrane.
INTRODUCTION
Gram-negative bacteria build a complex envelope architecture where biologically distinct inner and outer membranes (IM and OM) are separated by an aqueous periplasmic space. Several macromolecular assembly machines are involved in envelope biogenesis, and some can function in the absence of available energy sources (1). The complexity of coordinate envelope biogenesis during cell growth is underpinned by several stress response systems that sense envelope defects and alter gene expression to either alleviate or clear the damage (2). Some of these systems are specific: for example, σE responds primarily to OM membrane defects (3, 4). On the other hand, the Cpx stress response is invoked to respond to stress signals originating throughout the envelope (5). The phage-shock-protein (Psp) response is activated by IM damage that reduces the proton motif force (PMF) (6).
At the core of the Cpx two-component stress response system are the sensor kinase CpxA and response regulator CpxR (5, 7). CpxA is a polytopic IM protein with dual kinase and phosphatase activity that can detect stress signals via its periplasmic sensing domain (8). Activation of CpxA leads to autophosphorylation and then phosphotransfer to CpxR, enabling it to alter transcription of regulon members (8, 9).
A set of mutations in cpxA cause constitutive activation of Cpx (8). These dominant cpxA* alleles include cpxA17, causing an A188E substitution proximal to the site of autophosphorylation; and cpxA24, resulting in a deletion within the periplasmic sensing domain. These mutations have proven valuable in identifying members of the CpxR regulon (10). One of the most highly upregulated genes is cpxP, located immediately upstream from cpxRA. CpxP is a periplasmic protein that completes a negative-feedback loop by inhibiting CpxA activation, likely by interacting with the sensing domain (9, 11, 12). However, CpxA* proteins are refractory to CpxP inhibition (13). A second highly upregulated CpxR target is degP, which encodes a periplasmic protein with dual chaperone and protease function (14).
cpxA* alleles suppress a variety of envelope toxicities, including a toxicity caused by tethering LamB to the IM by its uncleaved signal sequence (15), a jamming toxicity of the Sec machine with folded LamB-LacZ (15), a periplasmic toxicity that occurs when LamB-LacZ is fully translocated from the cytoplasm (15), and a distinct periplasmic toxicity caused by misfolded P-pilus subunits (16).
The lamB(A23D) mutation alters the signal peptidase cleavage site of the OM maltoporin and causes delayed release of the mature protein (17). Accordingly, LamB(A23D) is efficiently translocated into the periplasm through the Sec translocon but remains tethered to the IM via its signal peptide rather than being released and assembled into the OM (17). Expression of lamB(A23D) is induced by maltose, and the resultant high-level production is toxic. cpxA* alleles suppress this toxicity (15). DegP is required but not sufficient for suppression. It is not known why LamB(A23D) is toxic or which additional Cpx regulon members are required to suppress toxicity (15).
Jamming toxicity is caused by the LamB-LacZ42-1 fusion protein (18). The LamB sequence targets the fusion to the Sec translocon and initiates secretion; however, the LacZ sequence folds rapidly in the cytoplasm, and the folded fusion cannot pass through Sec. The fusion jams the translocon, prompting the FtsH protease to degrade SecY (19). cpxA* alleles suppress LamB-LacZ42-1 toxicity by inducing production of YccA, which acts to stabilize SecY against proteolysis, allowing time for the hybrid protein to clear the translocator (19).
LamB-LacZ jamming toxicity can also be relieved either by mutations in the signal sequence that allow cotranslational secretion (H*LamB-LacZ) or by mutations that prevent LacZ folding (LamB-LacZX90). However, efficient translocation of these fusions then causes a periplasmic toxicity because the cysteine-rich LacZ misfolds and aggregates in the oxidizing environment of the periplasm (20–22). cpxA* mutations fully suppress the toxicity of these fusions because they overexpress degP, and the periplasmic protease degrades the fusion proteins. In fact, heterologous overproduction of DegP is sufficient to suppress periplasmic LacZ toxicity (21).
The PapE and PapG subunits of the uropathogenic Escherichia coli P-pilus are chaperoned in the periplasm by PapD and brought to the PapC usher for assembly (23). In E. coli K-12, the absence of PapD causes pilin subunits to misfold, aggregate, and stimulate the Cpx stress response (24). CpxP acts as an adaptor that binds misfolded pilins and delivers them to DegP for proteolytic degradation along with CpxP itself (16, 25). Indeed, misfolded pilins sequester CpxP from CpxA to relieve inhibition and allow activation of the Cpx stress response (16, 24). Unlike periplasmic LacZ toxicity, resistance against PapE/G toxicity requires both cpxP and degP (16).
Recent work in Salmonella identified an Hfq-stabilized small RNA (sRNA) product, named CpxQ, that is derived from the 3′ untranscribed region (UTR) of the cpxP mRNA (26). Cpx inducing conditions strongly activate cpxP transcription and so increase production of CpxQ, suggesting this sRNA may play a direct role in response to stress. In this work, we assess the effect of CpxQ on the production of CpxP in E. coli and explore its contribution to combating the different stresses alleviated by Cpx. We show that production of CpxQ can lower the levels of CpxP. Moreover, we find that although CpxP and CpxQ originate from the same mRNA, they mature to combat unique stresses at different sites of the cell envelope.
RESULTS
CpxQ negatively regulates production of CpxP.
Since CpxQ and CpxP are products of the same mRNA transcript, it was possible the sRNA was produced at the expense of the transcript, causing lowered CpxP production (26). In wild-type cells, CpxP abundance is low and undetectable by immunoblotting. Hence, we employed a multicopy plasmid system to investigate any effect of CpxQ on CpxP. We used the previously described pCpxP plasmid that expresses cpxP from a heterologous trc promoter (13). The plasmid also encodes 44 bp of native 5′ cpxP sequence, including the native transcriptional start site but no sequence from the 3′ UTR region; transcription of cpxP is terminated by the plasmid-carried rrnB terminator.
CpxP is abundant and readily detectable by immunoblotting in cells carrying pCpxP (Fig. 1A). We then created a derivative plasmid that introduced the entire 145-bp cpxP 3′ intergenic region (spanning CpxQ), creating pCpxPQ. The cellular levels of CpxP were markedly reduced by the presence of the cpxQ sequence (Fig. 1A). We concluded that cpxQ causes reduced production of CpxP.
FIG 1 .

CpxQ reduces production of CpxP. (A) Anti-CpxP immunoblot of whole-cell CpxP levels in ΔcpxP::kan strains carrying multicopy plasmids. The upper band is a cross-reactive protein that serves as a loading control. (B) Recombination schematic for constructing ΔcpxQ and cpxQ+ strains at the native cpxPQ locus. (C) Anti-CpxP immunoblot of whole-cell CpxP levels produced from the cpxPQ wild-type locus (wt) or from bla-marked recombineered ΔcpxQ locus (Δ) and the isogenic control that contains cpxQ (+). The upper band is a cross-reactive protein that serves as a loading control. (D) Whole-cell β-galactosidase activity expressed from transcriptional LacZ reporters. The cpxP reporter is located away from the native locus and includes 410-nt upstream and 214-nt downstream sequences of the cpxP gene, relative to the translation start site. (cpxQ is not present in the reporter.) The data presented are means ± standard deviations from three experiments.
We have been unable to predict likely sites of RNA-RNA interaction between CpxQ and the cpxP transcript. However, sRNA regulation often involves binding to the 5′ end of mRNA and can include binding to sequences that specify the signal peptide (27, 28). Therefore, we altered the 5′ end of the cpxP transcript by replacing the native signal peptide-encoding sequence, but not the ribosome-binding site, with one from MalE, creating plasmids pMal-CpxP and pMal-CpxPQ. Because the signal peptide is cleaved after translocation, the mature CpxP produced from each of the plasmids remained unaltered. The pMal-CpxP plasmid produced levels of CpxP that were comparable to those produced from pCpxP (Fig. 1A). Notably, a heterologous signal sequence region abrogated the negative effect of CpxQ on CpxP production, and CpxP levels from pMal-CpxPQ were much higher than from pCpxPQ and comparable to those from pCpxP (Fig. 1A). Each of the plasmids expressed comparable amounts of cpxP transcript, as determined by quantitative reverse transcription-PCR (qRT-PCR) (see Fig. S1 in the supplemental material). These results suggested that CpxQ reduces CpxP production in a manner that does not increase cpxP mRNA degradation and relies on the native 5′ mRNA sequence. Most likely, CpxQ exerts translational control over CpxP production.
CpxQ lowers CpxP levels but does not alter CpxA activation.
The reduction in CpxP levels caused by CpxQ led us to consider whether this regulation contributes to the low levels of CpxP in wild-type cells by reducing mRNA or inhibiting cpxP mRNA translation directly or by some combination of direct and indirect effects. To address these questions under more physiological conditions, we recombineered the pCpxP and pCpxPQ constructs at the native cpxPQ locus to generate a ΔcpxQ deletion strain (pCpxP recombinant) and an isogenic control cpxQ+ strain (pCpxPQ recombinant). This recombination scheme preserved the native cpxP promoter and introduced a heterologous rrnB transcription terminator and a bla ampicillin resistance marker (Fig. 1B).
To monitor the indirect effect of CpxQ on CpxP-mediated CpxA activity, we used a degP′-lacZ+ transcriptional fusion that reports on the level of Cpx activation. CpxQ did not affect the extent of CpxA activation since we observed no changes in a degP expression from the degP′-lacZ+ reporter (Fig. 1D). Thus, the changes in CpxP levels are not the indirect result of changes in CpxA activity.
Consistent with previous reports, we were unable to detect CpxP in the wild-type cpxA+ background, but CpxP was easily detectable in the cpxA17 background (16). The recombineered cpxQ+ strain produced CpxP levels equivalent to the wild type (Fig. 1C). However, we observed increased CpxP protein levels in the recombineered ΔcpxQ cells (Fig. 1C). This suggests that CpxQ downregulates cpxP directly.
We used a LacZ transcriptional reporter to determine whether CpxQ was acting directly to destabilize the cpxP mRNA. The cpxP′-lacZ+ transcriptional reporter is located at a heterologous site in the chromosome and measures mRNA levels expressed from the cpxP promoter. As noted above, the signal sequence coding region (nucleotides [nt] 1 to 63 of the cpxP gene) was required for CpxQ to lower CpxP levels. The lacZ reporter includes 410 nt of sequence upstream of cpxP as well as the first 214 nt of the cpxP gene (8). We observed that cpxQ had no effect on the amount of LacZ produced from the reporter either in cpxA+ or in cpxA17 strains, suggesting that the abundance of the reporter mRNA is unchanged between strains (Fig. 1D). The results in this section confirm results obtained with plasmid constructs and demonstrate that CpxQ reduces CpxP levels by decreasing translation, not by destabilizing cpxP mRNA.
CpxQ is not involved in combating misfolded pilin stress.
The pilin subunits PapE and PapG are toxic when they are produced without their dedicated chaperone, PapD, and this periplasmic toxicity is suppressed by the cpxA* alleles. In this case, suppression requires Cpx regulon members DegP and CpxP (16). The increased production of CpxP that we observed in ΔcpxQ strains suggested that the sRNA could be involved in combating stress caused by misfolded pilin subunits. We envision two possibilities in a ΔcpxQ background: elevated CpxP levels could inhibit timely activation of CpxA to exacerbate toxicity, or alternatively, elevated CpxP levels could enhance clearance of misfolded pilins at the onset of stress. We constructed cpxA+ strains with or without cpxQ and carrying either pHJ8 or pHJ13 plasmids, which contain isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible papG or papE, respectively. Strains were inoculated in media containing IPTG (10 µM) to overproduce PapE or PapG, and growth was monitored. We could not detect a difference in sensitivity to either misfolded PapE or PapG when cpxQ was deleted (Fig. 2). This result seemed consistent with the modest increase of CpxP levels in ΔcpxQ strains (Fig. 1C). Surprisingly, CpxQ regulation of CpxP levels does not affect the ability of cells to combat a periplasmic stress in which CpxP directly participates. What, then, is the physiological role of CpxQ?
FIG 2 .

CpxQ does not combat misfolded pilin stress. Shown is growth of cpxA+ strains with either a deletion in cpxQ at the native chromosomal locus (ΔcpxQ) or an isogenic control locus that contains cpxQ (cpxQ+) during pilin subunit overexpression. The strains carry plasmids containing IPTG-inducible papE (pHJ13) and papG (pHJ8) and were cultured in media with (gray) or without (black) supplemented IPTG (10 µM).
CpxQ contributes to combating the IM stress caused by tethered LamB.
Induction of lamB(A23D) expression by maltose is toxic and causes cell death. While cpxA* mutations suppress the toxicity of tethered LamB(A23D), neither the underlying mechanisms nor the Cpx regulon members involved are clear (17, 29). To investigate if CpxQ contributes to cpxA* suppression, we constructed isogenic ΔcpxQ and cpxQ+ strains in the lamB(A23D) background, with and without the cpxA* suppressor, and performed maltose disc diffusion assays to measure the zones of growth inhibition caused by the inducer. As expected, the cpxA+ strains were highly sensitive to inducer, and the absence of CpxQ sRNA did not alter maltose sensitivity (Table 1). However, cpxA* ΔcpxQ strains displayed an intermediate maltose sensitivity phenotype, while the control cpxA* cpxQ+ strains remained fully maltose resistant (Table 1). Specifically, we observed zones of growth inhibition in cpxA* ΔcpxQ strains that were the same size as those in cpxA+ strains, but with less cell death within the zone (Fig. 3A). We confirmed that this effect of CpxQ was not due to lowered production or increased degradation of LamB(A23D) following maltose induction (Fig. 3B).
TABLE 1 .
Maltose sensitivity profile of lamB(A23D) strains
| Relevant strain background | Zone of inhibition (mm)a |
|||||
|---|---|---|---|---|---|---|
|
cpxA+ |
cpxA17 |
cpx24 |
||||
| cpxQ+ | ΔcpxQ | cpxQ+ | ΔcpxQ | cpxQ+ | ΔcpxQ | |
| lamB(A23D) | 21 | 20 | 0 | (21) | 0 | (21) |
| lamB(A23D) pTrc99A | 20 | 21 | 0 | (21) | NT | NT |
| lamB(A23D) pCpxP | 27 | 26 | 0 | (20) | NT | NT |
The zone of inhibition is the diameter of growth clearance minus the 6-mm disc. Zones of inhibition with incomplete clearance are given within parentheses. NT, not tested.
FIG 3 .
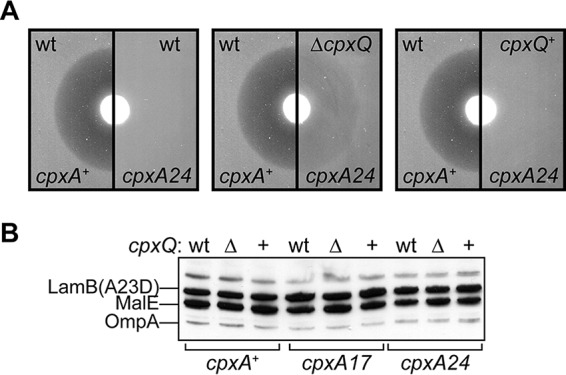
CpxQ is required for full suppression of lamB(A23D) by cpxA*. (A) Representative maltose disc diffusion assay images demonstrating zones of growth inhibition around a maltose disc. wt, native cpxPQ locus, compared to the deletion (ΔcpxQ) and its isogenic control (cpxQ+). (B) Combined anti-LamB and anti-MalE immunoblot of whole-cell levels of LamB(A23D) in cells grown in LB to mid-log phase and induced with 0.2% maltose for 60 min. Δ, bla-marked ΔcpxQ recombineered chromosomal deletion; +, the isogenic bla-marked recombineered control locus where cpxQ is present. OmpA is cross-reactive with anti-LamB and serves as a loading control. MalE serves as a control that measures the intensity of maltose induction.
CpxQ does not alleviate LamB(A23D) toxicity by reducing CpxP.
Overproduction of CpxP is known to exacerbate lamB(A23D) maltose sensitivity in cpxA+ strains (13). We observed above that ΔcpxQ strains produce increased levels of CpxP in a cpxA17 background. Perhaps this effect of CpxQ was responsible for increased maltose sensitivity in cpxA* ΔcpxQ strains? To test this, we introduced pCpxP and control pTrc99A plasmids into lamB(A23D) strains. The cpxA+ strains carrying pCpxP were indeed more maltose sensitive than the control strains (Table 1). However, in a cpxA17 background, pCpxP did not alter maltose sensitivity phenotypes: the cpxA17 cpxQ+ strain remained fully resistant, while the cpxA17 ΔcpxQ strain still displayed an intermediate maltose sensitivity (Table 1). Hence, elevated CpxP levels in cpxA* ΔcpxQ cells cannot account for the loss of suppression that occurs due to the absence of CpxQ. We conclude that cpxA*-mediated suppression of LamB(A23D) toxicity requires another CpxQ-regulated target.
CpxQ is not required to combat toxicity caused by periplasmic LacZ.
Though it is tethered to the IM, the LamB(A23D) protein is localized in the periplasm. The periplasmic chaperone protease DegP contributes to, but is not sufficient for, cpxA* suppression of lamB(A23D) (15). As noted above, in the case of the periplasmic toxicity caused by misfolded P-pilus subunits, DegP is required for suppression, but the misfolded subunits must be presented to the protease by CpxP for degradation to occur (16). We wondered if CpxQ might regulate additional regulators of DegP activity. DegP overproduction is both necessary and sufficient for suppression of the H*LamB-LacZ or the LamB-LacZX90 fusion proteins that cause periplasmic stress. Hence, we used these fusions as sensitive reporters of DegP activity. We introduced ΔcpxQ or control cpxQ+ alleles to cpxA+ and cpxA* strains expressing each fusion. Measuring inducer sensitivity, we observed that ΔcpxQ had no effect on the maltose sensitivity profiles of either fusion in the sensitive cpxA+ background or in the suppressed cpxA* backgrounds (Table 2). These data suggest that CpxQ involvement in lamB(A23D) suppression does not occur by regulating DegP.
TABLE 2 .
Maltose sensitivity profile of lamB-lacZ fusion strains
| Fusion | Zone of inhibition (mm)a |
|||||
|---|---|---|---|---|---|---|
|
cpxA+ |
cpxA17 |
cpx24 |
||||
| cpxQ+ | ΔcpxQ | cpxQ+ | ΔcpxQ | cpxQ+ | ΔcpxQ | |
| lamB-lacZX90 | 26 | 25 | 0 | 0 | 0 | 0 |
| H*lamB-lacZ | 13 | 13 | 0 | 0 | 0 | 0 |
| lamB-lacZ42-1 | 23 | 23 | 0 | 0 | 0 | 0 |
The zone of inhibition is the diameter of growth clearance minus the 6-mm disc.
CpxQ does not contribute to alleviating translocon jamming stress.
Jamming of the Sec translocon by the LamB-LacZ42-1 fusion protein is another stress that is suppressed by cpxA* alleles, and it is known that Cpx regulon member YccA contributes to this suppression by inhibiting the IM protease FtsH. To determine if these factors play a role in suppressing LamB(A23D), we constructed strains that expressed the LamB-LacZ42-1 fusion in both cpxA+ and cpxA* strains, each either lacking chromosomal cpxQ (ΔcpxQ) or with the control cpxQ+ locus. We then measured the maltose sensitivity of the strains. cpxA* alleles strongly suppress LamB-LacZ42-1 toxicity compared to cpxA+, but loss of cpxQ does not appreciably change the maltose sensitivity profile in either background (Table 2). We conclude that CpxQ is not involved in relieving jamming toxicity and that YccA and FtsH are not likely involved in the suppression of LamB(A23D).
CpxQ combats LamB(A23D) IM stress by regulating the periplasmic chaperone Skp.
During the course of this work, Chao and Vogel had identified the cellular mRNA targets of CpxQ in Salmonella that had not been detected in microarray experiments (30). Notable among these targets was the Na+/H+ antiporter, NhaB, and the periplasmic chaperone Skp. Removing NhaB from any of the lamB(A23D) strains neither increased nor decreased toxicity regardless of the presence or absence of CpxQ (data not shown).
To determine if CpxQ-dependent regulation of Skp was required for alleviation of LamB(A23D) stress, we proceeded to delete skp from the lamB(A23D) strains. We observed that cpxA* cpxQ+ cells remained fully maltose resistant in the absence of Skp (Table 3). Strikingly, however, Δskp restored full maltose resistance to cpxA* ΔcpxQ cells (Table 3; Fig. 4A) and also partially suppressed LamB(A23D) toxicity in cpxA+ strains (Table 3; Fig. 4A). Importantly, the effect of removing Skp in alleviating LamB(A23D) toxicity was not simply due to the loss of a periplasmic chaperone: loss of surA, which encodes the major periplasmic chaperone for outer membrane proteins (OMPs), had no effect on the maltose sensitivity of cpxA+ cells or the resistance of cpxA* cells (data not shown).
TABLE 3 .
Maltose sensitivity profile of lamB(A23D) strains lacking Skp
| Relevant strain background | Zone of inhibition (mm)a |
|||||
|---|---|---|---|---|---|---|
|
cpxA+ |
cpxA17 |
cpx24 |
||||
| cpxQ+ | ΔcpxQ | cpxQ+ | ΔcpxQ | cpxQ+ | ΔcpxQ | |
| lamB(A23D) | 21 | 20 | 0 | (21) | 0 | (21) |
| lamB(A23D) Δskp::kan | (18) | (18) | 0 | 0 | 0 | 0 |
The zone of inhibition is the diameter of growth clearance minus the 6-mm disc. Zones of inhibition with incomplete clearance are given within parentheses.
FIG 4 .
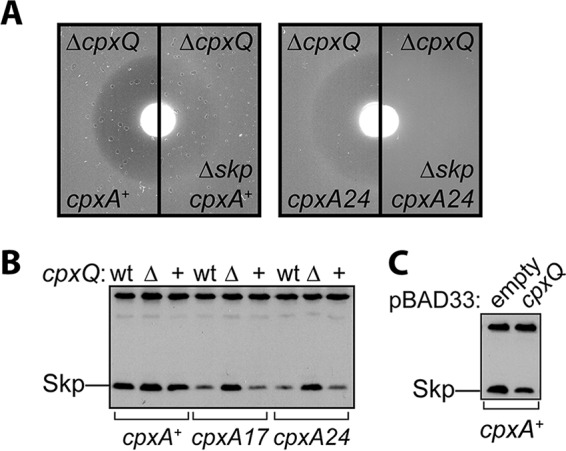
Loss of Skp suppresses LamB(A23D) toxicity and CpxQ acts by lowering Skp levels. (A) Representative maltose disc diffusion assay images demonstrating zones of growth inhibition around a disc containing maltose. (B) Immunoblotting of whole-cell Skp levels in lamB(A23D) strains. wt, wild-type cpxPQ locus; Δ, ΔcpxQ recombineered chromosomal deletion; +, isogenic recombineered control where cpxQ is present. (C) Immunoblotting of whole-cell Skp levels in cpxA+ plasmid carrying strains grown in the presence of 0.2% arabinose to induce expression of cloned cpxQ.
The fact that Δskp suppressed maltose sensitivity demonstrated that Skp is involved in promoting LamB(A23D) toxicity. Furthermore, the finding that Δskp restored maltose resistance to cpxA* ΔcpxQ cells strongly suggested that CpxQ contributes to combating LamB(A23D) toxicity by negatively regulating Skp. To test this hypothesis directly, we assessed Skp levels in lamB(A23D) strains by immunoblotting with anti-Skp antisera. In cpxA+ cells, Skp levels were abundant with or without cpxQ (Fig. 4B). On the other hand, the cpxA* mutations (where transcription from the cpxPQ promoter is strongly activated) resulted in markedly reduced Skp levels when cpxQ was present (Fig. 4B). In contrast, in cpxA* strains that lacked cpxQ Skp levels remained elevated and comparable to levels in cpxA+ strains (Fig. 4B). Furthermore, we were able to lower Skp levels in cpxA+ cells by overproducing CpxQ from an arabinose-inducible pBAD33 plasmid (Fig. 4C). Our data implicate Skp in contributing to the toxicity of LamB(A23D) and show that CpxQ negatively regulates Skp production when Cpx is activated.
Skp promotes LamB(A23D)-mediated activation of the Psp response.
In comparison with other examples of envelope toxicity, LamB(A23D) is unique in activating the Psp stress response; induction of lamB(A23D) causes elevated levels of PspA (17). Our data showed that Skp promotes LamB(A23D) toxicity, and introduction of Δskp in cpxA+ cells partially suppresses inducer sensitivity. We wondered if this effect of Δskp also lowered activation of the Psp response. We induced lamB(A23D) expression by growing cpxA+ cells in media supplemented with 0.2% maltose for 1 h and then assessed levels of PspA. As a control, we grew the same strains in media with 0.2% glucose to repress lamB(A23D) expression. We detected increased levels of PspA produced in cpxA+ skp+ cells treated with maltose, in agreement with prior observations (Fig. 5). In comparison, PspA levels remained low in cpxA+ Δskp cells (Fig. 5). We conclude that Skp promotes LamB(A23D) toxicity in a manner that activates the Psp response.
FIG 5 .
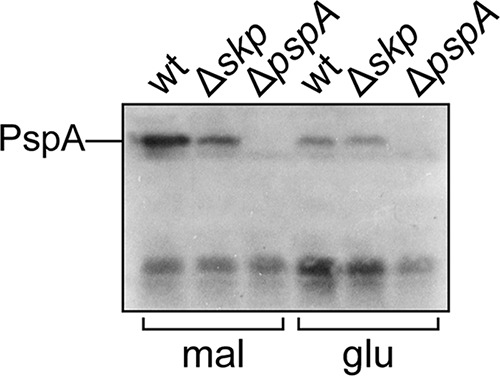
Skp is required for Psp activation in response to lamB(A23D) induction. Cultures of cpxA+ lamB(A23D) cells with (wt) or without (Δskp) skp were subcultured into media supplemented with either 0.2% maltose inducer (mal) or 0.2% glucose (glu) for 1 h. Cell lysates were then prepared and probed with anti-PspA antiserum.
DISCUSSION
Despite being produced from the same mRNA, our results show that CpxP protein and CpxQ sRNA mature to become effectors that combat distinct stresses in different compartments of the cell envelope. CpxP functions to alleviate P-pilus misfolding in the periplasm, but CpxQ combats stress at the IM modeled by the LamB(A23D) mutant protein. The lamB(A23D) mutation alters the signal peptidase cleavage site such that the translocated protein remains tethered to the IM by its uncleaved signal peptide. When production of LamB(A23D) is increased by maltose addition, toxicity is apparent. Importantly, toxicity requires production of the full-length LamB(A23D) protein. Amber mutations that cause C-terminal truncation of LamB abolish its ability to fold; when such mutations are combined with lamB(A23D), they fully relieve toxicity (17). Clearly, then, toxicity requires the C-terminal region either (i) because it is itself the direct cause of toxicity or (ii) because it is required for LamB(A23D) to remain folding competent. In support of the latter model, our findings demonstrate that the pro-folding factor Skp promotes toxicity. Hence, induction of the Psp response and toxicity likely arises from a Skp-dependent folding of tethered LamB(A23D) into the IM.
We suggest that Skp-dependent folding and insertion of LamB(A23D) into the IM creates an ion-conducting pore in the IM that disrupts the PMF. In support of this hypothesis, the Psp stress response, which is known to be activated by conditions that collapse the proton gradient across the IM (6, 31), is strongly stimulated by LamB(A23D) production (17). Indeed, filamentous phage produce pore-forming proteins that deplete PMF by this mechanism, and these are well-characterized inducers of the Psp response (6). Moreover, translocation-defective mutant OMPs or secretins lacking their pilotin also induce the Psp responses; in cells lacking the major effector PspA, these proteins are known to deplete PMF and cause toxicity (31–33). Since it is tethered to the IM, LamB(A23D) is perhaps uniquely potent in causing toxicity even in pspA+ cells.
It is clear that OMPs inherently possess the requisite structural information to fold and insert directly into membranes in vitro, provided that aggregation is prevented by denaturants such as urea (34). In vivo, following translocation, periplasmic chaperones such as SurA and Skp prevent the hydrophobic OMPs from aggregation and maintain them in a folding-competent state. From the periplasm, OMPs could fold and insert directly into the IM or the OM since either bilayer likely imposes the same energetic barrier (35). However, direct OMP folding and membrane insertion are slow; in vivo the BamABCDE complex within the OM lowers the kinetic barrier for OMP assembly and catalyzes folding and insertion into the correct membrane (35, 36).
In E. coli, SurA is the major chaperone for OMPs. Indeed, loss of SurA causes severe defects in OMP assembly, and cells survive the loss only because they are rescued by strong induction of the σE stress response. In contrast, there is no OMP that prefers Skp over SurA, and loss of Skp causes no OMP assembly defects and does not result in stress response induction. Skp does play a redundant role with FkpA in the assembly of LptD (37), but it functions primarily to rescue OMPs that have fallen off the normal assembly pathway (38). Since loss of Skp prevents folding and insertion of LamB(A23D) into the IM, we must conclude that this is a function that Skp does not share with SurA.
The fact that Skp can insert LamB(A23D) into the IM and SurA cannot is consistent with several experimental observations made previously. Most strikingly, Skp differs from SurA by its ability to promote OMP folding and insertion in vitro. SurA maintains OMPs in a folding-competent state, but membrane insertion requires the Bam complex (39–41). Skp, on the other hand, functions as a homotrimer with a large central cavity that can accommodate an entire unfolded OMP (42). Skp allows bound unfolded OMPs to undergo rapid conformational shifts, and it is sufficient to catalyze OMP insertion into membranes directly in vitro (41, 43–45).
Our findings indicate that OMPs can be assembled into membranes by Skp in vivo without participation from the OM Bam complex. In particular, our data suggest that Skp can assemble a β-barrel protein like LamB(A23D) that is tethered to the IM into the bilayer. Because high-level production of IM-tethered LamB is required to induce toxicity, it is likely that Skp cannot do this efficiently. We can detect it because only a few pores are required to inhibit cell growth. We do think it likely that Skp can assemble proteins directly into the OM as well. At present, we do not have an in vivo assay sensitive enough to detect this, but such an activity could rescue cells with defects in normal OMP biogenesis for one reason or another.
The Cpx response is an envelope stress response, and it seems counterintuitive that it would seek to lower Skp levels—why reduce the abundance of a chaperone that prevents aggregation of misfolded OMPs? We believe the râison d’être of such regulation is more apparent when considered in the context of mounting evidence that the primary responsibility of the Cpx response is to maintain IM homeostasis (5). Conditions that impede efficient OMP assembly into the OM trigger the σE response to promote recovery of OMP folding and assembly by overproducing the Bam complex and chaperones, including SurA and Skp (46). However, our findings suggest that Skp-OMP interactions can result in toxic OMP folding into the IM, and this mistargeting likely increases if Bam function is compromised. We suggest that this presents a challenge met by the Cpx response. To protect the IM, the Cpx response directly downregulates the abundant OMPs (47, 48) and negatively regulates the σE response (49), and as we show here, the Cpx response directly reduces the production of Skp via the CpxQ sRNA. It is tempting to think that this Cpx-σE cross-regulatory axis is designed to allow an initial attempt at recovery from OMP stress, which can then overridden by Cpx seeking to protect IM integrity so that energy generation can continue. Notably, a ΔcpxR mutation causes conditional lethality in strains where OMP biogenesis is compromised (47).
Chao and Vogel demonstrate in Salmonella that CpxQ represents the first bacterial trans-acting global regulatory sRNA that is produced from the 3′ UTR of an mRNA (30). We have no reason to believe that biogenesis of CpxQ differs in E. coli. However, we did observe that ΔcpxQ results in higher levels of CpxP in E. coli, an effect not seen in Salmonella. Apparently there are differences in sRNA regulation of the Cpx regulon between E. coli and Salmonella. This is not uncommon; an instructive example is the loss of the RNA chaperone Hfq, which triggers activation of Cpx responses in enteropathogenic E. coli strains but has no effect in E. coli K-12 strains (50). Our data suggest that the 5′ sequence of the cpxPQ mRNA is required for CpxQ-mediated regulation of CpxP production. However, the physiological significance of this regulation is not immediately clear. In fact, in response to pilin misfolding, a stress condition known to require CpxP, we show that ΔcpxQ has no effect. More work is required to understand why E. coli strives to keep the levels of CpxP so low.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
All strains and plasmids used in this study are listed in Table S1 in the supplemental material. Strains were routinely maintained in Luria medium (Miller), except for the lamB-lacZ and lamB(A23D) strains, which were maintained in M63 minimal medium supplemented with 0.2% (wt/vol) glucose at all times. cpxA* strains were maintained at 30°C in the presence of amikacin (1.5 µg/ml). The Δskp::kan allele was obtained from the Keio collection (51).
Plasmid construction.
All oligonucleotides used in this study are listed in Table S2 in the supplemental material. The plasmid pCpxP was constructed by cloning cpxP amplified with primers cpxP5′Eco and mg_pCpxP into the EcoRI and HindIII sites of pTrc99A (13). The cpxP 3′ UTR was cloned by amplifying cpxPQ with primers CpxP_intF_BglII and cpxP_3_HindIII and cloning the product into the BglII and HindIII sites of pCpxP. The malE signal sequence was amplified with primers OE_MalE_F and ssMalE_R and cloned by overlap-extension PCR with pMG95. The mal-cpxP construct was then subcloned from the pMG95 derivative, using EcoRI and HindIII, into the same sites of pTrc99A, creating pMal-CpxP.
Chromosomal cpxPQ constructs.
To facilitate recombineering, a pCpxPQ equivalent plasmid was constructed with a shorter 3′ UTR region (to the promoter of fieF) that was cloned from an amplicon of cpxP5′Eco and cpxP_3s_HindIII.
To generate double-stranded DNA (dsDNA) for recombineering, the cpxPQ locus was PCR amplified from pCpxP and pCpxPQ with primers cpxP_5 and primer cpxP_3. The PCR products were used to transform strain DK10 (DY378 ΔcpxP::kan), selecting for Ampr transformants and then screening for Kans. Successful recombination generated cpxP::rrnB-bla and cpxPQ::rrnB-bla alleles. Kanamycin-resistant alleles were generated by targeting the bla gene for a second recombination when the constructed strains were transformed with dsDNA from an amplification of pKD4 (52) with primers Chr_Amp2Kan_p1 and Chr_Amp2Kan_p2. Recombinants were selected for Kanr and screened for Amps.
Both Ampr and Kanr alleles were moved routinely by P1vir transduction. Because the cpxA and cpxP loci are tightly linked, to facilitate strain construction involving cpxA* alleles, the Ampr or Kanr constructs were first linked to a cpxA::cam allele for to cotransduction. Transductants of cpxA* strains were selected for Ampr/Kanr and then screened for Cams.
β-Galactosidase assays.
Overnight cultures subcultured 1:50 into LB. Cultures were grown for 1 to 2 h to mid-log phase. Equivalent A600 cell densities were taken, pelleted, and permeabilized using chloroform and SDS. Measurement of β-galactosidase activity for ortho-nitrophenyl-β-d-galactopyranoside (ONPG) hydrolysis was performed in triplicate (53), measuring spectrophotometric readings each minute during a 15-min time course, and the Vmax was calculated.
Maltose disc diffusion assay.
Overnight cultures were mixed with 3 ml of molten M63 medium top agar (agar at 0.75% wt/vol) supplemented with 0.2% (wt/vol) glycerol, spread onto a plate of M63 glycerol (agar at 1.5% wt/vol), and allowed to solidify. Filter discs infused with 10 ml of 20% maltose were placed in the center to the plate. Plates were incubated upright at 30°C overnight, and the diameters of zones of growth inhibition were measured.
Immunoblotting.
Cultures were grown to mid-log phase, and samples were standardized by A600. Aliquots were taken, pelleted, and resuspended in Laemmli buffer. Samples were first boiled and then resolved by SDS-PAGE before being transferred to nitrocellulose membranes. Immobilized samples were probed with polyclonal rabbit anti-CpxP (1:5,000), anti-LamB (1:30,000), or anti-Skp (1:8,000) antisera or anti-PspA (raised against the Yersinia enterocolitica protein [1:5,000]) as indicated. Membranes were subsequently washed, incubated with donkey anti-rabbit secondary antibody conjugated to horseradish peroxidase (used at 1:10,000), and developed with enhanced chemiluminescence substrate (Amersham). Blots were visualized by exposure to X-ray film (Denville).
SUPPLEMENTAL MATERIAL
Relative abundance of cpxP mRNA produced from plasmid constructs. Plasmid-carrying ΔcpxP::kan strains were grown to the mid-log phase. Total RNA was isolated (RNeasy; Qiagen) and used to prepare cDNA. qRT-PCR was performed using primers cpxP_qRT_F (5′ CTCCTGTTAATGTTAGCGAACTGG 3′) and cpxP_qRT_R (5′ ATTCGTTGTTGATGTTTCTCGTTT 3′), which amplify a 208-bp product within the cpxP open reading frame (ORF). Fold change was calculated by the threshold cycle (ΔΔCT) method. Data represent two biological replicates. Download
Strains and plasmids used in this study.
Oligonucleotides used in this study.
ACKNOWLEDGMENTS
We thank Jörg Vogel for generously sharing data prior to publication, Andrew Darwin for kindly providing anti-PspA antisera, Tracy Raivio for critical reading of the manuscript, and members of the Silhavy lab for their comments on the manuscript.
This work was supported by National Institute General Medical Sciences grant GM34821 (to T.J.S).
Footnotes
Citation Grabowicz M, Koren D, Silhavy TJ. 2016. The CpxQ sRNA negatively regulates Skp to prevent mistargeting of β-barrel outer membrane proteins into the cytoplasmic membrane. mBio 7(2):e00312-16. doi:10.1128/mBio.00312-16.
REFERENCES
- 1.Ruiz N, Kahne D, Silhavy TJ. 2006. Advances in understanding bacterial outer-membrane biogenesis. Nat Rev Microbiol 4:57–66. doi: 10.1038/nrmicro1322. [DOI] [PubMed] [Google Scholar]
- 2.Ruiz N, Silhavy TJ. 2005. Sensing external stress: watchdogs of the Escherichia coli cell envelope. Curr Opin Microbiol 8:122–126. doi: 10.1016/j.mib.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Ades SE. 2008. Regulation by destruction: design of the σ envelope stress response. Curr Opin Microbiol 11:535–540. doi: 10.1016/j.mib.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 4.Lima S, Guo MS, Chaba R, Gross CA, Sauer RT. 2013. Dual molecular signals mediate the bacterial response to outer-membrane stress. Science 340:837–841. doi: 10.1126/science.1235358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raivio TL. 2014. Everything old is new again: an update on current research on the Cpx envelope stress response. Biochim Biophys Acta 1843:1529–1541. doi: 10.1016/j.bbamcr.2013.10.018. [DOI] [PubMed] [Google Scholar]
- 6.Darwin AJ. 2005. The phage-shock-protein response. Mol Microbiol 57:621–628. doi: 10.1111/j.1365-2958.2005.04694.x. [DOI] [PubMed] [Google Scholar]
- 7.Hunke S, Keller R, Müller VS. 2012. Signal integration by the Cpx-envelope stress system. FEMS Microbiol Lett 326:12–22. doi: 10.1111/j.1574-6968.2011.02436.x. [DOI] [PubMed] [Google Scholar]
- 8.Raivio TL, Silhavy TJ. 1997. Transduction of envelope stress in Escherichia coli by the Cpx two-component system. J Bacteriol 179:7724–7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fleischer R, Heermann R, Jung K, Hunke S. 2007. Purification, reconstitution, and characterization of the CpxRAP envelope stress system of Escherichia coli. J Biol Chem 282:8583–8593. doi: 10.1074/jbc.M605785200. [DOI] [PubMed] [Google Scholar]
- 10.Price NL, Raivio TL. 2009. Characterization of the Cpx regulon in Escherichia coli strain MC4100. J Bacteriol 191:1798–1815. doi: 10.1128/JB.00798-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raivio TL, Laird MW, Joly JC, Silhavy TJ. 2000. Tethering of CpxP to the inner membrane prevents spheroplast induction of the Cpx envelope stress response. Mol Microbiol 37:1186–1197. doi: 10.1046/j.1365-2958.2000.02074.x. [DOI] [PubMed] [Google Scholar]
- 12.Tschauner K, Hörnschemeyer P, Müller VS, Hunke S. 2014. Dynamic interaction between the CpxA sensor kinase and the periplasmic accessory protein CpxP mediates signal recognition in E. coli. PLoS One 9:e107383. doi: 10.1371/journal.pone.0107383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raivio TL, Popkin DL, Silhavy TJ. 1999. The Cpx envelope stress response is controlled by amplification and feedback inhibition. J Bacteriol 181:5263–5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Danese PN, Snyder WB, Cosma CL, Davis LJ, Silhavy TJ. 1995. The Cpx two-component signal transduction pathway of Escherichia coli regulates transcription of the gene specifying the stress-inducible periplasmic protease, DegP. Genes Dev 9:387–398. doi: 10.1101/gad.9.4.387. [DOI] [PubMed] [Google Scholar]
- 15.Cosma CL, Danese PN, Carlson JH, Silhavy TJ, Snyder WB. 1995. Mutational activation of the Cpx signal transduction pathway of Escherichia coli suppresses the toxicity conferred by certain envelope-associated stresses. Mol Microbiol 18:491–505. doi: 10.1111/j.1365-2958.1995.mmi_18030491.x. [DOI] [PubMed] [Google Scholar]
- 16.Isaac DD, Pinkner JS, Hultgren SJ, Silhavy TJ. 2005. The extracytoplasmic adaptor protein CpxP is degraded with substrate by DegP. Proc Natl Acad Sci U S A 102:17775–17779. doi: 10.1073/pnas.0508936102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carlson JH, Silhavy TJ. 1993. Signal sequence processing is required for the assembly of LamB trimers in the outer membrane of Escherichia coli. J Bacteriol 175:3327–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Silhavy TJ, Shuman HA, Beckwith J, Schwartz M. 1977. Use of gene fusions to study outer membrane protein localization in Escherichia coli. Proc Natl Acad Sci U S A 74:5411–5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Stelten J, Silva F, Belin D, Silhavy TJ. 2009. Effects of antibiotics and a proto-oncogene homolog on destruction of protein translocator SecY. Science 325:753–756. doi: 10.1126/science.1172221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Snyder WB, Silhavy TJ. 1995. β-Galactosidase is inactivated by intermolecular disulfide bonds and is toxic when secreted to the periplasm of Escherichia coli. J Bacteriol 177:953–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bowers CW, Lau F, Silhavy TJ. 2003. Secretion of LamB-LacZ by the signal recognition particle pathway of Escherichia coli. J Bacteriol 185:5697–5705. doi: 10.1128/JB.185.19.5697-5705.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dwyer RS, Malinverni JC, Boyd D, Beckwith J, Silhavy TJ. 2014. Folding LacZ in the periplasm of Escherichia coli. J Bacteriol 196:3343–3350. doi: 10.1128/JB.01843-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sauer FG, Remaut H, Hultgren SJ, Waksman G. 2004. Fiber assembly by the chaperone-usher pathway. Biochim Biophys Acta 1694:259–267. doi: 10.1016/j.bbamcr.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 24.Hung DL, Raivio TL, Jones CH, Silhavy TJ, Hultgren SJ. 2001. Cpx signaling pathway monitors biogenesis and affects assembly and expression of P pili. EMBO J 20:1508–1518. doi: 10.1093/emboj/20.7.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou X, Keller R, Volkmer R, Krauss N, Scheerer P, Hunke S. 2011. Structural basis for two-component system inhibition and pilus sensing by the auxiliary CpxP protein. J Biol Chem 286:9805–9814. doi: 10.1074/jbc.M110.194092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chao Y, Papenfort K, Reinhardt R, Sharma CM, Vogel J. 2012. An atlas of Hfq-bound transcripts reveals 3′ UTRs as a genomic reservoir of regulatory small RNAs. EMBO J 31:4005–4019. doi: 10.1038/emboj.2012.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bossi L, Figueroa-Bossi N. 2007. A small RNA downregulates LamB maltoporin in Salmonella. Mol Microbiol 65:799–810. doi: 10.1111/j.1365-2958.2007.05829.x. [DOI] [PubMed] [Google Scholar]
- 28.Gogol EB, Rhodius VA, Papenfort K, Vogel J, Gross CA. 2011. Small RNAs endow a transcriptional activator with essential repressor functions for single-tier control of a global stress regulon. Proc Natl Acad Sci USA 108:12875–12880. doi: 10.1073/pnas.1109379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cosma CL, Crotwell MD, Burrows SY, Silhavy TJ. 1998. Folding-based suppression of extracytoplasmic toxicity conferred by processing-defective LamB. J Bacteriol 180:3120–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chao Y, Vogel J. 2016. A 3′ UTR-derived small RNA provides the regulatory noncoding arm of the inner membrane stress response. Mol Cell 61:352–363. doi: 10.1016/j.molcel.2015.12.023. [DOI] [PubMed] [Google Scholar]
- 31.Kleerebezem M, Crielaard W, Tommassen J. 1996. Involvement of stress protein PspA (phage shock protein A) of Escherichia coli in maintenance of the protonmotive force under stress conditions. EMBO J 15:162–171. [PMC free article] [PubMed] [Google Scholar]
- 32.Kleerebezem M, Tommassen J. 1993. Expression of the pspA gene stimulates efficient protein export in Escherichia coli. Mol Microbiol 7:947–956. doi: 10.1111/j.1365-2958.1993.tb01186.x. [DOI] [PubMed] [Google Scholar]
- 33.Guilvout I, Chami M, Engel A, Pugsley AP, Bayan N. 2006. Bacterial outer membrane secretin PulD assembles and inserts into the inner membrane in the absence of its pilotin. EMBO J 25:5241–5249. doi: 10.1038/sj.emboj.7601402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Surrey T, Jähnig F. 1992. Refolding and oriented insertion of a membrane protein into a lipid bilayer. Proc Natl Acad Sci U S A 89:7457–7461. doi: 10.1073/pnas.89.16.7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gessmann D, Chung YH, Danoff EJ, Plummer AM, Sandlin CW, Zaccai NR, Fleming KG. 2014. Outer membrane β-barrel protein folding is physically controlled by periplasmic lipid head groups and BamA. Proc Natl Acad Sci USA 111:5878–5883. doi: 10.1073/pnas.1322473111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ricci DP, Silhavy TJ. 2012. The Bam machine: a molecular cooper. Biochim Biophys Acta 1818:1067–1084. doi: 10.1016/j.bbamem.2011.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwalm J, Mahoney TF, Soltes GR, Silhavy TJ. 2013. Role for Skp in LptD assembly in Escherichia coli. J Bacteriol 195:3734–3742. doi: 10.1128/JB.00431-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sklar JG, Wu T, Kahne D, Silhavy TJ. 2007. Defining the roles of the periplasmic chaperones SurA, Skp, and DegP in Escherichia coli. Genes Dev 21:2473–2484. doi: 10.1101/gad.1581007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hagan CL, Kim S, Kahne D. 2010. Reconstitution of outer membrane protein assembly from purified components. Science 328:890–892. doi: 10.1126/science.1188919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hagan CL, Kahne D. 2011. The reconstituted Escherichia coli Bam complex catalyzes multiple rounds of β-barrel assembly. Biochemistry 50:7444–7446. doi: 10.1021/bi2010784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McMorran LM, Bartlett AI, Huysmans GH, Radford SE, Brockwell DJ. 2013. Dissecting the effects of periplasmic chaperones on the in vitro folding of the outer membrane protein PagP. J Mol Biol 425:3178–3191. doi: 10.1016/j.jmb.2013.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walton TA, Sousa MC. 2004. Crystal structure of Skp, a prefoldin-like chaperone that protects soluble and membrane proteins from aggregation. Mol Cell 15:367–374. doi: 10.1016/j.molcel.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 43.Patel GJ, Behrens-Kneip S, Holst O, Kleinschmidt JH. 2009. The periplasmic chaperone Skp facilitates targeting, insertion, and folding of OmpA into lipid membranes with a negative membrane surface potential. Biochemistry 48:10235–10245. doi: 10.1021/bi901403c. [DOI] [PubMed] [Google Scholar]
- 44.Burmann BM, Wang C, Hiller S. 2013. Conformation and dynamics of the periplasmic membrane-protein-chaperone complexes OmpX-Skp and tOmpA-Skp. Nat Struct Mol Biol 20:1265–1272. doi: 10.1038/nsmb.2677. [DOI] [PubMed] [Google Scholar]
- 45.Bulieris PV, Behrens S, Holst O, Kleinschmidt JH. 2003. Folding and insertion of the outer membrane protein OmpA is assisted by the chaperone Skp and by lipopolysaccharide. J Biol Chem 278:9092–9099. doi: 10.1074/jbc.M211177200. [DOI] [PubMed] [Google Scholar]
- 46.Rhodius VA, Suh WC, Nonaka G, West J, Gross CA. 2006. Conserved and variable functions of the σ stress response in related genomes. PLoS Biol 4:e2. doi: 10.1371/journal.pbio.0040002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gerken H, Leiser OP, Bennion D, Misra R. 2010. Involvement and necessity of the Cpx regulon in the event of aberrant β-barrel outer membrane protein assembly. Mol Microbiol 75:1033–1046. doi: 10.1111/j.1365-2958.2009.07042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Batchelor E, Walthers D, Kenney LJ, Goulian M. 2005. The Escherichia coli CpxA-CpxR envelope stress response system regulates expression of the porins OmpF and OmpC. J Bacteriol 187:5723–5731. doi: 10.1128/JB.187.16.5723-5731.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Wulf P, McGuire AM, Liu X, Lin EC. 2002. Genome-wide profiling of promoter recognition by the two-component response regulator CpxR-P in Escherichia coli. J Biol Chem 277:26652–26661. doi: 10.1074/jbc.M203487200. [DOI] [PubMed] [Google Scholar]
- 50.Vogt SL, Raivio TL. 2014. Hfq reduces envelope stress by controlling expression of envelope-localized proteins and protein complexes in enteropathogenic Escherichia coli. Mol Microbiol 92:681–697. doi: 10.1111/mmi.12581. [DOI] [PubMed] [Google Scholar]
- 51.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Relative abundance of cpxP mRNA produced from plasmid constructs. Plasmid-carrying ΔcpxP::kan strains were grown to the mid-log phase. Total RNA was isolated (RNeasy; Qiagen) and used to prepare cDNA. qRT-PCR was performed using primers cpxP_qRT_F (5′ CTCCTGTTAATGTTAGCGAACTGG 3′) and cpxP_qRT_R (5′ ATTCGTTGTTGATGTTTCTCGTTT 3′), which amplify a 208-bp product within the cpxP open reading frame (ORF). Fold change was calculated by the threshold cycle (ΔΔCT) method. Data represent two biological replicates. Download
Strains and plasmids used in this study.
Oligonucleotides used in this study.